Миоклонус-эпилепсия MERRF: причины, диагностика, лечение
Добавил пользователь Владимир З. Обновлено: 21.01.2026
Что такое юношеская миоклоническая эпилепсия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Аграновича Андрея Олеговича, невролога со стажем в 12 лет.
Над статьей доктора Аграновича Андрея Олеговича работали литературный редактор Вера Васина , научный редактор Татьяна Гаврилова и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Юношеской миоклонической эпилепсией (синдромом Янца) называют эпилептический синдром, который проявляется внезапными подёргиваниями в мышцах — миоклоническими приступами (от греч. "myos" — мышца, "klonos" — беспорядочное движение). Заболевание обычно развивается в подростковом возрасте.
Подёргивания в первую очередь возникают в мышцах верхнего плечевого пояса и рук. Сначала пациенты не обращают на них внимания, но со временем эпизоды возникают всё чаще и ухудшают качество жизни. Например, во время приступов из рук могут выпадать предметы. В дальнейшем появляются подёргивания ног, из-за которых человек может упасть.
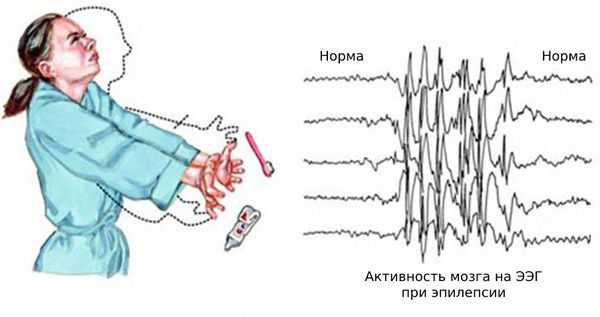
Нередко к этим эпизодам присоединяются генерализованные судорожные приступы — судороги возникают по всему телу и пациент теряет сознание. Также возникают абсансы — бессудорожные приступы с отключением сознания и амнезией на этот период. Как правило, частота генерализованных приступов невысокая: от одного за всю жизнь до раза в месяц. Подёргивания обычно случаются утром после пробуждения. Ярким провоцирующим фактором может стать недосыпание или вынужденное пробуждение. Также в трети случаев отмечается фотосенситивность — чувствительность к ритмическим вспышкам света.
Распространённость
Юношеская миоклоническая эпилепсия составляет 5-10 % среди всех эпилепсий и чуть больше четверти среди генетических генерализованных эпилепсий [5] . Заболевание проявляется в возрасте от 7 до 21 года, чаще в 11-15 лет, и более распространено среди женщин (61 %) [2] .
Причины заболевания
По классификации Международной противоэпилептической лиги за 2017 год, юношеская миоклоническая эпилепсия относится к генетическим болезням [1] . Заболевание имеет полигенное наследование, то есть контролируется двумя или более генами. Его развитие связано с локусами (участками ДНК): 6p11-12 (EJM1), 15q14 (EJM2), 6р21 (EJM3), 5q12-q14 (EJM4), 5q34-q35 (EJM5), 2q22-q23 (EJM6), 1p36 (EJM7), 3q26 (EJM8), 2q33-q36 (EJM9). Выделить ген, сильнее всего влияющий на развитие заболевание, пока не удалось [3] [4] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы юношеской миоклонической эпилепсии
Основным симптомом заболевания являются миоклонические вздрагивания (миоклонии) — патологические непроизвольные сокращения мышц или их групп [7] . Во время приступа пациенты испытывают ощущение, похожее на лёгкий удар током. В ответ на него возникают молниеносные движения в мышцах: от лёгкого подёргивания кончиков пальцев до патологического вздрагивания всего тела, которое может привести к падению.
Чаще всего подёргивания возникают в верхнем плечевом поясе: мышцах рук и плеч с обоих сторон. Из-за этого пациенты нередко выпускают предметы из рук, например разбивают кружки и роняют зубные щётки. Однако возможны различные вариации миоклоний.
Приступы учащаются в утренние часы, особенно при недосыпе или вынужденном пробуждении.
В 90 % случаев, помимо миоклонических эпизодов, отмечаются и генерализованные судорожные приступы [6] . После серии вздрагиваний в патологический процесс часто вовлекаются обе стороны тела.
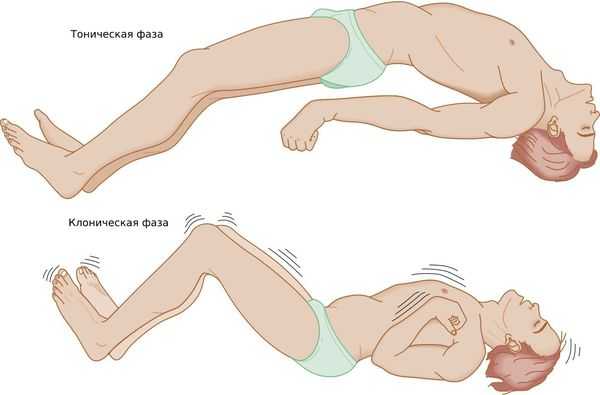
Генерализованный тонико-клонический приступ — состояние, при котором полностью отключается сознание. Приступ начинается с тонической фазы: напряжения в мышцах и специфического вскрикивания или хрипения. Руки полусогнуты и приподняты вверх или прижаты к телу. В этот момент из-за спазма дыхательной мускулатуры меняется цвет лица: оно синеет или сереет.
Далее развивается клоническая фаза, которая проявляется ритмичными подёргиваниями в конечностях. Она завершается полным мышечным расслаблением.
Третий вид эпилептических приступов при юношеской миоклонической эпилепсии — это абсансы [8] . Во время эпизода больной застывает, его взгляд устремлён в одну точку, сознание отключено. Состояние длится до 15 секунд и часто воспринимается окружающими как задумчивость. Сами пациенты могут не замечать эти приступы или воспринимать их как "провалы в памяти".
Патогенез юношеской миоклонической эпилепсии
Мозг человека состоит из двух основных типов клеток: нейронов и глии. Нейроны — это электрически возбудимые клетки, которые обрабатывают, хранят и передают информацию с помощью электрических и химических сигналов. Глиальные клетки играют в этом процессе вспомогательную роль.
Нейроны могут соединяться друг с другом и образовывать нервные сети. В пределах одного нейрона и его отростков информация передаётся в виде электрического возбуждения. В синапсе (месте контакта между нервными клетками) оно приводит к выделению различных химических веществ — нейромедиаторов.
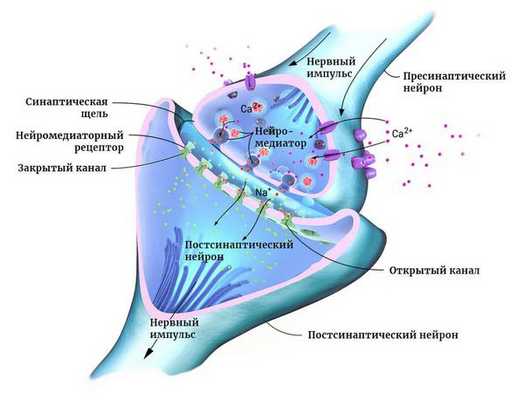
Нейромедиатор взаимодействует с рецепторами на мембране следующего нейрона. В результате в нём возникает электрическое возбуждение. Или не возникает — это зависит от конкретного нейромедиатора, активного в данный момент.
Чтобы заряд менялся в сторону возбуждения, в клетку должны поступать положительно и отрицательно заряженные ионы. Они проходят через ионные каналы в её мембране. Ионными каналами называют белки, образующие п о́ ру для обмена клетки с окружающей средой ионами K+, Na+ и другими [9] .
В нервных сетях между возбуждением и торможением работы нейронов поддерживается постоянный баланс. При сдвиге равновесия в сторону возбуждения происходит эпилептический приступ.
К юношеской миоклонической эпилепсии приводят мутации в генах ионных каналов. Однако выявлены нарушения и в других генах, также влияющих на процессы возбуждения в головном мозге [4] .
Классификация и стадии развития юношеской миоклонической эпилепсии
В Международной классификации болезней (МКБ-10) юношеская миоклоническая эпилепсия шифруется кодом G40.3 [10] .
В 2017 году Международная лига борьбы с эпилепсией (ILAE) обновила классификацию заболевания, выделив четыре уровня диагностики:
1. Определить тип приступа: фокальный (возникающий из одного очага), генерализованный и с неизвестным началом. Миоклонические, тонико-клонические приступы и абсансы относятся к генерализованным приступам.
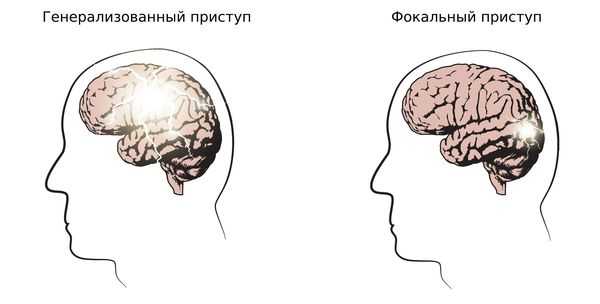
2. Установить тип эпилепсии: фокальная, генерализованная, сочетанная (фокальная + генерализованная) и неизвестная. Юношеская миоклоническая эпилепсия относится к генерализованной эпилепсии.
3. Определить эпилептический синдром. Юношеская миоклоническая эпилепсия как раз и является синдромом. Синдром включает типы приступов, возраст дебюта заболевания, характерные изменения на ЭЭГ, провоцирующие факторы и зачастую прогноз заболевания. Все эти факторы определяют лечебную тактику [11] .
4. Выявить причины заболевания: генетические, структурные, метаболические, иммунные, инфекционные и с неизвестной этиологией. Юношеская миоклоническая эпилепсия в большинстве случаев вызвана генетическими факторами.
Классификация юношеской миоклонической эпилепсии проводится в зависимости от течения заболевания. Главный критерий — это наличие миоклонических приступов. Также выделяют варианты течения с добавлением генерализованных судорожных приступов и/или абсансов.
Осложнения юношеской миоклонической эпилепсии
Пациенты часто не обращают внимания на патологические сокращения мышц, поэтому к неврологу и эпилептологу больной зачастую обращается после появления генерализованных тонико-клонических приступов. В результате противоэпилептические препараты назначают с опозданием. На фоне этого приступы могут учащаться и угрожать здоровью и жизни пациента травмами и утоплениями.
Серьёзным осложнением эпилепсии, в том числе и юношеской миоклонической эпилепсии, является внезапная смерть (SUDEP — Sudden Unexpected Death EPilepsy). Среди людей, страдающих эпилепсией, риск внезапной смерти в 20 раз выше, чем среди населения в целом [12] .
Точные причины SUDEP не установлены. Предполагается, что гибель пациентов связана с нарушением дыхания и развитием аритмии после приступа. Вероятность внезапной смерти при эпилепсии повышается при наличии генерализованных тонико-клонических приступов. Также важно, когда заболевание проявилось и сколько оно длится [12] .
При наличии дневных генерализованных приступов в течение предыдущего года риск развития SUDEP возрастает в 27 раз, ночных — в 15 раз. Проживание в одиночестве повышает риск внезапной смерти в 5 раз. Также SUDEP чаще встречается при злоупотреблении психоактивными веществами и алкоголем [13] .
Снизить риск внезапной смерти при эпилепсии можно, если придерживаться назначенного лечения: не пропускать приём противоэпилептических препаратов, не менять самостоятельно его частоту и дозировку [14] [15] .
Диагностика юношеской миоклонической эпилепсии
Основной диагностический критерий заболевания — это наличие миоклонических приступов.
Сбор анамнеза
На приёме врач спрашивает о необычных внезапных состояниях:
- вздрагиваниях в теле;
- дежавю — состоянии, при котором человек ощущает, что когда-то уже был в подобной ситуации или месте;
- потере сознания и т. д.
Пациенты могут не обращать внимания на такие симптомы и считать их своей особенностью. Абсансы и генерализованные тонико-клонические приступы с потерей сознания, особенно во сне, они могут и вовсе забывать. Поэтому при сборе анамнеза важно выяснить обстоятельства приступа не только у самих пациентов, но и у родственников и очевидцев.
Электроэнцефалограмма (ЭЭГ)
Основным способом диагностики эпилепсии является электроэнцефалограмма — метод исследования, при котором регистрируется суммарная электрическая активность клеток коры головного мозга.
Сейчас диагноз "эпилепсия" устанавливают с помощью длительного видео-ЭЭГ мониторинга — электроэнцефалограмма записывается параллельно с одной или несколькими видеокамерами, датчиком ЭКГ и при необходимости дополнительным контролем мышечной активности, частоты и глубины дыхания.

Основной фон биоэлектрической активности при юношеской миоклонической эпилепсии, как правило, соответствует возрастной норме. Патологическая активность проявляется короткими и генерализованными разрядами полиспайков (островолновых комплексов), которые регистрируются при миоклонических вздрагиваниях и полипик-волновыми комплексами между приступами.
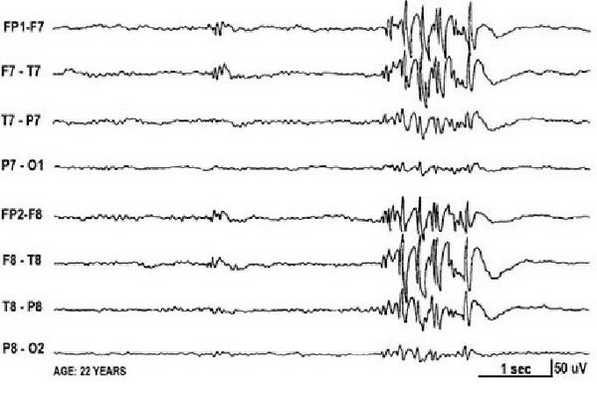
При заболевании часто встречается феномен фотосенситивности. Для её выявления во время ЭЭГ пациента просят закрыть глаза и проводят ритмичную фотостимуляцию с частотой около 15 Гц [16] .
Эпилептическая фотосенситивность — это предрасположенность к приступам под влиянием света. Может протекать бессимптомно или проявляться эпилептическими приступами под воздействием провоцирующих факторов: видеоигр, работы за компьютером, просмотра телевизора, мигающего освещения в ночных клубах и света природного происхождения.
На МРТ патологические изменения в головном мозге при юношеской миоклонической эпилепсии не выявляются [17] .
Интеллект и неврологический статус при заболевании находятся в норме. Выражена эмоциональная неустойчивость и признаки невротического развития личности: резкая смена настроения, вспыльчивость и повышенная тревожность
Лечение юношеской миоклонической эпилепсии
Образ жизни
При эпилепсии следует соблюдать режим сна и бодрствования, исключить алкоголь и избегать резких вспышек света. Также нужно по возможности уменьшить стрессы, переживания и тревоги [20] .
Антиэпилептические препараты
Приём антиэпилептических препаратов (АЭП) позволяет устранить до 90 % приступов. Монотерапия (лечение одним препаратом) при юношеской миоклонической эпилепсии применяется в 79 % случаев, дуотерапия (двумя препаратами) — 17 %, политерапия (несколькими препаратами) — 4 % [16] .
Прекращать приём лекарств рекомендуется не ранее чем через пять лет полной клинико-нейрофизиологической ремиссии. Но даже спустя 4-7 лет ремиссии рецидивы после отмены терапии возникают у 70 % больных. Поэтому пациентам с юношеской миоклонической эпилепсией может быть рекомендован пожизненный приём АЭП [21] .

Ранее лидерами в лечении юношеской миоклонической эпилепсии являлись препараты вальпроевой кислоты. Они эффективны для прекращения приступов, но вызывают много побочных эффектов:
Также выявлено, что они обладают повышенным тератогенным эффектом по сравнению с другими АЭП. Тератогенное действие — это нарушение эмбрионального развития ребёнка при приёме препаратов матерью. Поэтому назначение вальпроатов, в особенности у молодых женщин, ограничено [18] .
В настоящее время препаратом выбора стартовой терапии является "Леветирацетам". Он хорошо переносится и эффективно устраняет все три вида приступов, в том числе сопровождающихся фотосенситивностью [19] .
Также используется препарат "Ламотриджин". Он эффективно подавляет генерализованные тонико-клонические судороги и абсансы, но в половине случаев способствует учащению миоклоний. Его применение в монотерапии у пациентов с частыми миоклоническими приступами ограничено, но лекарство можно использовать в комбинированной терапии [21] .
Помимо перечисленных препаратов, могут применяться "Топирамат", "Зонисамид", "Перампанел" и "Фенобарбитал".
Чтобы избежать учащения приступов и усиления симптомов, важно ограничить приём "Карбамазепина", "Окскарбазепина", "Фенитоина", "Габапентина" и "Вигабатрина". Эти лекарства могут повышать гипервозбудимость мембраны клеток головного мозга, что приводит к обострению состояния [21] .
Прогноз. Профилактика
Прогноз определяется индивидуально в зависимости от частоты приступов, эффективности АЭП, возраста начала заболевания и т. д. Лечение часто не помогает пациентами с тремя видами приступов [21] .
Без приёма противоэпилептических препаратов (АЭП) приступы могут сохраняться всю жизнь. Их частота, как правило, снижается только после 40 лет [20] .
Эффективность АЭП в предотвращении приступов достигает 90 %. При отмене терапии часто возникают рецидивы, поэтому потребуется длительный приём препаратов, иногда пожизненный.
Качество жизни значительно ухудшается при частых миоклонических и генерализованных тонико-клонических приступах, при которых пациенты рискуют получить травмы.
Профилактика
Особое внимание стоит уделить образу и режиму жизни пациента. Самыми мощными провоцирующими факторами являются недосыпание и злоупотребление алкоголем. А учитывая, что дебют заболевания приходится на подростковый возраст, молодые люди часто нарушают эти рекомендации, особенно в студенческие годы.
Пациент, у которого выявили фотосенситивность, предрасположен к приступам под воздействием мерцающего света. Поэтому им необходимо ограничить просмотр телевизора и работу за компьютером, исключить видеоигры и избегать посещения ночных клубов.
У всех пациентов с эпилепсией имеются определённые социальные ограничения: они не могут работать в некоторых сферах, водить автомобиль и нести военную службу. Все они определяются индивидуально соответствующими комиссиями.
Миоклоническая эпилепсия
Миоклоническая эпилепсия — заболевание, основу которого оставляют миоклонические эпилептические пароксизмы. Эпизоды миоклонических судорог у больных сочетаются с генерализованными клонико-тоническими эпиприступами, абсансами. Сопутствующая неврологическая симптоматика зависит от формы эпилепсии. Диагностика включает сбор анамнеза, оценку неврологического и психического статуса, электроэнцефалографию, генеалогический анализ, биохимические исследования, нейровизуализацию. Лечение проводится антиконвульсантами, при резистентности — комбинацией противоэпилептических препаратов.

Общие сведения
Миоклонические судороги (миоклонии) представляют собой непроизвольные сокращения отдельной мышцы/мышечной группы. Соответственно, эпилепсия с преобладанием в клинической картине миоклоний получила название миоклоническая. Понятие «миоклоническая эпилепсия» (МЭ) включает ряд заболеваний, разнородных по этиопатогенезу, возрасту дебюта, особенностям симптоматики. В подавляющем большинстве случаев они характеризуется сочетанием миоклоний и генерализованных тонико-клонических судорожных приступов, имеют генетическую обусловленность. Встречаемость МЭ различна, некоторые нозологические формы являются настолько редкими, что в литературе по неврологии описано не более 100 клинических случаев.

Причины миоклонической эпилепсии
Обычно ведущим является генетический фактор. Чёткое аутосомно-доминантное наследование прослеживается при синдроме Драве, аутосомно-рецессивное — в отдельных случаях ранней миоклонической энцефалопатии. Некоторые заболевания имеют полигенное наследование. Локализация генетических дефектов установлена не для всех наследственных форм, исследования в этом направлении продолжаются. К генетически детерминированным патологиям относится и симптоматическая МЭ, возникающая вследствие дисметаболических процессов, обусловленных наличием дефектных генов. Образованию спонтанных мутаций в геноме способствуют:
- Внутриутробные инфекции. Инфекционный процесс неблагоприятно отражается на развитии плода. Особенно опасны вирусные инфекции, поскольку вирусы способны провоцировать аномальную перестройку отдельных генов.
- Хронические заболевания беременной. Сахарный диабет, сердечная недостаточность, хронические заболевания лёгких, эндокринная патология матери приводят к гипоксии, метаболическим расстройствам на ранних стадиях развития зародыша. В результате происходят сбои формирования ЦНС, отдельных механизмов обмена веществ.
- Повышенный радиоактивный фон. Радиация оказывает мутагенное влияние на живые организмы. Развивающийся плод наиболее подвержен подобному воздействию. Следствием является возникновение структурных, дисметаболических, функциональных аномалий, влекущих за собой повышенную эпилептическую активность.
- Прием тератогенных медикаментов. Самолечение, незнание о своей беременности в раннем периоде, медицинская необходимость фармакотерапии приводят к приёму опасных для плода медикаментов. Химические вещества оказывают повреждающее воздействие на отдельные гены, вносят изменения в существующие метаболические механизмы.
- Токсические воздействия на плод.Алкоголизм, наркомания, курение женщины в период беременности сопровождаются проникновением токсических веществ в организм плода. Подобно тератогенным фармпрепаратам они способны повредить отдельный локус генома, в результате возникает миоклоническая эпилепсия.
Патогенез
Идиопатические варианты МЭ развиваются вследствие генетически обусловленной повышенной возбудимости церебральных нейронов, приводящей к эпилептогенной активности мозга. Симптоматическая миоклоническая эпилепсия формируется в результате обменных нарушений, накопления в нервных клетках патологических соединений (полисахаридных включений, прионных белков).
При болезни Лафоры, миоклонической энцефалопатии младенцев повышенная эпиактивность обусловлена дисметаболизмом нейронов в условиях разрастания глиальных элементов (при гибели нейронов, нарушении апоптоза астроцитов). Нейрональная гипервозбудимость вызывает возникновение патологической нервной импульсации, идущей к мышечным волокнам. Результатом являются отдельные мышечные сокращения (миоклонии), тонические, клонические судороги. Различная локализация миоклоний отражает локальное возбуждение разных зон мозговой коры. При диффузном распространении гипервозбуждения возникает клонико-тонический пароксизм с тотальным вовлечением мышечных групп.
Классификация
В основе группировки отдельных видов МЭ лежит этиологический принцип. Согласно Международной классификации эпилепсии 1989 года выделяют 3 основные группы:
- Идиопатические — наследственно обусловленные формы. Характерна манифестация симптоматики в детском/подростковом возрасте. Идиопатическими являются доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ), юношеская миоклоническая эпилепсия (ЮМЭ), болезнь Унферрихта-Лундборга, синдром Драве.
- Криптогенные — не имеющие установленной этиологии. Отличаются выраженной резистентностью к фармакотерапии, наличием сопутствующей очаговой симптоматики, интеллектуального дефицита. К криптогенным относятся эпилепсия с миоклонически-астатическими приступами, эпилепсия с миоклоническими абсансами.
- Симптоматические — возникающие на фоне происходящих в организме патологических процессов. В большинстве случаев обусловлены метаболическими нарушениями. Симптоматическими считаются ранняя миоклоническая энцефалопатия, болезнь Лафоры, миоклонические пароксизмы при подостром склерозирующем панэнцефалите, болезни Крейтцфельдта-Якоба.
Впоследствии были выявлены генетические аспекты возникновения криптогенных форм МЭ. Учитывая результаты исследований, Международное общество неврологов предложило относить ранее считавшиеся криптогенными виды МЭ к идиопатическим.
Симптомы миоклонической эпилепсии
Базовым симптомом выступают пароксизмы миоклоний, затрагивающие различные мышечные группы конечностей, реже — лица, еще реже — туловища. Миоклонии выглядят как мышечные подёргивания, при вовлечении мышц одной группы сокращения приводят к непроизвольным двигательным актам, напоминающим гиперкинезы. Миоклонический эпилептический пароксизм происходит при сохранённом сознании, может протекать с перемещением сокращений по различным мышцам. Миоклоническая эпилепсия характеризуется комбинацией миоклоний с клонико-тоническими приступами и/или абсансами. В зависимости от нозологической формы наблюдаются задержка психического развития, атаксия, пирамидная недостаточность, расстройства мышечного тонуса, зрительные нарушения.
ДМЭМ дебютирует в возрастном периоде от 6 месяцев до 3 лет. Приступы захватывают верхние конечности, лицо, шею, могут имитировать наклон головы, моргание, кивки головой. Заболевание редко сопровождается интеллектуальным снижением. Миоклоническая эпилепсия юношеского возраста (манифестация в возрасте 12-18 лет) отличается присоединением тонико-клонических эпизодов, отсутствием неврологического дефицита. Синдром Драве клинически проявляется на первом году жизни, сопровождается олигофренией, нарушениями поведения, пирамидным дефицитом. Семейная миоклония Унферрихта-Лундборга начинается в 5-16 лет, сочетается с тремором, атаксией, дизартрией, психическими расстройствами.
Миоклонически-астатические пароксизмы отличаются возникающей на фоне миоклоний потерей устойчивости. Пациенты описывают приступ как «удар под коленки», «подкашивание ног», вынуждающее становиться на колени, падать. Миоклонические абсансы представляют собой эпизоды кратковременного отключения сознания с миоклоническими сокращениями плечевого пояса, мышц конечностей, периорбитальной области. Заболевание возникает у детей 2-12 лет.
Миоклоническая эпилепсия симптоматического характера отличается прогрессированием симптоматики, выраженным когнитивным дефицитом, прочими неврологическими нарушениями, наличием проявлений основного заболевания, клинико-лабораторных признаков метаболических расстройств.
Осложнения
Клонико-тонические, астатические приступы осложняются травмированием пациента вследствие падения. Генерализованные судороги с утратой сознания опасны западением языка, перекрытием дыхательных путей и асфиксией. Аспирация слюны, рвотных масс приводит к последующему развитию пневмонии. Длительный миоклонический пароксизм, непрерывно следующие кластерные сокращения перерастают в миоклонический эпистатус. В эпилептическом статусе возможны серьёзные дыхательные расстройства, остановка сердца, развитие отёка головного мозга.
Диагностика
Миоклоническая симптоматика входит в клинику многих болезней, эпилептических синдромов. Диагноз «миоклоническая эпилепсия» устанавливается только при превалировании миоклонических приступов над другими клиническими проявлениями. Диагностика направлена на верификацию нозологической формы эпилепсии, при выявлении вторичного характера миоклоний — на поиск основной патологии. Основными диагностическими этапами являются:
- Сбор анамнестических данных. Большое значение имеет возраст дебюта, характер начала, порядок развития симптоматики.
- Неврологический осмотр. Проводится неврологом, направлен на выявление миоклонических сокращений, очагового дефицита, определение уровня психического развития, степени когнитивных расстройств, оценку психического статуса.
- Электроэнцефалография. У большинства пациентов регистрируются диффузные интериктальные симметричные эпилептогенные разряды, иктальные высокоамплитудные спайки. В ряде случаев для выявления эпиактивности требуется суточный ЭЭГ-видеомониторинг, проведение провокационных проб (ЭЭГ при вспышках света, гипервентиляции, резких звуковых сигналах). Результаты исследований оцениваются нейрофизиологом, эпилептологом.
- Нейровизуализация. До закрытия родничков осуществляется путём нейросонографии, у детей старше года — при помощи МРТ головного мозга. Взрослым может проводиться МСКТ. Морфологические изменения церебральных тканей характерны для симптоматических МЭ.
- Лабораторные исследования. Производятся при подозрении на наличие обменных расстройств. Включают биохимический анализ крови и мочи, специфические анализы.
- Консультация генетика. Сбор семейного анамнеза, составление генеалогического древа позволяют определить наследственный характер эпилепсии, установить тип наследования.
Дифференциальная диагностика осуществляется с неэпилептическим миоклонусом, отличительной особенностью которого выступает фокальный характер миоклоний, отсутствие реакции на провокацию, нормальная ЭЭГ-картина. Дифференцировка МЭ необходима также с судорожным синдромом инфекционной этиологии, фебрильными судорогами, синдромом Леннокса-Гасто, мозжечковой миоклонической диссинергией Ханта.
Лечение миоклонической эпилепсии
Терапия базируется на антиконвульсантах. Подбор фармпрепарата и дозировки осуществляется индивидуально. Препаратами выбора выступают производные вальпроевой кислоты, обладающие противоэпилептическим эффектом в равной степени в отношении миоклонических, клонико-тонических, абсансных пароксизмов. В фармакорезистентных случаях показано комбинированное лечение вальпроатами, бензодиазепинами, этосуксимидом, барбитуратами, антиконвульсантами нового поколения (топираматом, леветирацетамом). Важным моментом является исключение провоцирующих приступы факторов: резких звуков, вспышек света, эмоциональных всплесков, физических перегрузок, перегреваний.
Прогноз и профилактика
Наиболее прогностически неблагоприятна ранняя миоклоническая энцефалопатия, смертность составляет половину случаев заболевания, остальные дети являются глубокими инвалидами. Миоклоническая эпилепсия при болезни Лафоры, Крейтцфельдта-Якоба плохо поддаётся противоэпилептической терапии, сопровождается прогрессирующим интеллектуальным распадом. ДМЭМ и ЮМЭ отличаются доброкачественным течением, редко приводят к когнитивному дефициту. Более 50% случаев ДМЭ заканчиваются спонтанным выздоровлением.
МЭ не имеет специфических мер профилактики. К мероприятиям, способным предупредить рождение больного ребёнка, относятся планирование беременности, ранняя постановка на учёт, исключение неблагоприятных воздействий на плод. Ведение беременности должно включать разъяснительные беседы с женщиной по поводу необходимости охранительного режима, тератогенной опасности лекарственных средств, пагубного воздействия на будущего ребёнка вредных привычек.
Юношеская миоклоническая эпилепсия
Юношеская миоклоническая эпилепсия — это форма генерализованной эпилепсии, основу клинической картины которой составляют миоклонические приступы — асинхронные мышечные сокращения, кратковременно возникающие в симметричных участках тела, преимущественно в руках и плечевом поясе. Наряду с миоклоническими эпизодами в клинике могут наблюдаться абсансы и клонико-тонические генерализованные эпиприступы. Юношеская миоклоническая эпилепсия диагностируется на основании клиники заболевания и результатов электроэнцефалографии, при исключении органической церебральной патологии по данным неврологического осмотра и МРТ. Лечение проводится преимущественно препаратами вальпроевой кислоты. Как правило, необходимо пожизненное наблюдение эпилептолога.
МКБ-10

Юношеская миоклоническая эпилепсия (ЮМЭ) составляет до 12% от всех форм этого заболевания и около 23% случаев идиопатической генерализованной эпилепсии. ЮМЭ является одной из разновидностей миоклонической эпилепсии — генерализованной эпилепсии, протекающей с миоклоническими приступами. В эту группу заболеваний также входят: детская доброкачественная миоклония, синдром Веста (миоклоническая энцефалопатия детского возраста с церебральной гипсаритмией), болезнь Лафоры и др.
Первое описание ЮМЭ датировано 1867 г. Однако в качестве отдельной нозологической единицы юношеская миоклоническая эпилепсия была выделена лишь в 1955 г. по предложению группы германских врачей во главе с Янцем, после чего она стала упоминаться как синдром Янца. В научной литературе по неврологии и эпилептологии можно встретить также термин «импульсивный petit mal».

Причины
Юношеская миоклоническая эпилепсия является наследственной. Случаи развития этой формы эпилепсии вследствие органического поражения головного мозга не зафиксированы. У половины пациентов в семейном анамнезе имеются родственники 1-й или 2-й линии, у которых происходят различного типа эпиприступы. Гены, ответственные за развитие заболевания, пока точно не установлены. Предполагают несколько возможных вариантов - хромосома 15q, один из локусов короткого плеча хромосомы 6, гены C6orf33 и BRD2 и др. Большинство генетиков склоняются к мнению о полигенном механизме наследования миоклонической эпилепсии. Специфический патогенез ЮМЭ не идентифицирован.
Симптомы
Юношеская миоклоническая эпилепсия манифестирует в возрасте от 8 до 24 лет. Наиболее часто дебют заболевания приходится на возрастной отрезок 12-18 лет. Патогномоничным симптомом ЮМЭ выступают миоклонические приступы — короткие, внезапно возникающие, непроизвольные мышечные сокращения, имеющие асинхронный характер.
Как правило, в начале заболевания пароксизмы отмечаются в утренние часы при пробуждении больного. Сокращения мышц происходят симметрично в обеих половинах тела, чаще охватывают только плечевой пояс и руки, реже — распространяются на нижние конечности или на все тело. Во время пароксизма пациенты могут автоматически отбрасывать или ронять удерживаемые в руках предметы, при вовлечении нижних конечностей происходит падение.
Пароксизмы ЮМЭ могут иметь одиночный характер или возникать кластерами. В редких случаях наблюдается т. н. миоклонический эпилептический статус. Отличительной чертой является полная сохранность сознания пациента во время миоклонического пароксизма, даже в тех случаях, когда речь идет о миоклоническом статусе.
В 3-5% случаев юношеская миоклоническая эпилепсия протекает с наличием только миоклонических пароксизмов. В подавляющем большинстве наблюдений (около 90%) через некоторое время после дебюта заболевания (в среднем через 3 года) у пациента возникают генерализованные тонико-клонические эпиприступы. Они могут начинаться с серии нарастающих миоклонических подергиваний, затем переходящих в клонико-тонические судороги. Примерно у 40% пациентов отмечаются абсансы — кратковременные эпизоды «выключения» сознания.
Установить диагноз ЮМЭ в ее начальном периоде весьма затруднительно. Зачастую возникающие на фоне пробуждения миоклонические эпизоды расцениваются как нервозность ребенка, а сами дети обычно не обращают внимания на подобные мелкие симптомы. Как правило, родители обращаются к неврологу, когда у ребенка появляются тонико-клонические приступы. Исследование неврологического статуса не определяет каких-либо нарушений. Инструментальные методы включают:
- МРТ головного мозга. Проводят для исключения церебральной патологии (внутримозговой опухоли, абсцесса мозга, церебральной кисты, энцефалита, внутримозговой гематомы) и органического происхождения эпиприступов. При необходимости исключения аневризмы сосудов головного мозга пациента направляют на МР-ангиографию.
- Электроэнцефалография. В 75% ЭЭГ выявляет наличие интериктальных эпилептиформных паттернов. Регистрируются билатерально-симметричные пароксизмальные разряды, состоящие из комплексов полиспайк-волна с частотой 4-6 Гц. В 17% случаев наблюдаются комплексы частотой 3 Гц. Иктальная ЭЭГ выявляет высокие и среднеамплитудные спайки частотой 10-16 Гц, после которых регистрируются медленные волны нерегулярного характера. Число спайков одного пароксизма варьирует от 5 до 20 и зависит скорее не от длительности, а от интенсивности приступа. Для ранней диагностики ЮМЭ может потребоваться проведение ЭЭГ при пробуждении, суточный ЭЭГ-видеомониторинг, провоцирующие пробы (депривация сна, фотостимуляция).
Дифференциальная диагностика
Массивность и билатеральная синхронность отличает пароксизмы ЮМЭ от неэпилептического миоклонуса, эпизоды которого носят спорадический и фокальный характер. Юношеская миоклоническая эпилепсия также требует дифференцировки от других форм эпилепсии, протекающих с миоклоническими эпизодами. Так, в отличии от ЮМЭ, при эпилепсии с генерализованными судорожными пароксизмами пробуждения или при юношеской абсанс эпилепсии миоклонические пароксизмы не являются доминирующими в клинической картине заболевания.
Эпилепсия с миоклонически-астатическими пароксизмами, синдром Леннокса-Гасто и эпилепсия с миоклоническими абсансами дебютируют в более раннем детском возрасте и сопровождаются задержкой психического развития. Последняя характеризуется приступами, в которых миоклонические судороги сочетаются с абсансами, в то время как пароксизмы ЮМЭ протекают без нарушения сознания.
Режимные мероприятия
Важное значение имеет не только фармакотерапия эпилепсии, но и соблюдение пациентом некоторых жизненных норм, позволяющих избегать провоцирования приступов. Как и при других видах эпилепсии, при ЮМЭ приступы могут быть вызваны нарушением режима, психической и физической перегрузкой, стрессом, недосыпанием, приемом содержащих алкоголь напитков. Поэтому пациенту следует избегать подобных провоцирующих факторов. Положительно влияет на течение заболевания спокойный, простой и неторопливый уклад жизни, пребывание на природе, вдали от городской суеты. В связи с этим некоторые семьи, где у ребенка диагностирована ЮМЭ, переезжают и живут в сельской местности.
Фармакотерапия
Медикаментозная терапия ЮМЭ проводится вальпроатами. Монотерапия данными препаратами оказалась эффективной в отношении всех видов приступов, сопровождающих клинику ЮМЭ, — миоклонических, тонико-клонических и абсансов. При недостаточности монотерапии возможно комбинированное лечение. Купирование резистентных абсансов достигается сочетанием вальпроатов с этосуксимидом, резистентных клонико-тонических приступов — сочетанием вальпроатов с примидоном или фенобарбиталом.
Для контроля миоклонических пароксизмов эффективен клоназепам, однако его действие не распространяется на тонико-клонические генерализованные приступы. При этом полное купирование миоклонических приступов лишает пациента возможности заранее знать о приближающемся тонико-клоническом приступе по возникающим перед ним миоклоническим проявлениям. Поэтому назначение клоназепама оправдано лишь при стойких миоклонических пароксизмах и должно сочетаться с препаратом вальпроевой кислоты.
Результаты лечения ЮМЭ противоэпилептическими препаратами нового поколения (леветирацетамом, ламотриджином, топираматом) пока проходят проверку в клинических условиях. Отмечена высокая перспективность леветирацетама.
Прогноз
Юношеская миоклоническая эпилепсия считается хроническим заболеванием, которое продолжается в течение всей жизни пациента. Случаи спонтанной ремиссии редки. У 90% больных, по различным причинам прервавших лечение антиэпилептическим препаратом, отмечалось возобновление эпиприступов. Однако имеются указания на то, что в отдельных случаях у пациентов после отмены препарата наблюдалась длительная ремиссия.
В целом, при правильно подобранной терапии приступы контролируются у большинства больных, хотя у половины из них на фоне лечения могут наблюдаться отдельные пароксизмы. Относительно резистентное к терапии течение наблюдается редко, в основном в случаях, когда у пациента отмечаются все 3 разновидности пароксизмов ЮМЭ.

Миоклоническая ювенальная эпилепсия - это такая форма заболевания, которая характерна для детей, но встречается и у взрослых, симптомы незаметны на первых этапах, так что распознают ее рано далеко не все врачи.
Болезнь является врожденной, как правило, - наследуемой. У таких больных в клетках мозга находят аномальные вкрапления углеводов. Дебют, первые проявления, ожидаются в 10-19 лет.
Отличительная черта - пациенты, как правило, не теряют сознания при приступах. Второй момент - без надлежащего лечения, эпилептический очаг отрицательно влияет на состояние мозга, приводя к отставанию психики, интеллекта, деменции.
Причины возникновения
В развитии эпилепсии с миоклоническими приступами у детей, решающую роль играет генетика.
Важен как фактор наследования генов предрасположенности к приступам от родителей, так и вредное влияние в период вынашивания плода.
Распространенными причинами являются:
- Хронические серьезные заболевания матери. Сердечная недостаточность, сахарный диабет, болезни легких приводят к нехватке кислорода для плода на ранней стадии развития, в период формирования ЦНС.

- Токсическое воздействие. При употреблении алкоголя, наркотиков, курения, проживания в местности с плохой экологией, попадающие в организм матери вредные вещества достаются зародышу, отравляя формирующийся организм, приводя к генным мутациям.
- Внутриутробные инфекции - одно из опаснейших явлений при беременности. Вирусная инфекция на ранних сроках может нанести непоправимый вред, спровоцировав аномалии в формировании органов и систем.
- Прием медикаментов. В первом триместре, попадание в организм матери даже привычных лекарств, чревато мутациями у плода. Поэтому важно после состоявшегося зачатия не заниматься самолечением, а перед приемом таблеток консультироваться с наблюдающим врачом.
- Повышенный радиоактивный фон также приводит к развитию мутаций.
Эпилепсия данного типа бывает не только наследственной, но и приобретенной из-за негативного воздействия на плод.
Однако, если хотя бы один из родителей болеет таким заболевание, шансы получить у ребенка судороги значительно повышаются.
Тип болезни прописывается в диагнозе исходя из тяжести проявившихся симптомов.
Миоклонус эпилепсия делится на 4 условных типа, каждый из которых требует своего лечения.

- Доброкачественный миоклонус младенцев. Легкая форма, проявляющаяся в возрасте от полугода. Установить ее происхождение сложно, приступы практически незаметные. Несколько раз на протяжении дня у младенца случаются резкие сокращения мышц головы, шеи, конечностей.
Длится приступ не больше нескольких секунд, поэтому редко воспринимается родителями, как повод для беспокойства. Поддается лечению, однако без необходимой терапии, приводит к отставанию психомоторных функций.
- Синдром Драве. Редкий тяжелый тип патологии, вызванный воспалительными процессами в ЦНС. Проявляется в виде сильных приступов в возрасте до года у младенцев только женского пола. Вылечивается с трудом.
- Болезнь Унферрихт-Лундборга. Как правило - наследственный вариант заболевания, медленно развивающаяся, но тяжело поддающаяся медикаментозному лечению. Применяется симптоматичная терапия, позволяющая снизить количество эпиприступов. Развивается незаметно, первые проявления становятся видны в возрасте 5-16 лет в виде легких судорог мышц лица.
Болезнь редко успевают распознать на начальной стадии. Подергивания переходят на плечевой пояс, а со временем становятся полноценными эпилептическими припадками. Отсутствие медицинской помощи приводит к нарушениям речи, тремору.
- MERRF или с красными рваными волокнами. Передающаяся по наследству патология митохондрий. Ее уникальность в том, что проявляется миоклоническая форма эпилепсии не только у детей, но и у взрослых, быстро прогрессируя, демонстрируя усиливающиеся симптомы. На фоне эпиприступов с судорогами ухудшается память, начинает развиваться деменция. При своевременном приеме препаратов прогрессирование замедляется.
Для любого из описанных вариантов судорожного синдрома есть разные степени тяжести приступов. Сильнее они становятся на фоне психоэмоциональных нагрузок, при сопутствующей психиатрической патологии.
Ювенальная миоклоническая эпилепсия проявляется разнообразно, требует лечения, без которого прогноз становится неутешительным.
Базовым симптомом для этого типа заболевания является пароксизмы напряжения и расслабления групп мышц - миоклонии.

Подергивания мышц лица редко воспринимаются как опасное явление, из-за этого постановка правильного диагноза также затягивается. Однако по совокупности всех проявлений понять, что у человека развивается опасная болезнь можно.
- Полное сохранение сознания - пациент осознает, что с ним происходит.
- Время проявления судорог - утро, в течение первых 2-3 часов после пробуждения.
- Миоклонические припадки. Легкое подергивание одной мышцы или всей конечности, лица. Спровоцировать их может как пробуждение, так и физическая усталость, алкоголь, наркотические вещества, громкие звуки, яркий свет, эмоциональное перенапряжение
- Тонико-клонические приступы в тяжелой форме встречаются как результат прогрессирования болезни почти у 50% пациентов. Человек теряет сознание, в судороги вовлекается все тело, после чего происходит полное расслабление мышц, что приводит к непроизвольной дефекации, мочеиспусканию. Кризис продолжается до 5 минут.
- Абсансы или малые припадки. Временное отключение сознания на 5-30 секунд. Человек замирает, переставая воспринимать окружающий мир. Несмотря на кратковременность, абсансы способны нанести серьезный вред сознанию. Потому что могут следовать пачками, не давая ребенку усваивать программу.



Диагностика и лечение
Некоторые виды юношеской миоклонической эпилепсии считаются излечимыми. Поэтому при подозрении на такую болезнь, пациент проходит диагностику у невролога и психиатра.
В списке необходимых процедур:
- , подтверждающая неадекватную активность нейронов коры головного мозга;
- эхоэнцефалография, дающая характеристику анатомической структуры мозга;
- МРТ, позволяющая точно исключить другие причины приступов, локализовать патологический очаг.
Пациентам назначаются исследования крови, освидетельствование психологом, позволяющее увидеть отклонения в развитии.
Лечение комплексное, так как одними медикаментами в столь сложном случае обойтись не получится.
Дополнительно к противосудорожным лекарствам используют:

- периферическую магнитную стимуляцию;
- и взрослых;
- биорезонансную терапию;
- ;
- диету с низким содержанием углеводов;
- строгий режим дня.
Несколько лет больному придется прожить в режиме ограничений, а некоторые из них соблюдать до конца жизни. Однако это обеспечит отсутствие приступов и сохранность психики и интеллекта.
Список использованной литературы
Была ли эта статья полезна?
Вы можете подписаться на нашу рассылку и узнать много интересного о лечение заболевания, научных достижений и инновационных решений:
Если у вас остались вопросы, задайте их врачам на нашем форуме!
Клиника восстановительной неврологии проводит консультации, диагностику, комплексное лечение с индивидуальным подходом для активации и стимуляции мозга.
123182 г. Москва, ул. Маршала Василевского, д. 13, корп. 3, под. 2
357501 г. Пятигорск, ул. Козлова, д. 8 ОФИС 1
Информация на сайте носит исключительно ознакомительный характер. Все материалы и цены, размещенные на сайте, не являются публичной офертой, определяемой положениями ст. 437 ГК РФ. Для получения точной информации обратитесь к сотрудникам клиники или посетите нашу клинику.
18+ Информация, представленная на сайте, не может быть использована для постановки диагноза, назначения лечения и не заменяет прием врача.
Синдром MERRF ( Миоклонус-эпилепсия с рваными красными волокнами )
Синдром MERRF - это редкое генетическое заболевание, которое вызвано структурными и биохимическими дефектами митохондрий, характеризуется тяжелым поражением центральной нервной системы и мышечной ткани. Клиническая картина может отличаться даже внутри одной семьи. Симптомы включают различные виды эпилептических припадков, нарушение координации, мышечную слабость. Подтверждающими диагностическими методами являются гистологическое исследование мышечного биоптата и ДНК-анализ. Специфического лечения не существует, проводится симптоматическая терапия нейрометаболическими, противосудорожными средствами, препаратами, улучшающими митохондриальную функцию.

Синдром MERRF относится к группе наследственных митохондриальных энцефаломиопатий. MERRF представляет собой аббревиатуру, означающую «миоклонус (М) эпилепсия (Е) с рваными (R) красными (R) волокнами (F)». Заболевание впервые было описано японским неврологом Н. Фукухара в 1980 году. По различным эпидемиологическим данным, распространенность синдрома MERRF составляет от 0,25:100000 до 1,5:100000 населения. Наследование происходит только по материнской линии, при этом оба пола страдают с одинаковой частотой.
Возникновение синдрома MERRF вызвано точечными мутациями митохондриальной ДНК, а именно, изменениями нуклеотидной последовательности. Наиболее частыми считаются замена аденина на гуанин в положении 8344 транспортной РНК лизина или замена тимина на цитозин в положении 8356. Это приводит к нарушению образования митохондриальных белков.
Клиническое развитие синдрома MERRF зависит от количества дефектных ДНК, т.е. чем больше митохондриальных ДНК, имеющих мутацию, тем выше риск заболевания. Также возможно абсолютно бессимптомное носительство мутантных генов. Какие-либо значимые факторы риска, способные спровоцировать манифестацию данной патологии, отсутствуют.
В результате генетической мутации в митохондриях происходит сбой синтеза ферментов дыхательной цепи (цитохромоксидазы, сукцинатдегидрогеназы и др). Нарушаются процессы окислительного фосфорилирования - наиболее важного этапа в энергетическом обмене клеток, который обеспечивает образование основного количества молекул АТФ (универсального источника энергии для клеток).
Данные явления протекают практически во всех клетках организма. Наиболее сильно поражающий эффект сказывается на органах с высокой энергетической потребностью - центральной нервной системе и мышцах. Вследствие тканевой гипоксии в мышцах накапливается избыток молочной кислоты, что приводит к повреждению мышечных волокон.
В современной неврологии четких официальных разделений синдрома MERRF на отдельные формы не принято. В зависимости от количества точечных мутаций и клинических проявлений можно выделить следующие разновидности:
- Бессимптомная - имеется генетическая мутация с возможностью передачи по наследству при полном отсутствии симптомов.
- Латентная - «мягкое течение» без вовлечения нервной системы. Пациентов может беспокоить умеренная мышечная слабость.
- Манифестная - яркая клиническая картина с тяжелым течением и неблагоприятным прогнозом.
Синдром MERRF может дебютировать практически в любом возрасте. Начало в детском возрасте ассоциировано с более неблагоприятным прогнозом. Ранними признаками считаются повышенная мышечная утомляемость, ухудшение переносимости физических нагрузок, ноющие боли в икроножных мышцах. Из-за постепенного прогрессирования миопатии мышечная слабость приобретает более выраженный характер - пациент испытывает трудности при подъеме по лестнице, ходьбе. В тяжелых случаях больной с большим усилием может подняться с постели.
Типичны эпилептические припадки, которые могут быть разнообразными - непроизвольные подергивания мышц лица или рук без потери сознания (миоклонии), пароксизмы по типу кивков головой, генерализованные тонико-клонические припадки с потерей сознания. Миоклонии часто провоцируются резкими звуками или вспышкой света. Нарушается координация, возникает неустойчивость при ходьбе, стоянии.
Существенно страдают когнитивные функции, снижается память, концентрация внимания. Постепенно ухудшается зрение и слух. В ряде случаев наблюдаются периферические невропатии, проявляющиеся онемением, ощущением жжения, покалывания (парестезиями) в конечностях. Очень редко встречаются кожные образования - липомы, гемангиомы, бородавчатые невусы.
Синдром MERRF считается тяжелым заболеванием с большим числом осложнений. Наиболее неблагоприятными из них являются эпилептический статус и отек головного мозга. Вследствие тяжелой миопатии мышц глотки возможно нарушение глотания и попадание пищи в дыхательные пути, что приведет к развитию аспирационной пневмонии. Также из-за слабости дыхательных мышц возникает дыхательная недостаточность.
Атрофия зрительных нервов и пигментная дегенерация сетчатки у части пациентов вызывает полную потерю зрения. При ранней клинической манифестации ребенок значительно отстает в физическом и нервно-психическом развитии. Нарушение равновесия повышает риск падений и переломов. Очень редко наблюдается хронический панкреатит и сахарный диабет.
Пациентов с синдромом MERRF курируют врачи-неврологи. Больные детского возраста находятся под совместным наблюдением детских невропатологов и педиатров. При осмотре обращается внимание на общее снижение мышечного тонуса, ослабление сухожильных и наличие патологических рефлексов (Бабинского, Оппенгейма), невыполнение координационных тестов - позы Ромберга, пяточно-коленной, пальце-носовой проб. Дополнительное обследование включает:
- Лабораторные исследования. В биохимическом анализе крови отмечается увеличение концентрации молочной кислоты. В ликворе выявляется высокое содержание белка.
- ЭЭГ. На электроэнцефалограмме удается обнаружить эпилептиформную активность - генерализованные комплексы спайк-волна, диффузные медленные волны.
- Томография. На МРТ головного мозга видна атрофия коркового и белого вещества больших полушарий и мозжечка, кальцификация базальных ганглиев.
- ЭНМГ. При проведении электронейромиографии отмечается снижение амплитуды и длительности потенциалов двигательных единиц, что свидетельствует о поражении мышечной ткани.
- Гистологическое исследование. Один из главных диагностических тестов синдрома MERRF. В мышечном биоптате выявляются признаки атрофии - уменьшение размеров мышечных волокон, их бледное окрашивание, склероз эндомизия и перимизия. При окраске гистологических срезов по методу Гомори более чем в 5% мышечных волокон обнаруживается наличие «рваных красных волокон».
- ДНК-анализ. Основной метод для верификации диагноза. При молекулярно-генетическом исследовании находят мутации в митохондриальной ДНК - A8344G или Т8356С.
Синдром MERRF следует дифференцировать с другими митохондриальными заболеваниями (MELAS-синдром), а также с наследственными метаболическими расстройствами, поражающими мышечные ткани и нервную систему:
- миоклонус-эпилепсией Унферрихта-Лундборга;
- болезнью Лафоры;
- болезнями накопления (болезнь Гоше, лейкодистрофии, ганглиозидозы).
Лечение синдрома MERRF
На сегодняшний день эффективных способов терапии данной патологии не существует. Все мероприятия носят симптоматический и паллиативный характер, направлены на улучшение состояния пациента. Для замедления атрофических процессов в мышечной ткани обязательны занятия лечебной физкультурой. Регрессу симптоматики способствует диета с ограничением углеводов.
Детям с умственной отсталостью рекомендуются индивидуальные консультации дефектолога, психолога, логопеда. Лекарственная терапия проводится по общим принципам лечения митохондриальных болезней и включает 2 основные группы медикаментов:
- Метаболические препараты. С целью улучшения процессов клеточного дыхания применяется комплекс из энерготропных препаратов («митохондриальный коктейль»), в который входят кофакторы ферментов (витамины группы В), средства, стимулирующие перенос электронов в дыхательной цепи (коэнзим Q10) и антиоксидантов (аскорбиновая кислота, токоферол).
- Антиконвульсанты. Для предупреждения эпилептических припадков назначаются противосудорожные лекарственные средства - клоназепам, ламотриджин, топирамат. Вальпроевая кислота и ее производные строго противопоказаны, так как они угнетают митохондриальную функцию. Их применение приводит к резкому ухудшению состояния больного.
Продолжительность жизни, вероятность летального исхода при синдроме MERRF может сильно варьировать у разных пациентов. При малосимптомном течении (отсутствие поражения нервной системы, умеренная миопатия), систематическом проведении нелекарственного и лекарственного лечения продолжительность жизни может не отличаться от таковой в общей популяции.
При развернутом течении прогноз крайне неблагоприятный. Летальный исход наступает в течение 10-15 лет от начала заболевания. Основными причинами смерти выступают эпилептический статус, аспирация, дыхательная недостаточность. Единственным специфическим методом профилактики является пренатальная диагностика и прерывание беременности.
1. Наследственные болезни нервной системы: Рук. для врачей/ Под ред. Ю.Е. Вельтищева, П.А. Темина. - 1998.
4. Молекулярно генетическая характеристика болезней дыхательной цепи митохондрий у детей: Автореферат диссертации/ Цыганкова П.Г. - 2012.
Читайте также:
- Шоковые реакции. История определения шоковой реакции
- Гигантоклеточный артериит (болезнь Хортона): лечение, симптомы, причины возникновения
- Трубная стерилизация под местной анестезией. Лекарства и особенности
- Синдромы близкие инфантильным спазмам и их характеристика
- Определение объема замещения стекловидного тела при витрэктомии