Нервная система при нарушениях в цикле Кребса
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Для цитирования: Зиновьева О.Е., Емельянова А.Ю., Кожев А.И. и др. Неврологические проявления дефицита витамина В12 // Эффективная фармакотерапия. 2021. Т. 17. № 6. С. 22-28.
- Аннотация
- Статья
- Ссылки
- Английский вариант
На основании анализа отечественной и зарубежной литературы рассматриваются распространенность в популяции и причины развития дефицита витамина В12, патогенетические механизмы и клинические проявления патологии нервной системы, обусловленной таким дефицитом. Проанализированы наиболее информативные методы диагностики и схемы лечения неврологических расстройств при дефиците витамина В12.
- КЛЮЧЕВЫЕ СЛОВА: витамин, дефицитарные состояния, миелоз, энцефалопатия, деменция, гипергомоцистеинемия, диагностика, лечение, Нейробион
На основании анализа отечественной и зарубежной литературы рассматриваются распространенность в популяции и причины развития дефицита витамина В12, патогенетические механизмы и клинические проявления патологии нервной системы, обусловленной таким дефицитом. Проанализированы наиболее информативные методы диагностики и схемы лечения неврологических расстройств при дефиците витамина В12.
Витамин B12 - водорастворимый витамин, который в природе вырабатывается микроорганизмами, а в организме человека не синтезируется. Средняя суточная потребность в витамине B12 составляет 2,4 мкг для мужчин и небеременных женщин, 2,6 мкг для беременных, 2,8 мкг - для кормящих [1].
Основной источник витамина B12 - белки животного происхождения (мясо, печень, почки, рыба, молочные продукты, яичные желтки). Запасы витамина В12 в организме взрослого человека составляют примерно 2000-5000 мкг. Витамин B12 депонируется в основном в печени (1 мкг в 1 г ткани печени), в меньшем количестве - в почках и скелетных мышцах. В12 выделяется с желчью и калом, в сутки теряется 0,1% общего количества депонированного витамина. Существует кишечно-печеночный кругооборот витамина В12 - около 3/4 выделенного с желчью витамина вновь реабсорбируется в кишечнике через систему воротной вены печени (энтерогепатическая циркуляция). Этим объясняется развитие клинических проявлений дефицита В12 через 1-3 года после полного прекращения его поступления в организм [2].
В продуктах питания В12 обычно содержится в форме кофермента дезоксиаденозилкобаламина или метилкобаламина и связан с белками. На первом этапе метаболизма витамина В12 происходит его протеолиз в желудке при низком pH, в результате чего он высвобождается из пищевого белка и связывается с белком R желудочного сока. Париетальные клетки желудка вырабатывают внутренний фактор Касла - гликопротеид, который вместе с белковым комплексом «кобаламин - R» поступает в двенадцатиперстную кишку. Комплекс внутреннего фактора Касла и кобаламина в подвздошной кишке связывается с рецепторами внутреннего фактора кобаламина, расположенными на микроворсинках клеток слизистой оболочки подвздошной кишки. При нейтральном рН и в присутствии ионов кальция комплекс «кобаламин - фактор Касла» распадется, кобаламин попадает в энтероцит, переносится на транскобаламин II (голотранскобаламин) и в комплексе с ним попадает в кровоток. При наличии большого количества витамина В12 около 1% может проникать в кровь за счет пассивной диффузии [1, 2].
B12 участвует во многих ключевых процессах метаболизма липидов, углеводов и белков, играет центральную роль в гемопоэзе. В организме человека витамин В12 служит кофактором для работы двух ферментов - метилмалонил-коэнзим A (CoA)-мутазы в митохондриях и метионинсинтетазы (MeCbl) в цитоплазме. Витамин В12 участвует в синтезе нуклеиновых кислот: связанный с MeCbl, он облегчает удаление метильной группы из метилфолата с образованием гомоцистеина (HCYS), который превращается в метионин. Витамин B12 необходим для клеточного дыхания и поддержания энергии, поскольку задействован в цикле Кребса. B12 участвует в процессах миелинизации и нормального формирования нервной трубки на этапе эмбриогенеза, синтезе нуклеиновых кислот, белковых и липидных структур миелиновой оболочки нервного волокна, холина и нейротрансмиттеров [2, 3].
Причины развития дефицита витамина В12
Причин развития дефицита витамина В12 много. Их можно разделить на несколько групп.
Недостаточное поступление витамина В12 с пищей
К дефициту В12 может привести недостаточное употребление белков животного происхождения в силу различных причин (пожилой возраст, низкий социальный статус, в частности у лиц, страдающих хроническим алкоголизмом). Большинство вегетарианцев получают только 0,25-0,5 мкг кобаламина в день. Не случайно распространенность дефицита витамина B12 среди них достигает 40-80% [3].
Нарушение всасывания и метаболизма витамина В12 в желудочно-кишечном тракте
Подобные состояния имеют место у пациентов после операций на желудке или тонком кишечнике, в том числе после бариатрических операций по поводу патологического ожирения, лиц, страдающих атрофическим гастритом (чаще пожилых людей), пациентов с заболеваниями тонкой кишки, недостаточностью поджелудочной железы и синдромом мальабсорбции, целиакией, глистными инвазиями, дивертикулярной болезнью. Дефицит В12 нередко развивается при ожоговой болезни или тяжелых травмах на фоне заместительной почечной терапии, а также при хроническом воздействии токсигенной плесени и микотоксинов (чаще встречаются в поврежденных водой зданиях). К нарушению всасывания и метаболизма витамина В12 могут приводить генетические дефекты, при которых нарушается синтез переносчиков кобаламина [2, 4].
Повышенная потребность в витамине В12
Беременные, кормящие женщины, дети раннего возраста, пациенты с ВИЧ/СПИДом, гемолитической анемией испытывают повышенную потребность в витамине B12.
Снижение содержания витамина B12 относится к этиологическим факторам ряда врожденных пороков. Речь прежде всего идет о дефектах формирования нервной трубки.
Низкие концентрации витамина В12 ( 12 мкмоль/л и MMК > 0,4 мкмоль/л. Однако ориентироваться только на эти показатели не следует, поскольку повышенный уровень HCYS имеет место при дефиците фолатов, витамина В6, гипотиреозе, а повышенный уровень ММК - при печеночной и почечной недостаточности [22, 24].
Еще одним методом диагностики дефицита В12 является определение уровня холотранскобаламина в сыворотке крови. В норме концентрация холотранскобаламина составляет от 19-42 до 134-157 пмоль/л. К преимуществу метода определения концентрации холотранскобаламина относится то, что его уровень в крови достаточно стабилен и практически не подвержен колебаниям при различных физиологических состояниях организма (беременность, прием некоторых лекарственных препаратов) [22].
Таким образом, на текущий момент золотого стандарта лабораторной диагностики дефицита витамина В12 не существует. Для точной постановки диагноза его дефицита необходима комплексная оценка клинических проявлений и лабораторных данных (общий уровень В12 в плазме и ликворе, уровень сывороточного холотранскобаламина, гомоцистеина и ММК).
На рисунке представлен возможный алгоритм диагностики дефицита витамина В12 у взрослых [25].
Терапия неврологических проявлений дефицита витамина В12
Целями терапии В12-дефицитных состояний являются нормализация лабораторных показателей крови и постепенный регресс неврологического дефицита. Одновременно с медикаментозным восполнением дефицита витамина В12 необходимы обследование, тщательный сбор анамнеза для выявления причины развития дефицитарного состояния и ее устранения или коррекции. Прогноз функционального восстановления зависит от исходной степени поражения нервной системы: при легких нарушениях и раннем начале лечения возможно полное или практически полное восстановление, при более тяжелых остаточный неврологический дефицит неизбежен.
Пациенты с доказанным дефицитом витамина В12 могут получать терапию в виде пероральных форм витамина, а также парентерально (главным образом внутримышечно), что зависит от тяжести неврологических проявлений, остроты развития заболевания и причины дефицита.
Как правило, пациентам в отсутствие нарушений всасывания в желудочно-кишечном тракте (ЖКТ) в случае постепенного развития неврологического дефекта и легкой или средней тяжести его выраженности показан прием пероральных препаратов, содержащих 1 мг кобаламина, ежедневно. При тяжелой степени неврологического дефекта и остром развитии заболевания, а также при состояниях, потенциально способных нарушать всасывание витамина В12 в ЖКТ, целесообразно начинать терапию с парентеральных форм витамина и его внутримышечного введения в дозе 1 мг [26, 27].
Единого мнения об оптимальных дозах витамина В12, направленных на коррекцию неврологических и гематологических проявлений дефицита, не существует.
В исследовании сравнивали эффективность лечения различными дозами перорального витамина B12 у пожилых больных с дефицитом В12. Установлено, что оптимальная доза перорального витамина B12, необходимая для снижения уровня ММК на 80-90%, составляет 647-1032 мкг/сут [28].
Начальные схемы лечения и продолжительность витаминотерапии в разных исследованиях, медицинских учреждениях и странах различаются.
Одна из предлагаемых схем лечения предполагает внутримышечные инъекции кобаламина - 8-10 инъекций в течение двух месяцев с последующими ежемесячными инъекциями по 1 мг препарата. Другие схемы включают пероральное введение кобаламина ежедневно в течение десяти дней, затем еженедельно в течение четырех недель с последующим ежемесячным приемом препарата внутрь. Британский национальный формуляр рекомендует использовать 1 мг витамина B12 внутримышечно три раза в неделю в течение двух недель, а затем один раз каждые три месяца для пациентов с мегалобластной анемией без неврологических синдромов. При наличии неврологических симптомов целесообразно внутримышечное введение 1 мг В12 через день в течение трех недель. Лечение может быть продолжено в зависимости от клинической ситуации. Пациентам с дефицитом витамина В12, причины которого невозможно устранить, как правило, требуется пожизненное лечение для предотвращения рецидива заболевания [26].
В руководстве по гематологии [27] для лечения В12-дефицитной анемии предложена следующая схема: цианокобаламин 1000 мкг внутримышечно ежедневно в течение 4-6 недель, после нормализации показателей крови 1000 мкг внутримышечно один раз в неделю, затем пожизненно 1000 мкг один раз в месяц, если устранить причину дефицита витамина В12 невозможно. Подобная схема применима и при тяжелых неврологических нарушениях, обусловленных дефицитом витамина В12. Пациенты, получающие лечение по поводу дефицита витамина B12, каждые несколько месяцев проходят лабораторный контроль уровня гемоглобина и витамина В12 [27].
При приеме цианокобаламина перорально в высоких дозах (1000-2000 мкг) возможно пассивное (за счет диффузии) всасывание 1% потребляемой дозы. В ряде исследований показано, что прием препаратов витамина В12 внутрь в высоких дозах по эффективности не уступает внутримышечному введению [24, 28]. В руководстве по гематологии также предложена альтернативная внутримышечному введению витамина В12 схема лечения: ежедневный прием препарата в дозе 2000-4000 мкг в течение 4-6 недель, затем 1000 мкг цианокобаламина один раз в месяц пожизненно [27].
В России единственной лечебной формой витамина В12 является цианокобаламин, который входит в состав комбинированных поливитаминных препаратов в таблетированной форме в дозах 200-500 мкг. Неинъекционных средств для специфического лечения дефицита витамина В12 в настоящее время нет.
Среди препаратов, содержащих витамин В12, следует отметить Нейробион (Австрия), который более 50 лет применяется в разных странах для лечения пациентов с заболеваниями нервной системы. Нейробион представляет собой комбинацию нейротропных витаминов: тиамина (витамин В1), цианокобаламина (витамин В12) и пиридоксина (витамин В6). Препарат выпускается в двух лекарственных формах - пероральной (таблетки) и парентеральной (раствор для инъекций). Одна таблетка Нейробиона содержит тиамина дисульфид 100 мг, пиридоксина гидрохлорид 200 мг и цианокобаламин 240 мкг. Одна ампула Нейробиона также содержит три витамина группы B: тиамин 100 мг, пиридоксин 100 мг и цианокобаламин 1 мг.
Как правило, лечение тяжелых форм поражения центральной и периферической нервной систем начинается с парентерального введения комплекса витаминов группы В. Инъекции Нейробиона проводят глубоко внутримышечно по 3 мл (одна ампула) один раз в сутки в течение десяти дней. Затем в качестве поддерживающей терапии Нейробион применяется в таблетированной форме. Стандартный лечебный курс для взрослых и детей старше 15 лет предполагает прием одной таблетки три раза в сутки на протяжении 1-3 месяцев в зависимости от тяжести клинических проявлений [29].
Пациенты с неврологическими нарушениями, обусловленными дефицитом витамина В12, помимо восполнения его дефицита в зависимости от конкретной патологии нуждаются в ноотропной и метаболической терапии, антиоксидантах (препараты альфа-липоевой кислоты). Им также назначаются реабилитационные мероприятия (массаж, лечебная физкультура, физиопроцедуры). Описана эффективность ритмической транскраниальной магнитной стимуляции при поражении спинного мозга на фоне фуникулярного миелоза [20].
Скорее всего распространенность дефицита витамина В12 в популяции значительно выше, чем принято считать. Это обусловлено увеличением продолжительности жизни и соответственно доли лиц пожилого и старческого возраста, высокой популярностью различных видов диет и вегетарианства, широким применением препаратов, способных нарушать всасывание и метаболизм витамина В12, и рядом других причин. Клинические проявления дефицита витамина В12 весьма разнообразны и включают в себя широкий спектр нарушений - от классической макроцитарной анемии до выраженных когнитивных и психических расстройств. Из-за отсутствия четких схем диагностики и недоступности в ряде случаев лабораторных исследований дефицит витамина В12 часто остается недиагностированным.
Ранняя диагностика неврологических проявлений дефицита витамина В12 и своевременное начало терапии крайне важны, поскольку позволяют сделать процесс неврологического дефицита обратимым.
Таким образом, проблема дефицита В12 требует дальнейшего изучения. Необходимо разработать четкие алгоритмы диагностики и лечения данной патологии.
Окислительный стресс и его коррекция при неврологических болезнях. Обзор литературы
Одним из универсальных механизмов жизнедеятельности клеток и процессов, происходящих в межклеточном пространстве, является образование свободных радикалов (СР). СР составляют особый класс химических веществ, различных по своему атомарному составу, но характеризующихся наличием в молекуле непарного электрона. СР являются непременными спутниками кислорода и обладают высокой химической активностью.
Процессы свободнорадикального окисления нужно рассматривать как необходимое метаболическое звено в окислительном фосфорилировании, биосинтезе простагландинов и нуклеиновых кислот; иммунных реакциях. Оксид азота выполняет роль нейромедиатора и принимает участие в регуляции кровотока. СР образуются при перекисном окислении ненасыщенных жирных кислот с регуляцией физических свойств биологических мембран.
С другой стороны, свободнорадикальное окисление является универсальным патофизиологическим феноменом при многих патологических состояниях. Кислород для любой клетки, особенно для нейрона, является ведущим энергоакцептором в дыхательной митохондриальной цепи. Связываясь с атомом железа цитохромоксидазы, молекула кислорода подвергается четырех-электронному восстановлению и превращается в воду. Но в условиях нарушения энергообразующих процессов при неполном восстановлении кислорода происходит образование высокореактивных, а потому токсичных СР или продуктов, их генерирующих.
Образованию СР способствуют многие процессы, сопровождающие жизнедеятельность организма: стрессы, экзогенные и эндогенные интоксикации, влияние техногенных загрязнений окружающей среды и ионизирующего излучения. По данным некоторых авторов, СР участвуют в патогенезе более 100 различных заболеваний. Патологическое действие СР связано прежде всего с их влиянием на структурное состояние и функции биологических мембран. Установлено, что гипоксия и ишемия тканей сопровождаются активацией перекисного окисления липидов. Как известно, в состав клеточных мембран входит большое количество фосфолипидов. При появлении в мембране СР вероятность его взаимодействия с жирной кислотой нарастает по мере увеличения числа кратных связей. Поскольку ненасыщенные жирные кислоты обеспечивают мембранам большую подвижность, то их изменения в результате процессов перекисного окисления липидов приводят как к увеличению вязкости мембран, так и к частичной утрате барьерных функций.
Головной мозг особо чувствителен к гиперпродукции СР и к так называемому окислительному стрессу. Окислительный стресс, ведущий к гиперпродукции СР и деструкции мембран, связанной с активацией фосфолипазного гидролиза, играет в патогенетических механизмах ишемии мозга особо значимую роль. В этих случаях основным фактором, повреждающим митохондриальные, плазматические и микросомальные мембраны, является высокоактивный гидроксильный радикал ОН. Повышенная продукция СР, инициируемая при ишемии мозга арахидоновой кислотой, является одной из причин длительного спазма сосудов и срыва церебральной ауторегуляции, а также прогрессирования постишемического отека и набухания за счет дезинтеграции нейронов и повреждения мембранных насосов. В процессе ишемии вследствие энергодефицита снижается активность ферментов антиоксидантной защиты: супероксиддисмутазы, каталазы и глутатионпероксидазы. Одновременно уменьшается количество практически всех водо- и жирорастворимых антиоксидантов.
| Как показано в исследованиях, выполненных на кафедре неврологии ФДПО РНИМУ им. Н.И.Пирогова, окислительный стресс играет значимую и неблагоприятную роль в патогенезе инфаркта мозга, субарахноидального кровоизлияния, внутримозговой гематомы и хронической ишемии мозга. |
Как показано в исследованиях, выполненных на кафедре неврологии ФДПО РНИМУ им. Н.И.Пирогова, окислительный стресс играет значимую и неблагоприятную роль в патогенезе инфаркта мозга, субарахноидального кровоизлияния, внутримозговой гематомы и хронической ишемии мозга.
В последние годы окислительный стресс также рассматривается как один из наиболее значимых факторов патогенеза таких нейродегенеративных заболеваний, как болезнь Альцгеймера и другие типы деменций, болезнь Паркинсона, боковой амиотрофический склероз, эпилепсия и рассеянный склероз.
| В настоящее время продолжается изучение использования производных янтарной кислоты с целью уменьшения выраженности ишемических повреждений головного мозга. Самым изученным на сегодняшний день препаратом является Мексидол © . |
Наряду со свободнорадикальным окислением в процессе функционирования биологических объектов из групп радикалов вырабатываются вещества, обладающие антиоксидантным действием, которые называют стабильными радикалами. Такие радикалы не способны отрывать атомы водорода от большинства молекул, входящих в состав клетки, но могут совершать эту операцию с особыми молекулами, имеющими слабо связанные атомы водорода. Рассматриваемый класс химических соединений получил название антиоксидантов (АО), поскольку механизм их действия основан на торможении свободнорадикальных процессов в тканях. В отличие от нестабильных СР, оказывающих повреждающее действие на клетки, стабильные СР тормозят развитие деструктивных процессов.
Существующая в организме физиологическая антиоксидантная система представляет собой совокупную иерархию защитных механизмов клеток, тканей, органов и систем, направленных на сохранение и поддержание в пределах нормы реакций организма, в том числе в условиях ишемии и стресса. Сохранение окислительно-антиоксидантного равновесия, являющегося важнейшим механизмом гомеостаза живых систем, реализуется как в жидкостных средах организма (кровь, лимфа, межклеточная и внутриклеточная жидкость), так и в структурных элементах клетки, прежде всего в мембранных структурах (плазматических, эндоплазматических и митохондриальных, клеточных мембранах). К антиокислительным внутриклеточным ферментам относятся супероксиддисмутаза, осуществляющая инактивацию супероксидного радикала, и каталаза, разлагающая пероксид водорода.
Известные к настоящему времени биологические и химически синтезированные АО подразделяются на жирорастворимые и водорастворимые.
Жирорастворимые АО локализуются там, где расположены субстраты-мишени атаки СР и пероксидов — наиболее уязвимые для процессов перекисного окисления биологические структуры. К числу таких структур относятся прежде всего биологические мембраны и липопротеины крови, а основными мишенями в них являются ненасыщенные жирные кислоты.
Следует отметить, что для того, чтобы набрать физиологически необходимый минимум АО из продуктов растительного происхождения, удельный их вес при ежедневном питании должен существенно превосходить все остальные компоненты пищи.
В рационе современного питания преобладают рафинированные и технологически обработанные продукты, лишенные ценных природных качеств. Если принять во внимание постоянно увеличивающуюся потребность в АО вследствие воздействия неблагоприятных факторов внешней среды, то становится понятной причина хронического дефицита АО у значительной части населения.
В последние годы изучается действие янтарной кислоты, ее солей и эфиров, представляющих собой универсальные внутриклеточные метаболиты. Янтарная кислота, содержащаяся в органах и тканях, является продуктом 5-й реакции и субстратом 6-й реакции цикла трикарбоновых кислот. Окисление янтарной кислоты в 6-й реакции цикла Кребса осуществляется с помощью сукцинатдегидрогеназы. Выполняя каталитическую функцию по отношению к циклу Кребса, янтарная кислота снижает в крови концентрацию других интермедиатов данного цикла — лактата, пирувата и цитрата, продуцируемых на ранних стадиях гипоксии. Феномен быстрого окисления янтарной кислоты сукцинатдегидрогеназой, сопровождающийся АТФ-зависимым восстановлением пула пиримидиновых динуклеотидов, получил название «монополизация дыхательной цепи», биологическое значение которого заключается в быстром ресинтезе АТФ. В нервной ткани функционирует так называемый аминобутиратный шунт (цикл Робертса), в ходе которого янтарная кислота образуется из аминомасляной кислоты (ГАМК) через промежуточную стадию янтарного альдегида. В условиях стресса и гипоксии образование янтарной кислоты возможно также в реакции окислительного дезаминирования кетаглутаровой кислоты в печени.
Антигипоксическое действие янтарной кислоты обусловлено ее влиянием на транспорт медиаторных аминокислот, а также увеличением содержания в мозге ГАМК при функционировании шунта Робертса. Янтарная кислота в организме в целом нормализует содержание гистамина и серотонина и повышает микроциркуляцию в органах и тканях, прежде всего в тканях мозга, не оказывая влияния на артериальное давление и показатели работы сердца. Противоишемический эффект янтарной кислоты связан не только с активацией сукцинатдегидрогеназного окисления, но и с восстановлением активности ключевого окислительно-восстановительного фермента дыхательной митохондриальной цепи — цитохромоксидазы.
В настоящее время продолжается изучение использования производных янтарной кислоты с целью уменьшения выраженности ишемических повреждений головного мозга. Самым изученным на сегодняшний день препаратом является Мексидол © .
Мексидол © (2-этил-6-метил-3-гидроксипиридина сукцинат), отечественный оригинальный антиоксидант и антигипоксант, создан в НИИ фармакологии РАМН в середине 80-х годов. За разработку и внедрение Мексидола © в клиническую практику группе специалистов в 2003 году присуждена премия правительства РФ.
Мексидол © состоит из двух связанных и функционально значимых соединений: 2-этил-6-метил-3-гидроксипиридина и янтарной кислоты. Наличие 3-гидроксипиридина в структуре Мексидола © обеспечивает комплекс его антиоксидантных и мембранотропных эффектов, способность уменьшать глутаматную эксайтотоксичность, модулировать функционирование рецепторов, что принципиально отличает мексидол от других препаратов, содержащих янтарную кислоту. Наличие сукцината в структуре Мексидола © отличает его от эмоксипина и других производных 3-оксипиридина, поскольку сукцинат функционально значим для многих процессов, протекающих в организме и, в частности, является субстратом для повышения энергетического обмена в клетке.
Сочетание в структуре Мексидола © двух соединений с необходимыми свойствами обеспечивает его хорошую проходимость через гематоэнцефалический барьер, высокую биодоступность и воздействие на различные мишени, следствием чего является широкий спектр эффектов препарата и высокий терапевтический потенциал.
Эффективность действия антиоксиданта, как и других лекарственных веществ, определяется дозой, сроками и способами их введения. В связи с этим рекомендуется курсовое использование инъекционной и таблетированной форм Мексидола © , начиная с 250-500 мг/сутки в/в или в/м в течение 10-15 дней, с последующим переходом на таблетированную форму (125 мг) по 12 таблетке 2-3 раза в день не менее месяца.
Таким образом, Мексидол © обладает широким мультимодальным спектром эффектов, оказывает наряду с антиоксидантной активностью выраженный противоишемический эффект с повышением энергетического потенциала мозга и нейромодулирующее действие на рецепторный аппарат мозга.
Очень нервное возбуждение

Обзор
Автор
Редактор
Шестая (и последняя) статья цикла о нейромедиаторах будет посвящена глутамату. Это вещество больше знакомо нам как усилитель вкуса в продуктах, но оно играет важную роль в нашей нервной системе. Глутамат — это самый распространенный возбуждающий нейротрансмиттер в нервной системе млекопитающих вообще и человека в частности.
Молекулы и связи
Глутамат (глутаминовая кислота) является одной из 20 основных аминокислот. Кроме участия в синтезе белков он может выполнять функцию нейромедиатора — вещества, которое передает сигнал от одной нервной клетки к другой в синаптической щели. При этом нужно учитывать, что глутамат, который есть в пище, не проникает через гематоэнцефалический барьер, то есть не оказывает прямого влияния на мозг. Глутамат образуется в клетках нашего тела из α-кетоглутарата путем трансаминирования. Аминогруппа переносится с аланина или аспартата, заменяя кетоновый радикал α-кетоглутарата (рис. 1). В итоге мы получаем глутамат и пируват или щавелевоуксусную кислоту (в зависимости от донора аминогруппы). Два последних вещества участвуют во многих важных процессах: щавелевоуксусная кислота, например, — это один из метаболитов в великом и ужасном цикле Кребса. Разрушение глутамата происходит при помощи фермента глутаматдегидрогеназы, и в ходе реакции образуются уже знакомый нам α-кетоглутарат и аммиак.
У глутамата, как и у большинства других медиаторов, есть два типа рецепторов — ионотропные (которые открывают мембранную пору для ионов в ответ на присоединение лиганда) и метаботропные (которые при присоединении лиганда вызывают метаболические перестройки в клетке). Группа ионотропных рецепторов делится на три семейства: NMDA-рецепторы, AMPA-рецепторы и рецепторы каиновой кислоты. NMDA-рецепторы так называются, поскольку их селективным агонистом, веществом, избирательно стимулирующим эти рецепторы, является N-метил-D-аспартат (NMDA). В случае AMPA-рецепторов таким агонистом будет α-аминометилизоксазолпропионовая кислота, а каинатные рецепторы избирательно стимулируются каиновой кислотой. Это вещество содержится в красных водорослях и используется в нейробиологических исследованиях для моделирования эпилепсии и болезни Альцгеймера. В последнее время к ионотропным рецепторам стали также добавлять δ-рецепторы: они расположены на клетках Пуркинье в мозжечке млекопитающих. Стимуляция «классических» — NMDA-, AMPA- и каинатных — рецепторов приводит к тому, что калий начинает выходить из клетки, а кальций и натрий поступают в клетку. В ходе этих процессов в нейроне возникает возбуждение, и запускается потенциал действия. Метаботропные же рецепторы связаны с системой G-белков и участвуют в процессах нейропластичности [1]. Под нейропластичностью понимается способность нервных клеток образовывать новые связи друг другом или уничтожать их. Также в понятие нейропластичности включается способность синапсов изменять количество высвобождаемого нейромедиатора в зависимости от того, какие поведенческие акты и мыслительные процессы происходят в данный момент и с какой частотой.
Глутаматная система неспецифична: на глутаминовой кислоте «работает» почти весь мозг. Прочие, описанные в предыдущих статьях, нейромедиаторные системы имели более или менее узкую специфику — например, дофаминовая влияла на наши движения и мотивацию [2]. В случае с глутаматом такого не происходит — слишком широко и неизбирательно его влияние на процессы внутри мозга. Сложно выделить какую-то конкретную функцию, кроме возбуждающей. По этой причине приходится говорить о глутаматной системе как о совокупности большого количества связей в головном мозге. Такую совокупность называют коннектомом. Мозг человека содержит огромное количество нейронов, которые образуют между собой еще большее количество связей. Составить коннектом человека — задача, которая на сегодняшний день науке не под силу. Однако уже описан коннектом червя Caenorhabditis elegans [3] (рис. 2). Поклонники идеи коннектома утверждают, что в человеческих коннектомах записана наша идентичность: наши личность и память. По их мнению, в совокупности всех связей прячется наше «Я». Также «связисты» считают, что после описания всех нейронных связей мы сможем понять причину множества психических и неврологических расстройств, а значит и сможем их успешно лечить.

Как мне кажется, эта идея перспективна. В упрощённом виде связи между нейронами можно представить в виде проводов, сложных кабелей, соединяющих одни нейроны с другими. При поражении этих связей — искажении сигнала, обрыве проводов — может происходить нарушение слаженной работы головного мозга. Такие болезни, возникающие при сбое в нейронных каналах связи, называются коннектопатиями. Термин новый, но за ним скрываются уже известные ученым патологические процессы. Если вам хочется узнать о коннектомах больше, рекомендую прочесть книгу Себастьяна Сеунга «Коннектом. Как мозг делает нас тем, что мы есть» [4].
Перегрузка сети

Рисунок 3. Структура мемантина. Мемантин является производным углеводорода адамантана (не путайте с адамантом). Рисунок из «Википедии».
В нормально работающем мозге сигналы от нейронов равномерно распределены по всем другим клеткам. Нейромедиаторы выделяются в необходимом количестве, и нет поврежденных клеток. Однако после инсульта (острое поражение) или при деменции (длительно текущий процесс) из нейронов в окружающее пространство начинает выделяться глутамат. Он стимулирует NMDA-рецепторы других нейронов, и в эти нейроны поступает кальций. Приток кальция запускает ряд патологических механизмов, что в итоге приводит к гибели нейрона. Процесс повреждения клеток за счет выделения большого количества эндогенного токсина (в данном случае — глутамата) называется эксайтотоксичностью.
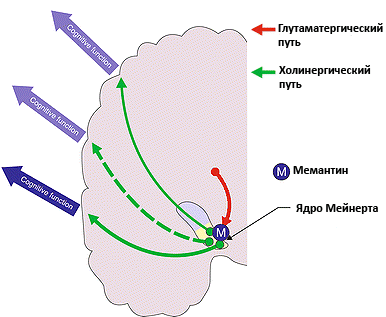
Рисунок 4. Действие мемантина при альцгеймеровской деменции. Мемантин снижает интенсивность возбуждающих сигналов, которые приходят от корковых нейронов на ядро Мейнерта. Ацетилхолиновые нейроны, составляющие эту структуру, регулируют внимание и ряд других когнитивных функций. Уменьшение избыточной активации ядра Мейнерта приводит к уменьшению симптомов деменции. Рисунок из [6].
Для того чтобы предотвратить развитие эксайтотоксичности или уменьшить ее влияние на течение болезни, можно назначить мемантин. Мемантин — очень красивая молекула-антагонист NMDA-рецепторов (рис. 3). Чаще всего этот препарат назначают при сосудистой деменции и деменции при болезни Альцгеймера. В норме NMDA-рецепторы заблокированы ионами магния, но при стимуляции глутаматом эти ионы высвобождаются из рецептора, и в клетку начинает проникать кальций. Мемантин блокирует рецептор и препятствует прохождению ионов кальция в нейрон — лекарство оказывает свое нейропротективное действие, снижая общий электрический «шум» в сигналах клетки. При альцгеймеровской деменции, помимо глутамат-опосредованных проблем, снижается уровень ацетилхолина — нейромедиатора, участвующего в таких процессах как память, обучение и внимание. В связи с этой особенностью болезни Альцгеймера психиатры и неврологи используют для лечения ингибиторы ацетилхолинэстеразы, фермента, который разрушает ацетилхолин в синаптической щели. Использование этой группы лекарств увеличивает содержание ацетилхолина в мозге и нормализует состояние пациента [5]. Специалисты рекомендуют совместное назначение мемантина и ингибиторов ацетилхолинэстеразы для более эффективной борьбы с деменцией при болезни Альцгеймера [6]. При совместном применении этих препаратов происходит воздействие сразу на два механизма развития болезни (рис. 4).
Деменция — это растянутое по времени поражение головного мозга, при котором гибель нейронов происходит медленно. А бывают заболевания, приводящие к быстрому и большому по объему поражению нервной ткани. Эксайтотоксичность — важный компонент повреждения нервных клеток при инсульте. По этой причине при нарушениях мозгового кровообращения применение мемантина может быть оправданно, однако исследования на эту тему только начинаются. В настоящее время есть работы, проведенные на мышах, где показано, что назначение мемантина в дозе 0,2 мг/кг в день уменьшает объем поражения мозга и улучшает прогноз инсульта [7]. Возможно, дальнейшие работы на эту тему позволят усовершенствовать терапию инсультов у людей.
Голоса в голове
Шизофрения — это еще одно заболевание, при котором воздействие на глутаматную систему мозга является новым и перспективным направлением терапии. В настоящее время главной причиной развития шизофрении считают нарушение дофаминовой передачи в мозге. Избыток дофамина в одних частях нервной системы приводит к бреду и галлюцинациям, а недостаток в других — к апатии, подавленности и отсутствию побуждений. Нейролептики — лекарства, блокирующие дофаминовые рецепторы — хорошо справляются с галлюцинациями и бредом, но с другой группой симптомов возникают проблемы. Ограниченность клинического эффекта нейролептиков указывает на то, что в развитие шизофрении могут быть вовлечены другие нейромедиаторные системы.
Если глутаматная система задействована при шизофрении, то можно проверить это даже на здоровых людях. Если здоровым испытуемым вводить препараты, блокирующие действие глутамата (кетамин, амфетамин), то у них развиваются симптомы шизофрении [8]. Введение кетамина больным шизофренией приводило к повторному возникновению психоза с повторением типичных для пациента симптомов, то есть кетамин вызывал не «просто психоз», а возвращал галлюцинации и бред, которые были раньше [9]. Это противоречие двух гипотез усложняет и без того непростую картину нейробиологических основ шизофрении. Психотическую симптоматику при введении кетамина можно объяснить его способностью влиять и на дофаминовые рецепторы. Другим объяснением может быть то, что дофаминовые и глутаматные нейроны способны оказывать влияние на полосатое тело. Эта часть мозга активно задействована в «производстве» галлюцинаций [10].
Самые частые галлюцинации у пациентов с шизофренией — слуховые: больной слышит «голоса» в своей голове. Голос может ругать, комментировать происходящее вокруг, в том числе и действия пациента. У одной из моих пациенток «голоса» читали вывески магазинов на улице, где она шла; другая услышала, как голос произнес: «Получишь пенсию, и пойдем в кафе». В настоящее время существует теория, объясняющая возникновение таких голосов. Представим, что пациент идет по улице. Он видит вывеску, а мозг автоматически «прочитывает» ее. При повышенной активности в височной доле, отвечающей за слуховое восприятие, у пациента возникают слуховые ощущения. Они могли бы подавляться за счет нормальной работы участков лобной коры, но этого не происходит из-за снижения их активности (рис. 5). Избыточная активность слуховой коры может быть вызвана гиперфункцией глутаматной (возбуждающей) системы или дефектом ГАМКергических структур, отвечающих за нормальное торможение в мозге человека. Вероятнее всего, недостаточная активность лобной доли в случае шизофрении также связана с нарушением нейромедиаторного баланса. Рассогласованность действий приводит к тому, что человек начинает слышать «голоса», которые явно соотносятся с окружающей обстановкой или передают его мысли. Очень часто свои мысли мы «проговариваем» в голове, что тоже может быть источником «голосов» в мозге человека, больного шизофренией [11].
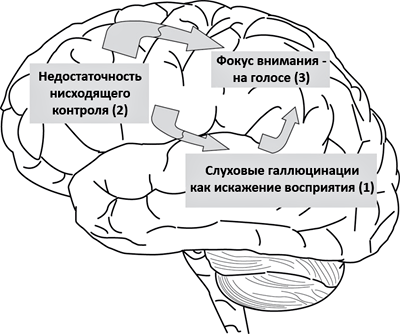
Рисунок 5. Возникновение слуховых галлюцинаций в мозге пациента с шизофренией. Первичное ощущение от автоматического «прочтения» вывесок или при возникновении мыслей, локализованное в височной коре (1), не подавляется лобной корой (2). Теменная кора (3) улавливает возникший паттерн активности в головном мозге и смещает на него фокус активности. В итоге человек начинает слышать «голос». Рисунок из [12].
На этом наше путешествие в мир нейромедиаторов закончено. Мы познакомились с мотивирующим дофамином, успокаивающей γ-аминомасляной кислотой и еще четырьмя героями нашего мозга. Интересуйтесь своим мозгом — потому что, как гласит название книги Дика Свааба, мы — это наш мозг.
Метаболомика биполярного аффективного расстройства
Метаболомика представляет собой науку , исследующую профиль низкомолекулярных метаболитов и предоставляет возможность характеризовать специфические метаболические фенотипы, связанные с заболеванием. Метаболомика имеет преимущество перед другими техниками «омикс» в том, что она напрямую анализирует метаболические изменения в организме и интегрирует информацию об изменениях на уровне генов, транскриптов и белков, а также посттрансляционных модификаций. Метаболический анализ с использованием времени пролетной масс-спектрометрии с капиллярным электрофорезом (CE-TOFMS) был использован для открытия биомаркеров психических расстройств, включая BD, шизофрению ( SZ) и расстройства аутистического спектра (ASD).
Некоторые авторы обнаружили, что уровни в CSF изоцитрата были значительно выше у пациентов с BD, чем у здоровых контролей, и что экспрессия мРНК и белка изоцитратдегидрогеназы (IDH3A) была значительно ниже в ткани мозга после смерти у пациентов с BD, чем в контрольных образцах. Эти данные свидетельствуют о том, что аномальный митохондриальный метаболизм изоцитрата с помощью IDH3A играет ключевую роль в патогенезе BD.
Представляет интерес оценка уровней основных метаболических соединений из следующих путей метаболического анализа: гликолитическая система, пентозофосфатный путь, цикл лимонной кислоты, цикл мочевины, путь метаболизма полиамина и креатина, путь метаболизма пурина, путь метаболизма глутатиона, путь метаболизма никотинамида, путь метаболизма холина и несколько путей метаболизма аминокислот. При биполярном аффективном расстройстве восемнадцать соединений, включая мочевую кислоту, CoA, глицерин-3-фосфат, пировиноградную кислоту (пируват), N- ацетилглутаминовую кислоту ( N-ацетилглутамат), 2-гидроксиглутаровую кислоту, 2-оксоглутаровую кислоту (α-кетоглутарат), лимонную кислоту (цитрат), цис-аконитовую кислоту (цис-аконитат), изоцитарную кислоту (изоцитрат), мочевину, β-аланин, серин, валин треонин, орнитин, глютамин и аргинин были изменены. Эти метаболиты представляют собой молекулы в цикле лимонной кислоты, в цикле мочевины и метаболизме аминокислот.
Метаболический путь цикла лимонной кислоты , цикла мочевины и метаболизма аминокислот
Аминокислотный обмен - это биохимические процессы , посредством которых различные аминокислоты производятся из других веществ. Пировиноградная кислота ( пируват) получается из глюкозы путем гликолиза . Пируват метаболизируется до оксалоацетата пируваткарбоксилазой. Ацетил - КоА также происходит из-за расщепления сахаров гликолизом , что приводит к образованию пирувата , который в свою очередь декарбоксилируется ферментом пируватдегидрогеназой ( PDH). Цикл лимонной кислоты , также известный как цикл трикарбоновых кислот ( TCA ) или цикл Кребса начинается с переноса двухуглеродной ацетильной группы из ацетил - Ко-А в четырехуглеродное акцепторное вещество оксалоацетат с образованием шестиуглеродного вещества ( цитрат). В цикле лимонной кислоты α-кетоглутарат синтезируется из изоцитратной кислоты ( изоцитрат) изоцитратдегидрогеназой ( IDH2) , N - ацетилглутаминовая кислота синтезируется из глутаминовой кислоты и ацетил - КоА с помощью N- ацетилглутаматсинтазы ( NAGS) . Аргинин синтезируется из цитруллина в цикле мочевины и играет роль в производстве оксида азота ( NO). β-аланин синтезируется из аспартата аспартат-1-декаи ацетил - рбоксилазой, а также является ограничивающим скорость предшественником карнозина.
Распространенность биполярного аффективного расстройства I и II в течение жизни оценивается от 1 до 3%. Пациенты с биполярным расстройством (BD) более склонны обращаться за медицинской помощью, когда они находятся в состоянии депрессии, чем когда они испытывают манию или гипоманию. Отметим, что монотерапия антидепрессантами увеличивает риск перехода к мании при BD и может иногда усиливать симптомы этого психического расстройства. Большинство специалистов рекомендуют назначение антидепрессантов только в качестве дополнительной терапии к стабилизаторам настроения, поэтому врачам необходимы биомаркеры для помощи в диагностике BD.
Исследователи предполагают, что воспаление, дисфункция митохондрий и окислительный стресс играют роль в патогенезе BD. Кроме того, нейротрофический фактор мозга (BDNF) и его предшественник proBDNF были предложены в качестве периферических биомаркеров для BD. Исследователи сообщали, что сывороточные уровни глутамина, глицина и D - серина были значительно выше у пациентов с BD, чем у здоровых людей, тогда как сывороточные уровни L - серина были значительно ниже у пациентов с BD. Однако , авторы не обнаружили изменений в этих аминокислотах в спинномозговой жидкости (CSF) у тех же пациентов с BD.
Сывороточные уровни пирувата, N-ацетилглутаминовой кислоты, α-кетоглутарата ( введение лития повышает сывороточные уровни α-кетоглутарата ) и аргинина значительно выше , а уровни β-аланина и серина значительно ниже у пациентов с биполярным аффективным расстройством (BD), чем у здоровых людей.
Накопленные исследователями данные подтверждают тот факт, что митохондриальная дисфункция играет ключевую роль в патогенезе биполярного аффективного расстройства.
Пируват
Пируват - конечный продукт гликолиза, продуцируется из дополнительных источников в клеточной цитоплазме и, в конечном итоге, предназначен для транспорта в митохондрии в качестве основного источника топлива, поддерживающего поток углерода цикла лимонной кислоты. Кроме того, пируват играет решающую роль для образования митохондриальной АТФ и для прохождения нескольких основных путей биосинтеза, пересекающих цикл лимонной кислоты. Пируват также превращается в ацетил-КоА с помощью комплекса пируватдегидрогеназы (PDH) и в оксалоацетат с помощью пируваткарбоксилазы. Обратим внимание читателя на то , что сывороточные уровни пирувата у пациентов с биполярным расстройством были значительно выше, чем у здоровых людей, в то время как сывороточные уровни ацетил-КоА и оксалоацетата не изменялись у пациентов с этим психическим расстройством.
Повышенные уровни пирувата играют роль в патогенезе BD. Поскольку пируват поставляет энергию живым клеткам через цикл лимонной кислоты, аномалия в цикле лимонной кислоты в митохондриях может играть роль и в патогенезе BD. Сообщалось, что у пациентов с расстройством аутистического спектра повышен уровень пирувата в сыворотке крови и спинномозговой жидкости , что свидетельствует о возможной дисфункции митохондрий также при расстройствах аутистического спектра.
α-кетоглутарат
Пациенты с BD имели более высокие сывороточные уровни α-кетоглутарата, чем здоровые люди. α-кетоглутарат (2-оксоглутарат) является ключевым метаболитом в цикле лимонной кислоты, но также и является обязательным субстратом для 2-оксоглутарат-зависимых диоксигеназ (2-OGDO). Семейство ферментов 2-OGDO включает основные ферменты метилирования ДНК и гистонов. Возможно, что измененные уровни α-кетоглутарата при BD приводят к эпигенетическим изменениям. Предполагается, что эпигенетические модификации играют важную роль в патогенезе ряда психических расстройств, включая BD. Предполагают, что препараты Li могут влиять на сывороточные уровни α-кетоглутарата у пациентов с BD, несмотря на то, что препараты Li , по - видимому , связаны с сывороточными уровнями α-кетоглутарата у пациентов с BD.
Изоцитрат
В литературе сообщалось, что уровни в CSF изоцитрата были значительно выше у пациентов с BD, чем у здорового контроля, и что экспрессия подтипа IDH3A изоцитратдегидрогеназы (IDH) была ниже в образцах мозга после BD, чем в контрольных образцах . Эти данные подтверждают гипотезу о дисфункции митохондрий при биполярном аффективном расстройстве. Тем не менее, исследователи не обнаружили изменений в уровне изоцитрата в сыворотке у тех же пациентов с BD . Таким образом, эти изменения могут быть специфичными для центральной нервной системы и не различимы в периферических образцах.
N- ацетилглутамат
N- ацетилглутамат биосинтезируется из глутаминовой кислоты (глутамата) и ацетил-КоА ферментом N- ацетилглутаматсинтазы (NAGS). NAGS млекопитающих обнаруживается преимущественно в митохондриальном матриксе клеток печени и кишечника. Известно также, что N- ацетилглутамат активирует карбамоилфосфат-синтетазу (CPSI) в митохондриальной матрице цикла мочевины . Исследователи обнаружили повышение уровня N-ацетилглутамата в сыворотке крови у пациентов с BD. Учитывая решающую роль N-ацетилглутамата в митохондриальном матриксе клеток, вполне вероятно, что изменение в уровня N -ацетилглутамата могут вызывать митохондриальную дисфункцию, поддерживая гипотезу митохондриальной дисфункции при BD. В CSF, однако, ранее исследователи не выявили, что N-ацетилглутамат находится ниже предела обнаружения. Следовательно, неизвестно, изменяются ли уровни N- ацетилглутамата в мозге у пациентов с BD.
Аргинин
Аргинин является условно незаменимой аминокислотой для взрослых млекопитающих. Эта аминокислота не только метаболически взаимозаменяема с аминокислотами пролином и глутаматом, но также служит предшественником для биосинтеза белков, креатина, полиамина, оксида азота (NO), агматина и мочевины. Аргинин в основном метаболизируется ферментами, аргиназой и NO-синтазой (NOS) с образованием мочевины и L-орнитина, а также NO и цитруллина соответственно. Исследователи обнаружили, что сывороточные уровни аргинина у пациентов с биполярным расстройтвом были значительно выше, чем у здоровых людей, что свидетельствует о нарушении цикла мочевины (или метаболизма аргинина) при биполярном расстройстве . Было выявлено , что сывороточные уровни аргинина связаны с анксиолитическими и антипсихотическими препаратами и, что уровни аргинина в CSF не различались между пациентами с BD и здоровым контролем . Одно одно из исследований исследование показало, что активность NOS в тромбоцитах от пациентов с BD была значительно ниже, чем у здоровых контролей, предполагая нарушение генерирования NO у пациентов с BD . Метаанализ показал, что уровни NO были изменены у пациентов с биполярным расстройством. таким образом, создается впечатление , что путь аргинин-NO может играть роль в патогенезе BD.
Серин
Сывороточные уровни общего серина ( l- серина и d- серина) значительно ниже у пациентов с BD, чем в контроле, однако, анализ метаболомики обычно не различал два энантиомера серина, хотя известно, что в крови человека существуют умеренные уровни d- серина . В то время как сывороточные уровни d- серина оказываются выше у пациентов с BD, сывороточные уровни l -серина , напротив ниже у пациентов с BD, чем в контрольной группе.Таким образом, можно предположить нарушение метаболизма серина у пациентов с биполярным аффективным расстройством . Интересно, что в литературе сообщалось о противоположной схеме при шизофрении, с повышением уровня l- серина и снижением уровня d- сыворотки в сыворотке. Последние данные позволяют предположить, что гипофункционирующие NMDA-рецепторы при шизофрении могут быть обусловлены снижением уровня d- серина . Уровень сывороточного общего серина, включая как l- серин, так и d- серин, у пациентов с биполярным расстройством значительно снижен , хотя оба энантиомера серина у пациентов с депрессией выше, чем у здоровых субъектов. Учитывая различную роль нейротрансмиссии NMDA-рецепторов в патогенезе шизофрении и BD, измерения сериновых энантиомеров ( d- и l- серина) могут представлять собой диагностические периферические биомаркеры для этих нарушений , позволяющих проводить дифференциальную диагностику. Серин-гидроксиметилтрансфераза 1 представляет собой фермент обратимого взаимопревращения между глицином и l -серином. l -серин также синтезируется из 3-фосфоглицерата 3-фосфоглицератдегидрогеназой, а d -серин синтезируется из l-серина сериновой рацемазой и метаболизируется с помощью d- аминооксидазы.
β-аланин
Исследователями фиксировалось снижение уровня β-аланина у пациентов с биполярным аффективным расстройством . Одно исследование показало, что уровни бета-аланина в спинномозговой жидкости не изменялись при биполярном расстройстве . β-аланин является естественной незаменимой аминокислотой. β-аланин может высвобождаться во время расщепления дипептида гистидина, такого как карнозин или ансерин. Или он может быть сформирован как вторичный побочный продукт реакции, которая превращает 1- аланин в пируват. Кроме того, β-аланин может продуцироваться во время процесса пищеварения, когда кишечные микробы удаляют атом углерода из l- аспартата, высвобождая как β-аланин, так и диоксид углерода . Точные механизмы, лежащие в основе синтеза / метаболизма β-аланина в патогенезе BD, в настоящее время неизвестны. Добавки β-аланина широко используются в качестве спортивного питания . Отсюда создается впечатление, что было бы интересно исследовать, могут ли добавки β-аланина влиять на симптомы у пациентов с BD.
Читайте также: