Поражение мотонейронов спинного мозга у детей. Синдромы Пенья-Шокейра и Мардена-Уокера
Добавил пользователь Владимир З. Обновлено: 01.02.2026
Спинальные амиотрофии — это генетические заболевания, проявляющиеся мышечной атрофией и обусловленные дегенеративными изменениями спинальных мотонейронов и моторных ядер ствола головного мозга. Общим симптомокомплексом выступают симметричные вялые параличи с атрофиями мышц и фасцикуляциями на фоне интактной чувствительной сферы. Диагностируются спинальные амиотрофии по данным семейного анамнеза, неврологического статуса, ЭФИ нервно-мышечного аппарата, МРТ позвоночника, ДНК-анализа и морфологического исследования мышечного биоптата. Лечение малоэффективно. Прогноз зависит от формы спинальной мышечной атрофии и возраста ее дебюта.

Общие сведения
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. Описаны в конце XIX века. Их частота составляет 1 случай на 6-10 тыс. новорожденных. Около 85% спинальных мышечных атрофий составляют проксимальные формы с более выраженной слабостью и атрофиями проксимальных мышечных групп конечностей. На долю дистальных форм приходится лишь 10% СМА. На сегодняшний день спинальные амиотрофии представляют практический интерес для целого ряда дисциплин: детской и взрослой неврологии, педиатрии, генетики.

Причины
Благодаря современной генетике установлено, что возникающие дегенеративные процессы двигательных нейронов обусловлены мутациями в генах SMN, NAIP, H4F5, ВTF2p44, расположенных на 5-ой хромосоме в локусе 5q13. Несмотря на то, что спинальные амиотрофии детерминируются аберрациями одного хромосомного локуса, они представляют собой группу разнородных нозологий, одни из которых проявляются в младенческом возрасте, а другие манифестируют у взрослых. В большинстве случаев амиотрофии наследуются аутосомно-рецессивно.
Патогенез
Генетические мутации приводят к развитию дегенеративных изменений в передних рогах спинного мозга. Нарушается иннервация и нейротрофика поперечно-полосатой мускулатуры. В результате постепенно возникает атрофия мышечной ткани. Преимущественное поражение отдельных групп мышц (проксимальных или дистальных частей верхних или нижних конечностей) у различных форм спинальных амиотрофий отличается. Характерно отсутствие нарушений чувствительности.
Классификация
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
- амиотрофией Верднига-Гоффманна;
- ювенильной формой Кугельберга-Веландера;
- хронической инфантильной СМА;
- синдромом Виалетто-ван Лэре (бульбоспинальная форма с глухотой);
- синдромом Фацио-Лонде.
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
- бульбоспинальная амиотрофия Кеннеди;
- скапулоперонеальная;
- лицелопаточноплечевая и окулофарингеальная формы;
- дистальная СМА;
- мономелическая СМА.
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Симптомы спинальных амиотрофий
Общим для спинальных мышечных атрофий является симптомокомплекс симметричного вялого периферического паралича: слабость, атрофия и гипотония мышечных групп одноименных конечностей (чаще вначале обеих ног, а затем и рук) и туловища. Пирамидные нарушения не типичны, но могут развиваться на поздних стадиях. Расстройства чувствительности отсутствуют, функция тазовых органов сохранена. Обращает внимание более выраженное поражение проксимальных (при проксимальных СМА) или дистальных (при дистальных СМА) мышечных групп. Типично наличие фасцикулярных подергиваний и фибрилляций.
Болезнь Верднига-Гоффмана
Встречается в 3-х клинических вариантах. Врожденный вариант дебютирует в первые 6 мес. жизни и является наиболее злокачественным. Его симптомы могут проявляться еще во внутриутробном периоде слабым шевелением плода. Дети с рождения имеют мышечную гипотонию, не способны переворачиваться и держать голову, при более позднем дебюте — не могут сидеть. Патогномонична поза лягушки — ребенок лежит с разведенными в стороны и согнутыми в коленях и локтях конечностями.
Амиотрофии имеют восходящий характер — вначале возникают в ногах, затем вовлекаются руки, позже — дыхательная мускулатура, мышцы глотки и гортани. Сопровождается задержкой психического развития. К 1,5 годам наступает смертельный исход.
Ранняя спинальная амиотрофия манифестирует до 1,5 лет зачастую после инфекционного заболевания. Ребенок утрачивает двигательные способности, не может стоять и даже сидеть. Периферические парезы сочетаются с контрактурами. После вовлечения дыхательных мышц развивается дыхательная недостаточность и застойная пневмония. Летальный исход обычно происходит в возрасте до 5-ти лет. Поздний вариант дебютирует после 1,5 лет, отличается сохранением двигательной способности до 10-летнего возраста. Летальный исход наступает к 15-18 годам.
Ювенильная спинальная амиотрофия Кугельберга-Веландера
Характеризуется дебютом в период от 2 до 15 лет. Начинается с поражения проксимальных мышц ног и тазового пояса, затем захватывает плечевой пояс. Около четверти пациентов имеют псевдогипертрофии, что делает клинику сходной с проявлениями мышечной дистрофии Беккера. В плане дифдиагностики большое значение имеет наличие мышечных фасцикуляций и данные ЭМГ. Течение амиотрофии Кугельберга-Веландера доброкачественное без костных деформаций, в течение ряда лет пациенты остаются способными к самообслуживанию.
Бульбоспинальная амиотрофия Кеннеди
Наследуется рецессивно сцеплено с Х-хромосомой, манифестирует только у мужчин после 30-летнего возраста. Типично медленное, относительно доброкачественное течение. Дебютирует с амиотрофии проксимальных мышц ног. Бульбарные расстройства появляются через 10-20 лет и благодаря медленному прогрессированию не вызывают нарушения витальных функций. Может наблюдаться тремор головы и рук. Патогномоничным симптомом выступают фасцикулярные подергивания в периоральных мышцах. Зачастую отмечается эндокринная патология: атрофия яичек, снижение либидо, гинекомастия, сахарный диабет.
Дистальная СМА Дюшенна-Арана
Может иметь как рецессивный, так и доминантный тип наследования. Дебют приходится чаще на 20-летний возраст, но может произойти в любой период до 50 лет. Амиотрофии начинаются в кистях рук и приводят к формированию «когтистой кисти», затем охватывают предплечье и плечо, в связи с чем рука приобретает вид «руки скелета». Парезы мышц голеней, бедер и туловища присоединяются гораздо позже. Описаны случаи манифестации заболевания монопарезом (поражением одной руки). Прогноз благоприятный, за исключением случаев сочетания данного вида СМА с торсионной дистонией и паркинсонизмом.
Скапуло-перонеальная СМА Вюльпиана
Манифестирует в период от 20 до 40 лет амиотрофиями плечевого пояса. Типичны «крыловидные лопатки». Затем присоединяется поражение перонеальной группы мышц (разгибатели стопы и голени). В ряде случаев вначале поражаются перонеальные мышцы, а затем плечевой пояс. Спинальная амиотрофия Вюльпиана отличается медленным течением с сохранностью способности передвигаться спустя 30-40 лет от ее дебюта.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем - эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Диагностика
Больных со спинальными амиотрофиями курируют врачи-неврологи, а при манифестации в детском возрасте - педиатры или неонатологи. При необходимости может потребоваться привлечение для консультации других специалистов - эндокринологов и генетиков. Для диагностики некоторых разновидностей амиотрофий большое значение имеют анамнестические данные, а именно возраст появления симптоматики (например, болезнь Верднига-Гоффмана всегда дебютирует у детей до 6 месяцев, а амиотрофия Кугельберга-Веландера - после 2 лет).
Во время осмотра пациента обращается внимание на снижение общего мышечного тонуса, ослабление или утрату сухожильных рефлексов, скелетно-мышечные деформации. У части больных отмечается псевдогипертрофия икроножных мышц. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
- Лабораторные исследования. В лабораторных анализах практически все показатели находятся в пределах нормальных значений. Исключение составляет концентрация креатинфосфокиназы, которая у небольшой части пациентов может быть высокой.
- ЭМГ. При проведении игольчатой электромиографии обнаруживаются признаки дегенерации двигательных спинномозговых нейронов - снижение скорости и амплитуды вызванных потенциалов действия, регистрация спонтанной биоэлектрической активности в покое (фасцикуляций, фибрилляций), «ритм частокола».
- Биопсия мышц. При гистологическом исследовании мышечной ткани отмечаются типичные для амиотрофии изменения: некроз миофибрилл, разрастание жировой и соединительной ткани, чередование атрофии и гипертрофии в сочетании с интактными участками мышечной ткани.
- Спирометрия. При поражении дыхательной мускулатуры во время выполнения функции внешнего дыхания выявляются рестриктивные нарушения в виде снижения жизненной емкости легких.
- Генетический анализ. Основной метод диагностики, позволяющий достоверно установить диагноз спинальной амиотрофии. С помощью полимеразной цепной реакции обнаруживаются генетические мутации.
Дифференциальная диагностика
Дифференциальный диагноз спинальных амиотрофий необходимо проводить с другими наследственными нейродегенеративными заболеваниями, поражающими мышечную ткань. К таким патологиям можно отнести:
- псевдогипертрофическую мышечную дистрофию Дюшенна;
- ювенильный боковой амиотрофический склероз;
- детский церебральный паралич.
Лечение спинальных амиотрофий
Немедикаментозная терапия
Все без исключения больные должны быть госпитализированы в стационар. В тяжелых ситуациях (например, при дыхательной недостаточности вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. Все мероприятия направлены на облегчение состояния пациента. Несмотря на это, комплексный подход и строгое соблюдение рекомендаций врачей позволяют улучшить качество жизни больного. Виды консервативной терапии, применяющиеся для терапии спинальных амиотрофий:
- Обеспечение питания. При значительном нарушении акта глотания особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение пациента вертикальным. Может понадобиться установка назогастрального зонда.
- ЛФК. С целью повышения тонуса мышц и замедления их атрофии рекомендуются регулярные физические нагрузки. Наиболее эффективно совмещение активных (выполняются специалистом) и пассивных упражнений (выполняются самим пациентом).
- Физиолечение. Для активации метаболизма в мышечных тканях назначаются сеансы электростимуляции модулированным током, грязевые аппликации, электрофорез.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах выполняются различные виды массажа - ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Ортопедическое лечение. Для предупреждения и коррекции костных деформаций и суставных контрактур рекомендуется использование ортопедических приспособлений: корсетов, ортезов, ортопедической обуви.
- Респираторная поддержка. Нередко возникает необходимость в устранении кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно проводиться непрерывно и подбираться индивидуально для конкретного пациента.
Медикаментозная терапия
Этиотропная терапия с доказанной эффективностью на сегодняшний день отсутствует. Лекарственные препараты, применяющие для лечения спинальных амиотрофий, следующие:
- Метаболические средства. Для улучшения обменных процессов в мотонейронах и мышечных тканях применяются препараты, стимулирующие функцию митохондрий (коэнзим Q10, янтарная кислота), ноотропы (пирацетам, гамма-аминомасляная кислота), L-карнитин.
- Вальпроаты и Кленбутерол. Исследования показали, что противоэпилептические препараты из группы производных вальпроевой кислоты и агонист бета-адренергических рецепторов Кленбутерол способны увеличивать образование белка выживаемости мотонейронов (SMN) и, соответственно, улучшать клиническое течение заболевания.
- ИПП и прокинетики. Пациентам с гастроэзофагальным рефлюксом, который нередко развивается при нарушении глотания, назначаются ингибиторы протонной помпы (эзомепразол) и средства, стимулирующие моторику желудочно-кишечного тракта (итоприд).
- Муколитики и отхаркивающие средства. У больных со слабостью дыхательной мускулатуры для устранения таких проблем как слабое отхаркивание и скопление в дыхательных путях густой мокроты применяются препараты, разжижающие мокроту (ацетилцисетин) и стимулирующие ее отхаркивание (терпингидрат).
- Гормональные препараты. Людям с возникшими эндокринологическими осложнениями показаны инсулин, сахароснижающие препараты (метформин), тестостерон и антиандрогены (дутастерид).
Хирургическое лечение
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. Лежачим больным, страдающим постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Экспериментальное лечение
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Профилактика
Специфических методов первичной профилактики не существует. Единственный способ предотвратить возникновение болезни - пренатальная диагностика (обнаружение мутаций в ворсинах хориона или амниотической жидкости) с прерыванием беременности. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Синдром фиксированного спинного мозга
Синдром фиксированного спинного мозга — это комплекс патологической симптоматики, вызванной натяжением спинного мозга вследствие фиксации его каудального отела. Состояние возникает в результате врожденных пороков развития, либо на протяжении жизни вследствие травм, новообразований. Синдром проявляется нарушениями чувствительности и двигательной функции нижних конечностей, дисфункцией органов таза, кожными симптомами в области поясницы. Для диагностики болезни назначается спинальное МР-сканирование, нейросонография с эхоспондилографией, ЭНМГ. Лечение оперативное: иссечение фиксирующих образований, шунтирование, краниовертебральная костнодуральная декомпрессия.
МКБ-10


СФСМ бывает врожденным и приобретенным. В первом случае он обусловлен аномалиями спинномозгового канала, к которым относят spina bifida, пояснично-крестцовые миеломенингоцеле, терминальные липомы медуллярного конуса, мальформации. У детей и взрослых может развиваться приобретенный синдром фиксации, который провоцируют следующие причины:
- Рубцово-спаечный процесс. Около 2/3 операций на спинном мозге и позвоночнике сопровождаются формированием соединительнотканных перетяжек, нарушающих нормальную структуру спинномозгового канала.
- Посттравматические изменения. СФСМ возникает как осложнение тяжелых механических повреждений спины при ДТП, бытовых или производственных травмах, вследствие которых происходят переломы позвонков, поражаются спинномозговые оболочки.
- Новообразования. Приобретенная аномалия формируется при кистах, других доброкачественных спинальных опухолях, а также является типичным последствием злокачественных образований нервной ткани или костно-хрящевых структур.
Среди факторов риска врожденного синдрома фиксированного спинного мозга выделяют дефицит фолиевой кислоты во время первого триместра беременности, что чревато серьезными дефектами формирования нервной трубки. Другие предрасполагающие факторы синдрома включают воздействие тератогенных факторов на эмбрион, наследственную предрасположенность, отягощенный акушерский анамнез.
В норме при сгибании туловища позвоночный канал удлиняется на 7%, при этом не происходит поражение спинальных тканей за счет его растяжения. Физиологически за функцию амортизации отвечает конечная нить, которая состоит из эластиновых, коллагеновых, ретикулярных волокон. Патофизиологической основой синдрома является тракция каудальной части спинного мозга, ее стойкая фиксация на уровне пояснично-крестцовых позвонков.
В случае СФСМ при растяжении спинного мозга ухудшается кровоток в его дистальных отделах, угнетается электрическая активность клеток. На биохимическом уровне аномалия проявляется снижением интенсивности окислительного фосфорилирования в митохондриях нейронов. Исследования демонстрируют функциональный характер возникших дисциркуляторно-метаболических нарушений, о чем свидетельствует сохранность спинномозговых проводящих путей.
Симптомы
Манифестация синдрома происходит в возрасте от 1 года до 4 лет, когда дети учатся ходить, у них происходит заметный скачок роста. Второй пик выявления заболевания приходится на период активного удлинения тела у детей подросткового периода: в 11-15 лет у девочек, в 13-18 лет — у мальчиков. Диагностика фиксированного спинного мозга осложняется из-за распространенности скрытого или малосимптомного течения.
Основную группу симптомов составляют неврологические нарушения, вызванные поражением дистальных спинномозговых отделов. У детей ощущается слабость в ногах, беспокоят неуверенная походка, неуклюжесть движений. Зачастую присоединяются нарушения температурной, тактильной, болевой чувствительности нижних конечностей. Со временем становится заметной гипотрофия мышц, непропорциональность строения тела.
Вторым по частоте встречаемости является кожный синдром, представленный видимыми изменениями в зоне поясницы (подкожная липома, усиленная пигментация, локальный гипертрихоз). Реже определяются гемангиома, дермальный синус. Постепенно у детей появляются и нарастают ортопедические нарушения: деформация стоп, укорочение конечностей, искривление позвоночника (сколиоз, кифоз). Нередко присоединяется дисфункция тазовых органов.
При отсутствии лечения неврологический дефицит у детей прогрессирует, возникают параличи в нижних конечностях, утрачиваются моторные навыки. Тазовые расстройства усугубляются, что проявляется недержанием кала и мочи, императивными позывами к мочеиспусканию, у девушек возможно чувство инородного тела во влагалище. До 37% пациентов сталкиваются с сильными постоянными или приступообразными болями в ногах и пояснице.
Опасным последствием болезни является гидроцефально-гипертензионный синдром, который обусловлен критическими нарушениями циркуляции цереброспинальной жидкости. У детей СФСМ обычно дополняется аномалией Арнольда-Киари. Патология проявляется мозжечковыми расстройствами (нистагм, атаксия, дизартрия), множественными поражениями черепных нервов (бульбарный, псевдобульбарный синдромы), нарушениями функций мозгового ствола.
После нейрохирургической коррекции в 5-50% случаев возможны рецидивы СФСМ, которые сложнее поддаются лечению. При неполном устранении фиксированного спинномозгового участка частота рецидивов достигает 80%. К ранним послеоперационным осложнениям относятся ликворея, краевой некроз кожного лоскута, смещение костных структур. Они требуют ревизии операционной раны.
Прием детского невролога начинается со сбора жалоб и анамнеза заболевания, детального клинического осмотра, проверки мышечной силы, поверхностной и глубокой чувствительности. При синдроме фиксированного спинного мозга симптомы у детей неспецифичны, поэтому для верификации диагноза применяются следующие диагностические методы:
- МРТ спинного мозга. Исследование демонстрирует низкое расположение и иммобилизацию конуса органа на фоне утолщенной или укороченной терминальной нити. К косвенным признакам относят незаращение задней стенки позвоночного канала, нарушения сегментации позвонков, патологические образования в канале позвоночника.
- Нейросонография. УЗИ головного и спинного мозга проводится у детей первого года жизни как скрининговый метод диагностики структурных аномалий ЦНС. Для дополнительного исследования позвоночника назначается высокоинформативная эхоспондилография.
- ЭНМГ. Электрофизиологическое исследование нервно-мышечной передачи необходимо для дифференциальной диагностики поражений центральной и периферической нервной системы у детей, уточнения объема и глубины поражения.
- Консультации специалистов. Помимо невролога, больного должны осмотреть другие профильные врачи: ортопед, уролог, специалист функциональной диагностики. Для решения вопроса о целесообразности оперативного лечения ребенка показано обследование у детского нейрохирурга.
- Исследование биологического материала. Ткани, иссеченные в ходе нейрохирургической операции, подлежат биохимическим, гистологическим, иммуногистохимическим исследованиям, чтобы уточнить природу патологического процесса, исключить злокачественные новообразования.
Лечение синдрома фиксированного спинного мозга
Основной метод лечения СФСМ — устранение фиксирующих компонентов, позволяющее высвободить спинной мозг, устранить его патологическое натяжение. Для повышения точности нейрохирургических операций используются системы интраоперационного нейрофизиологического мониторинга, в том числе регистрация ССВП, электростимуляционное картирование. Все хирургические методы объединяются в 2 группы:
- Полное устранение фиксации. Радикальный способ коррекции синдрома, который восстанавливает нормальные анатомические взаимоотношения в позвоночном канале. Применяется при врожденных пороках развития, небольших опухолях, посттравматических спайках.
- Неполное устранение фиксации. Такие операции выполняются, если фиксирующие ткани прочно соединены со структурами ЦНС, их разделение чревато неврологическим дефицитом. Это наблюдается при рубцово-спаечных процессах, крупных липомах с вовлечением спинного мозга.
При выраженном гидроцефально-гипертензионном синдроме (отек дисков зрительных нервов, нарушения сознания, индекс Эванса более 0,3) коррекция гидроцефалии производится до устранения фиксации спинного мозга. Проводятся ликворошунтирующие операции, эндоскопическая вентрикулоцистернотомия, вентрикулярный дренаж. При аномалии Арнольда-Киари детям показана костнодуральная краниовертебральная декомпрессия, пластика твердой мозговой оболочки.
Прогноз и профилактика
Вероятность излечения определяется степенью фиксации, причиной развития патологических изменений, наличием сопутствующих врожденных пороков или органических болезней ЦНС у ребенка. При радикальном нейрохирургическом устранении фиксирующих элементов прогноз благоприятный, удается достичь полной ликвидации неврологического дефицита. Менее оптимистичный прогноз для детей с осложненным течением заболевания, рецидивами после хирургического лечения.
Первичные превентивные меры: исключение тератогенных влияний на плод, всесторонняя акушерско-гинекологическая помощь беременным, предупреждение бытового и производственного травматизма. Вторичная профилактика заключается в диспансерном наблюдении пациентов у детского невролога с выполнением контрольных МРТ ежегодно в течение 3-х лет, а затем каждые 2-3 года.
1. Результаты хирургического лечения детей с синдромом фиксированного спинного мозга. Прогноз на основании данных спинальной ЗТл МРТ-трактографии/ К.В. Сысоев, А.Р. Тадевосян, Ю.В. Назинкина, В.А. Хачатрян// Вопросы нейрохирургии. — 2016. — №3.
2. Синдром фиксированного спинного мозга у детей (клиническое наблюдение и обзор литературы)/ К.В. Сысоев, Е.Н. Жарова, Ю.М. Забродская, В.А. Хачатрян// Нейрохирургия. — 2016. — №2.
3. Диагностика и лечение синдрома фиксированного спинного мозга у детей. Клинические рекомендации. — 2015.
4. Синдром фиксированного спинного мозга (клиника, диагностика, хирургическая коррекция, ближайшие и отдаленные результаты) в детском возрасте: автореферат диссертации/ А.А. Зябров. — 2012.
Синдром Денди-Уокера
Синдром Денди-Уокера - это врожденная патология нервной системы, для которой характерна триада признаков: гидроцефалия, гипоплазия или аплазия мозжечка, кисты задней черепной ямки. Болезнь имеет полиэтиологическую природу, среди провоцирующих факторов выделяют генетические аномалии, тератогенные влияния. Клиническая симптоматика включает классические признаки гидроцефалии, многообразные неврологические нарушения. Диагностика порока требует проведения нейросонографии, МРТ, эхокардиографии, также возможна пренатальная постановка диагноза при УЗИ-скрининге. Лечение заключается в консервативных и хирургических методах коррекции гидроцефалии.


Пороки ЦНС занимают второе место в группе врожденных болезней, уступая только сердечно-сосудистым аномалиям. До 80% всех патологий сопровождаются гидроцефалией, как при синдроме Денди-Уокера. Данное заболевание было описано американским нейрохирургом В. Денди в 1914 году, спустя 28 лет канадско-американский нейрохирург А. Уокер с коллегами предложил оперативный способ лечения порока. Патология встречается с частотой 1:25000-1:35000, девочки болеют чаще. Среди младенцев с врожденной гидроцефалией патология регистрируется в 3,5-12% случаев.
Этиологические факторы синдрома Денди-Уокера до сих пор точно не выяснены. По современным данным, патология имеет мультифакториальную природу, ее развитию способствуют как внутренние причины (хромосомные и генные мутации), так и воздействие экзогенных тератогенов. Провоцирующими факторами заболевания могут выступать:
- вирусные инфекции (цитомегаловирус, краснуха);
- гестационный диабет;
- употребление алкоголя матерью во время беременности.
Новейшие исследования показывают четкую связь изолированной формы аномалии Денди-Уокера с мутациями генов ZIC1 и ZIC4. Риск развития заболевания повышается у младенцев с врожденными нарушениями обмена веществ. Чаще всего синдром ассоциирован с одной из форм 3-метилглутаконовой ацидурии, которая проявляется ретинопатией, почечной дисфункцией, метаболическим ацидозом и повышением активности печеночных ферментов.
Выделяют три основные гипотезы структурных поражений головного мозга при болезни Денди-Уокера. Согласно первой из них, пороки ЦНС вызваны замедлением эмбрионального развития на раннем этапе при закладке ромбовидного мозга. Такая ситуация наблюдается под действием тератогенных факторов либо на фоне генетических мутаций. Вторая гипотеза указывает на заращение выходного отверстия 4 желудочка мозга и позднее открытие апертуры Мажанди.
Новые экспериментальные данные подтверждают вероятность третьей гипотезы: типичные анатомические изменения развиваются при возникновении сосудистого сплетения на тонкой крыше ромбовидного мозга. Патология сопровождается внутриутробной гидроцефалией, вызывает серьезные неврологические нарушения. На фоне этого развивается большая киста в задних отделах черепа, которая препятствует формированию мозолистого тела и червя мозжечка.
Специалисты выделяют открытую и закрытую формы синдрома. В первом случае наблюдается окклюзия отверстий Люшка и Мажанди, что сопровождается серьезными нарушениями ликвородинамики. Второй вариант не имеет таких патологий, поэтому протекает в более благоприятной форме. В нейрохирургии важное значение имеет анатомическая классификация порока, согласно которой выделяют 2 формы синдрома Денди-Уокера:
- Полная. Характеризуется отсутствием червя мозжечка, наличием связи между полостью четвертого желудочка и мозжечково-мозговой цистерной.
- Неполная. Проявляется частичным недоразвитием мозжечкового червя в его нижней части, за счет чего коммуникация с большой цистерной реализуется не на всем протяжении.
В 1989 г. американским педиатром и нейрорадиологом Джеймсом Барковичем была предложена альтернативная классификация, базирующаяся на результатах МР-диагностики. Выделена группа заболеваний, названная «комплексом Денди-Уокера», который включает 4 клинических формы: классическую аномалию (полная форма), вариант Денди-Уокера (менее грубые нарушения), кисту кармана Блейка и mega cisterna magna.
Основное проявление синдрома Денди-Уокера - гидроцефалия, возникающая в первые месяцы жизни ребенка. Степень выраженности симптоматики зависит от варианта синдрома, скорости прогрессирования нарушений, общего состояния новорожденного. К ранним признакам относят беспокойное поведение, монотонный крик, нарушение питания и частые срыгивания. Внешне обнаруживается выбухание родничков, расширение черепных швов, быстрое увеличение окружности головы.
Характерный признак гидроцефалии при синдроме Денди-Уокера - симптом заходящего солнца: при взгляде вниз радужка частично скрывается нижним веком, над ней появляется белая полоска склеры. Неврологические симптомы также представлены нистагмом, экзофтальмом, косоглазием. Нередко у новорожденных отмечается судорожный синдром, парезы и параличи конечностей. При прогрессировании болезни характерно отставание в психомоторном развитии.
В 65-68% случаев синдром Денди-Уокера сопровождается другими структурными неврологическими аномалиями. Чаще всего диагностируется агенезия мозолистого тела, стеноз Сильвиева водопровода, гетеротопия извилин коры мозжечка. При тяжелых формах заболевания определяется недоразвитие ствола головного мозга, что сопряжено с глубоким угнетением витальных функций и высоким риском младенческой летальности.
До 55% детей имеют сопутствующие врожденные синдромы: Уокера-Варбурга, PHACE, Ritscher-Schinzel. Характерны различные формы хромосомных аберраций: делеции, дупликации, трисомии и триплоидии. Поражение кардиоваскулярной системы проявляется септальными дефектами и нарушениями постнатальной гемодинамики. Реже определяются офтальмологические пороки: ретинальная дисплазия, микрофтальмия.
Первичное обследование ребенка с неврологическими нарушениями проводится врачом-педиатром или неонатологом. При гидроцефалии и подозрении на синдром Денди-Уокера к диагностике подключается детский невролог, нейрохирург. Физикальный осмотр выявляет неспецифические признаки внутричерепной гипертензии, нарушения моторного развития, сопутствующие аномалии. Для постановки окончательного диагноза проводится:
- Нейросонография. При ультразвуковом сканировании головного мозга определяется крупное кистозное образование в задней части черепной коробки. На УЗИ мозжечковый червь не визуализируется, недоразвитые полушария мозжечка раздвинуты, желудочковая система мозга расширена и деформирована.
- МРТ головного мозга. Магнитно-резонансная томография в сагиттальной и аксиллярной проекции демонстрирует расширение четвертого желудочка, грубые нарушения развития мозжечка, другие структурные аномалии.
- Эхокардиография. УЗИ сердца рекомендовано всем детям с синдромом Денди-Уокера, поскольку он нередко ассоциирован с врожденной кардиальной патологией. По результатам ЭхоКГ можно определить аномалии развития клапанного аппарата, дефекты внутрисердечной перегородки, патологии расположения крупных сосудов.
- Кариотипирование. При комбинированных врожденных пороках развития необходима консультация генетика и тщательное изучение генетического материала ребенка. Углубленное исследование направлено на диагностику хромосомных аберраций.
- Пренатальная диагностика. При проведении УЗИ плода удается предположить аномалию на сроке 15-16 недель, более четкая визуализация структур IV желудочка возможна после 22 недели гестации. При сочетании патологии с расщелинами лица эхосонография может быть недостаточно информативна.
При постановке диагноза необходимо дифференцировать синдром с гипоплазией мозжечка другой этиологии, ретроцеребральными кистами и расширениями большой мозговой цистерны. Патогномоничным признаком синдрома Денди-Уокера считается дефект червя мозжечка, который не возникает при других вариантах пороков ЦНС. Проводится дифференциальная диагностика с арахноидальными кистами, которая требует дополнительных инструментальных методов исследования.
Лечение синдрома Денди-Уокера
Консервативная терапия
Наибольшую опасность для жизни и здоровья младенца представляет гидроцефалия, для коррекции которой показано лечение в отделении интенсивной терапии. Назначается массивная дегидратационная терапия, которая включает разные типы диуретиков, гипертонические инфузионные растворы. Усилия врачей направлены на уменьшение ликворопродукции и нормализацию внутричерепного давления, чтобы предупредить отек мозга и компрессию церебральных структур.
При неэффективности консервативных методов и неуклонном прогрессировании гидроцефалии требуется помощь детских нейрохирургов. Основным методом коррекции при синдроме Денди-Уокера являются шунтирующие операции с имплантацией дренажных трубок или клапанных регулируемых систем. Существует несколько типов шунтирования: вентрикуло-перитонеальное, вентрикуло-атриальное, вентрикулоцистерностомия.
Для коррекции окклюзии ликворных путей применяются методы эндоскопической вентрикулостомии, которые восстанавливают нормальный отток цереброспинальной жидкости, устраняют острую неврологическую симптоматику. При гипертензионно-гидроцефальном кризе показана вентрикулярная разгрузочная пункция, которая относится к вариантам экстренного временного дренирования.
Исход заболевания зависит от глубины поражения ЦНС, скорости нарастания гидроцефалии, наличия сочетанной патологии. Нарастающая гипертензия и грубые неврологические пороки в 90% случаев становятся причиной смерти пациента в первые годы жизни, для выживших детей с неполным вариантом аномалии прогноз более благоприятный. Вследствие недостаточной изученности этиопатогенеза болезни эффективные меры профилактика пока не разработаны.
1. Случая мальформации Денди-Уокера с доношенной беременностью и родами / Е.В. Беляева, Л.В. Лапшина, Е.В. Шапошникова, А.А. Молгачев // Журнал неврологии и психиатрии. - 2018. - №2.
2. Аномалия Денди-Уокера - редкая причина сирингомиелии у взрослых /Г.Ю. Евзиков// Неврология, нейропсихиатрия, психосоматика. - 2017. - №9.
3. Синдром Денди-Уокера у новорожденных/ Л.А. Петрова, А.В. Розанов, Ю.И. Барашнев, В.О. Панов// Российский вестник перинатологии и педиатрии. - 2010. - №1.
Пирамидная недостаточность у детей и взрослых: причины, симптомы и лечение

Пирамидный синдром или синдром пирамидной недостаточности еще называют синдромом балерины, ходящей на цыпочках, либо эквинусной постановкой стоп.
Данное нарушение может быть следствием целого комплекса факторов. В подавляющем большинстве случаев оно сопровождается мышечной дистрофией разной степени, сопряженной с гипертонусом голени и стопы.
Поражение пирамидных путей с двухсторонней мышечной дистонией обычно диагностируют у новорожденных или деток на первом году жизни, реже оно проявляется у ребят двух - трех лет от роду.
Анатомическая справка
Продолговатый мозг, соединяющий головной мозг со спинным, отвечает за работу сложных рефлексов. Состоит данный орган из клеток особой пирамидальной формы, которые так и называют - пирамиды. Поэтому и синдром именуется пирамидным. Нарушение координации движений - следствие повреждения вышеозначенных клеток.
Любопытно, что официально симптома «пирамидальная недостаточность» не существует. Тем не менее, его ставят маленьким детям - и далеко не всегда оправданно.
Основная причина развития у младенца такой патологии - механическая либо ишемическая внутриутробная травма шейного отдела спинного мозга. При нарушении тока крови в шейном утолщении и стволе головного мозга мышечный тонус сгибателей конечностей претерпевает значительное отклонение от нормы.
Степень выраженности синдрома находится в прямой зависимости от тяжести поражения мозга. Легкая форма недостаточности проявляется в скованности рук, а более распространенная ишемия добавляет к симптоматике нарушения двигательной активности нижних конечностей: при попытке поставить малыша на ножки он упорно опирается только на носки, то есть работает дистальный отдел стопы.
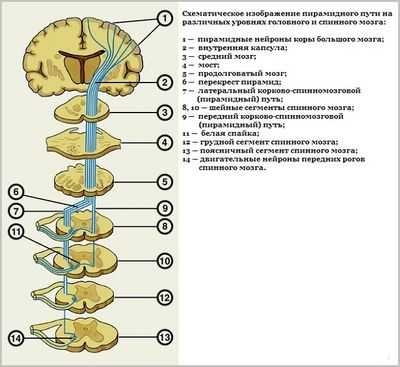
Синдром пирамидальной недостаточности у детей
У малышей первых месяцев жизни пирамидальный синдром обычно проявляется повышенным тонусом мышц ручек и ножек (они становятся сильно зажатыми), отсутствием возможности держать головку, подгибанием пальчиков при проверке шагового рефлекса (ходьба на пальчиках), и в других подобных признаках.

Детки старшего возраста, страдающие от пирамидального синдрома, могут все время стоять и - или ходить на цыпочках, быстро уставать и постоянно проситься «на ручки». Некоторые маленькие пациенты не желают ходить вовсе и жалуются на боль в ножках. Родителям трудно отделить подобного рода жалобы от простых капризов, потому что двух - трехлетние малыши не могут их понятно объяснить.
В вопросе, необходимо ли лечить данное нарушение, главное - тяжесть проблемы. При незначительном поражении малыша стоит просто наблюдать у участкового невролога. Также необходимо самостоятельно помогать ребенку массажем, ванночками и специально разработанным комплексом физических упражнений.
При более обширном поражении либо несвоевременном или неправильном лечении ребенок может начать отставать от норм развития моторики. В таком случае возможно появление не увиденных на первичном осмотре признаков неврологического дефицита. Таким деткам следует незамедлительно начать лечение.
Родителям младенцев с пирамидальной недостаточностью важно уяснить, что «простая» ходьба на цыпочках при отсутствии своевременной квалифицированной помощи может перерасти в устойчивые нарушения двигательных функций, иногда доходящие до полного их отказа.
О причинах и симптомах
Пирамидальная недостаточность в ногах у детей может развиваться на фоне следующих патологий:
- родовые травмы;
- врожденные поражения нервной системы (ДЦП, гидроцефалия и другие);
- гипоксия мозга;
- вирусные и бактериальные инфекции;
- гнойные воспаления спинного или головного мозга;
- опухоли;
- нарушения тока спинномозговой жидкости;
- менингит, энцефалит.
Внешне выраженные симптомы:
- дрожание подбородка, а также ножек и ручек;
- трудности с удержанием вещей;
- запрокидывание головы;
- плохое управление пальцами;
- ходьба на цыпочках;
- расстройство речи;
- пониженный интеллект.
Синдром пирамидной недостаточности у детей не может быть диагностирован со стопроцентной точностью исключительно по данным врачебного осмотра. Для получения исчерпывающих данных требуется сдать все назначенные врачом анализы. Только на основе их результатов возможна постановка правильного диагноза.
«Взрослые» причины
Пирамидная недостаточность - не болезнь, а синдром. Однако это не мешает многим врачам классифицировать патологию как заболевание и ставить однозначный диагноз. Медики часто обозначают ее как центральный паралич или парез.
Если синдром пирамидной недостаточности у самых маленьких детей может быть следствием родовых травм, гипоксии либо аномалий внутриутробного развития, то взрослом возрасте синдром чаще всего наблюдается у людей, страдающих онкологическими заболеваниями либо болезнями сосудов или сердца.
Пирамидная недостаточность у взрослых развивается вследствие таких причин:
- воспалительные процессы в тканях мозга (энцефалит, менингит или подобные заболевания);
- патологические изменения мозгового кровообращения (к примеру, вследствие перенесенного инсульта);
- механические повреждения черепа, повлекшие за собой нарушения процесса передачи нервных импульсов;
- доброкачественные или злокачественные опухоли головного мозга.
Пирамидальный синдром у взрослых пациентов - приобретенная патология, лечение которой подразумевает устранение не только симптомов, но и первопричины нарушений.
Клиническая картина и диагностические методы
Для выявления пирамидального синдрома у взрослых недостаточно простого осмотра невропатолога. Пациент должен пройти ряд современных диагностических процедур. Чтобы получить исчерпывающие сведения о состоянии здоровья больного, невропатологу потребуются результаты следующих обследований:
- магнитно-резонансной томографии (назначается при наличии судорог либо подозрении на эпилепсию);
- компьютерной томографии мозга;
- электроэнцефалографии, которая помогает проследить скрытые судороги (в большинстве случаев они имеют место во сне и поэтому не могут быть выявлены на осмотре невропатолога);
- электромиографии, позволяющей определить электрический потенциал мышц;
- УЗИ головного мозга (при подозрении на опухолевые образования).
Пирамидальная недостаточность во взрослом возрасте также проявляется и в более заметных симптомах, которые возможно выявить на врачебном осмотре:
- гипертонии;
- повышенном тонусе мышц рук и ног;
- судорогах;
- полном либо частичном параличе разных частей тела;
- нарушениях рефлекторной деятельности, в частности, снижении ее скорости;
- в некоторых случаях, особенно если поражение настигло гипоталамо-гипофизарную систему, могут наблюдаться половая дисфункция и повышенная масса тела.
Как лечить и нужно ли?
Если пирамидальная недостаточность не сопровождается другим, более серьезным диагнозом (из приведенных выше списков), для ее устранения достаточно процедур физиотерапии. Для лечения детей повсеместно используют массаж. Профессионал - массажист помогает укрепить мышцы и снять гипертонус. Родители же выполняют с ребенком в домашних условиях комплекс специальных профилактических упражнений.
Также помогают наладить двигательную активность и укрепить организм плавание, гимнастика, упражнения на координацию движений, бальнеотерапия (целебные ванны). Данная рекомендация актуальна как для детей, так и для взрослых.

В дополнение врач может назначить витамины, активирующие микроциркуляцию вазоактивные вещества или препараты для улучшения метаболизма нервных клеток (Ноотропил, Церебролизин, Энцефабол и другие). Нормализовать проведение нервных импульсов помогают Прозерин и Дибазол. Улучшить тонус мышц можно с помощью витаминов групп Е и В, а также препараты Баклофен, Лиоресал и Мидокалм.
Нейрохирургическая терапия назначается, если имеют место травмы спинного или головного мозга, либо опухоли. Оперативное вмешательство применяется при нарушениях кровообращения в мозгу, мальформациях церебральных сосудов, а также при внутримозговой гематоме и тромбозе экстрацеребральных артерий.
Лицам, страдающим от данной патологии, особенно малышам, которые только учатся ходить, необходимо носить специальную ортопедическую обувь. Ее отличает закрытый перед и жесткая задняя часть.
Также родителям малышей до года стоит запомнить: ребенка не нужно раньше времени ставить на ножки, не стоит сажать в ходунки - это может помешать нормальному развитию стопы. Когда придет время, малыш сам попробует ходить - и у него обязательно все получится!
Взрослым для лечения пирамидальной недостаточности назначаются ощутимые физические нагрузки: бег, плавание, прогулки, занятия лечебной физкультурой и спортом. Отличные результаты дает ЛФК, точечный массаж и рефлексотерапия.
Если же синдром возник на фоне другого заболевания, основной упор следует сделать на исцеление от него. Так же важно восстановить двигательную активность пациента, если имеется паралич. Однако не стоит забывать и о необходимости купировать симптомы описываемой патологии; в особенности это важно для маленьких пациентов.
Чтобы избежать такого неприятного и опасного своими осложнениями патологического состояния, как пирамидная недостаточность, необходимо:
Болезнь двигательного нейрона: симптомы, диагностика и лечение БАС и других форм БДН
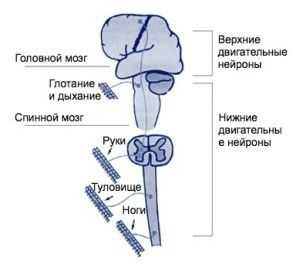
Верхние нейроны головного мозга, отвечающие за движение, расположены в полушарной коре, спускаются и контактируют в бульбарном отделе своими отростками с клетками спинного мозга.
Мотонейроны спинного мозга также распространены в поясничном, грудном и шейном отделах (исходя из типа мышц, куда направлены сигналы, регулирующие их сокращения). Двигательный нейрон проводит нервный импульс от спинного мозга к коре головного.
Болезнь двигательного нейрона (БДН) является нейродегенеративной патологией с нарастающим характером, в результате чего повреждаются мотонейроны спинного и головного мозга. В результате постепенного отмирания нервных волокон нарастает мышечная слабость с усилением симптоматики и летальным исходом.
Из-за повреждения моторных нейронов пропадают функции спинного мозга и позвоночника, постепенно наступает паралич, теряется речь, затрудняется глотание.
Уровень заболеваемости составляет 2-5 человека к 100 тысячам людей ежегодно. Заболевание проявляется в возрасте 50-70 лет, иногда у пациентов моложе 40 лет. В среднем пациенты живут 1-4 года после постановки диагноза.
Поражаемые области
С постепенным развитием заболевания двигательных нейронов у пациентов проявляется нарастание проблем с дыханием, глотанием, движением и общением с другими людьми. Могут проявляться следующие затруднения:
- Кисти, руки. Даже при выполнении ежедневных дел (мытья посуды, поворачивание крана, нажатие клавиш) будут проявляться все большие трудности с усилением слабости в ладонях и руках.
- Ноги. Из-за ослабления ног пациенту будет все труднее передвигаться.
- Эмоциональное состояние. Иногда могут поражаться зоны, которые отвечают за эмоциональные реакции, что проявляется непроизвольным смехом или плачем физиологического характера.
- Плечи, шея. Через определенное время пациенту становится трудно дышать из-за слабеющих мышц шеи.
- Дыхание. В основном заболевание распространяется на дыхательные мышцы, затрудняя дыхание.
- Глотание, речевые функции. Могут быть затруднения с приемом пищи, питьем и разговором.
У некоторых пациентов БДН вызывает нарушения памяти, сниженную концентрацию внимания, сложности с выбором слов и обучаемостью. То есть происходит незаметное и скрытое изменение психических функций.
При БДН остается не нарушенным интеллект, пациенты осознают все происходящее. У многих людей не поражается зрение, вкус, осязание и слух, остаются работоспособными функции мочевого пузыря, кишечника, половых органов (вплоть до поздних стадий) и сердечной мышцы.
Патогенез и патоморфология болезни
Болезнь мотонейрона считается патологией мультифакторного типа, развитие которой происходит от воздействия генетической наследственности с окружающими факторами.
В 10% ситуаций болезнь связана с семейными формами, где 25% — характеризуются мутированием гена СОД-1. Иногда спорадические виды патологии связаны с изменениями других генов (нейрофиламентов) в результате нарушений функций или структуры, при действии которых мотонейроны начинают дегенерировать. Механизм запускается при действии провокаторов (возраста, пола, тяжелых металлов и пр.). Маловероятной является взаимосвязь появления заболевания от инфекционных агентов.
Клинически нейроны дегенерируют при разрушении от 80% клеточных структур, что затрудняет своевременную терапию патологии и нуждается в диагностических исследованиях во время доклинического этапа.
Возле лобных долей, в 3, 5 слоях центральных извилин, двигательных ядрах 5, 7, 9 и 11 нервов черепа в стволе мозга определяется дегенерация нейронов и изменение кортикоспинальных путей. На различных этапах изменения мотонейронов определяются эозинофильные, базофильные включения и тельца Буниной.
Аксональные сфероиды образуются а проксимальных отделах аксонов. На основании полученных данных, отмечаются нарушения перемещения и деградации белков. В мышечных структурах определяется денервационная атрофия.
Что является причиной гибели двигательных нейронов?
По мнению специалистов, основными причинами БДН является наследственность и средовые факторы, повышающие риск патологии.

Примерно у 5-10% пациентов с болезнью моторного нейрона была выявлена семейная наследственная форма. В основном БДН была диагностирована (90-95% случаев) при спорадической форме, которая появляется по неизвестным причинам.
По эпидемиологическим исследованиям была выявлена возможная связь действия шоковых, механических травм, высоких спортивных нагрузок, вредных веществ на развитие заболевания.
Сейчас проводятся обследования механизмов повреждения внутриклеточных процессов, из-за которых погибают двигательные нейроны с окружающими клетками.
К возможным причинам относят забастовку и сбой работы «редакторов» в обработке РНК, изменения химических коммуникативных связей спинного мозга, нарушения транспортировки продуктов метаболизма, питательных веществ, скопления агрегатов белка в нейронах с нарушением нормальных функций и склеиванием, появление свободных радикалов кислорода, нарушение энергетического фона клеток и недостаток питательных веществ для мотонейронов. Также гибель двигательных нейронов может возникать из-за окружения глиальными клетками, которые могут быть токсичными.
Основные и редкие виды БДН
Заболевание двигательных нейронов имеет несколько основных форм:
- БАС (боковой амиотрофический склероз) развивается у 80% людей с болезнью двигательного нейрона, вызывает в основном слабость, судороги и истощение в мышцах, скованность верхних и нижних конечностей;
- ПБП (бульбарный паралич прогрессирующего типа) поражает около 10-25% людей, распространяется на нейроны спинного и головного мозга, проявляется затруднениями в жевании, глотании, неразборчивости речи;
- мышечная прогрессирующая атрофия (8%) - редкий тип БДН, распространяется на двигательные нейроны в нижних конечностях, развивается слабость, атрофия мышц, фасцикуляции и потеря веса;
- ПЛС (латеральный первичный склероз) (2%) поражает преимущественно двигательные нейроны в верхних конечностях, развивая спастику мышц, неустойчивую ходьбу, затрудненную речь.
Симптоматика и клиника БДН в зависимости от формы
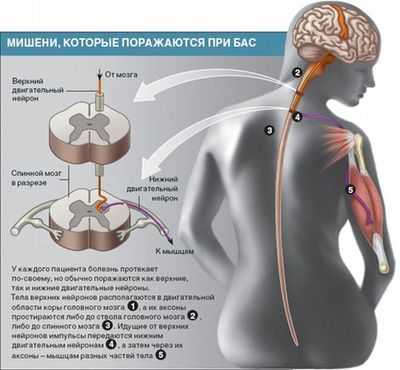
На моторные нейроны влияет целая группа болезней. В состав нейронов входят две группы клеток, необходимые для произвольных движений.
В первую группу входят двигательные нейроны, расположенные в прецентральной мозговой извилине, а во вторую - нейроны, расположенные в стволе спинного и головного мозга.
В зависимости от локализации погибшего двигательного нейрона будут зависеть форма болезни и ее симптомы.
При спастической параплегии поражается двигательный нейрон первой группы, при спинальной мышечной атрофии, детском параличе - вторая группа, а при боковом амиотрофическом склерозе обе группы нейронов.
Формы БДН и их проявления:
- Спастическая параплегия определяется повышением спастического тонуса ног с нарушением походки. Это вызывается генетическими мутациями.
- Для спинальной мышечной атрофии характерна слабость и уменьшение мышц, отсутствие их активации или иннервирования нервными волокнами. В зависимости от времени появления первой симптоматики и характеру распределения слабости мышц, осуществляется классификация спинальной мышечной атрофии.
- Более распространенной патологией опорно-двигательной системы является БАС, частота развития которого повышается с возрастом. В результате прогрессирует омертвление нервных клеток, нарастает слабость и мышечная атрофия.
- Детский церебральный паралич, полиомиелит является вирусной болезнью нижних нейронов.
Дифференциальная диагностика
Диагноз БДН подтверждается элеткронейромиографией, которая выявляет генерализованный денервационный процесс. Для выявления спонтанности, продолжительности двигательных единиц, фасцикуляций, фибрилляций, выполняется трехуровневая игольчатая ЭМГ.
Стимуляционная электронейромиография позволяет выявлять замедление двигательных волокон, пирамидную недостаточность.
Транскраниальная магнитная стимуляция выявляет уменьшение возбудимости в центральных мотонейронах. МРТ позволяет исключать остальные патологии с похожей клиникой (компрессию грыжи или опухолью).
Возможности лечения
Болезнь двигательного нейрона неизлечима, а целью терапии является замедление развития процесса, сокращение выраженности симптоматики, так как не существует методов для полного излечения. Доказана эффективность Рилутека - пресинаптического ингибитора.
Как и при БАС, могут применяться антиоксидантные средства. Также специалисты назначают креатин, карнитин, витамины. Другим лекарственным средством является Рилузол, но средство официально недоступно для пациентов в РФ. Как и Рилутек, препарат снижает объем глутамата, освобождающегося при передаче нервных импульсов.
К полиативной терапии относится устранение болей от мышечных спазмов, для чего назначается Карбамазепин в дозе около 100-200 мг несколько раз в сутки. При увеличенном тонусе мышц нужно принимать миорелаксанты (Сирдалуд, Баклофен).
Дыхательная поддержка - это основной фактор, влияющий на качество жизни и прогноз. С прогрессированием патологии наблюдается ослабление и атрофия мышц диафрагмы и других мышечных групп, требуется неинвазивное вентилирование легких.
Это нормализует сон, снижает утомляемость и одышку. Используется аппарат со специальной маской.
Если нарушены функции глотания, то кормление проводится назогастральным зондом, установкой гастростомы. Назначается специальное питание, восполняющее недостаток калорий.
При заболевании БДН трудно говорить про прогноз. В редких случаях люди живут десятки лет. Человек может умереть через несколько месяцев. Но в среднем длительность жизни равна 2-5 года от начала выявления болезни. Не существует специфических профилактических мер.
БАС — двигательные нейроны отмирают полностью
Во время БАС сочетается равномерно выраженная симптоматика центрального и периферического паралича, преобладание одних симптомов над другими (сегментарно-ядерный или пирамидный варианты). На поздних этапах болезни преобладают показатели периферического паралича.
Во время БАС начинается повреждение мотонейронов в передних рогах спинового мозга. С прогрессированием патологии проявляются бульбарные нарушения в основном у мужчин.
Выделяется грудной, шейный, диффузный и поясничный дебют. Классическим вариантом является шейный, первая симптоматика которого характеризуется слабостью и атрофией кисти, нарушенными мелкими движениями, наличием фасцикулярных подергиваний пораженных мышц.
Стивен Хокинг — самый известный пациент с БАС
В дальнейшем появляется слабость и атрофия мышц плечевого пояса, пациентам сложно одеваться, поднимать руки выше горизонтальной плоскости. Сначала поражается одна сторона тела, затем симптомы проявляются на другой руке с развитием верхнего смешанного парапареза.
Развивается слабость и скованность нижних конечностей, больным сложно ходить на большие расстояния, перемещаться по лестнице.
Повреждаются разгибательные мышцы с проявлением смешанного нижнего парапареза. На поздних стадиях проявляются псевдобульбарный и бульбарный синдромы, появление дисфагии.
Первой симптоматикой грудного дебюта будут фасцикуляции, атрофия мышц в животе, спине и слабость. Пациентам становится сложно стоять и нагибаться. На следующих этапах появляется смешанный односторонний гемипарез с вовлечением мускулатуры противоположной части тела. На начальном этапе выпадают брюшные рефлексы. В результате дыхательных нарушений наступает летальный исход.
Диффузный дебют проявляется признаками нарушения периферических мотонейронов с резко выраженным утомлением, дыхательными нарушениями. Снижается масса тела до появления дисфагии, в результате дыхательной недостаточности наступает смерть.
Обычно поражение жевательных и мимических мышц наступает позже, пациенту сложно высовывать язык, вытягивать в трубочку губы, надувать щеки.
К осложнениям БАС относят параличи, парезы шейных мышц и конечностей, нарушенное глотание, дыхательную недостаточность, контрактуры конечностей, аспирационную пневмонию, уросепсис, депрессию, болезненные спазмы мышц, кахексию. С прогрессированием двигательных нарушений пациент умирает.
Проведение диагностики и лечение
Диагностика БАС основывается на детальном анализе клиники. Проводится электронейромиография (ЭМГ обследование) для подтверждения нарушений мотонейрона.
Не существует эффективного лечения патологии. Можно отсрочить летальный исход на несколько месяцев приемом Рилузола в дозировке 50 мг 2 раза в день. Основой терапии является лечебная гимнастика, физическая активность, диета и пр.
Болезненные спазмы мышц устраняются Дифенином, Хинина сульфат, Тегретолом, Финлепсином, витамином Е400 мг, Изоптином или препаратами магния. При слюнотечении назначается Атропин, Бускопн, при спастичности - Сирдалуд, Баклосан, Мемантин и пр.
Болевые симптомы устраняются анальгетиками. Временное улучшение может быть от антихолинэстеразных средств. Также специалисты назначают антидепрессанты (Амитриптилин, Серталин).
Пациент должен поддерживать физическую активность, при прогрессировании патологии использовать специальные приспособления. В результате дисфагии может потребоваться питание через зонд или гастростомия. В последнее время проводятся исследования лечения патологии стволовыми клетками.
БАС считается фатальной патологией, средняя продолжительность жизни при которой составляет 3-5 лет. Только около 10% пациентов живут в течение 10 лет. К негативным прогностическим показателям относят бульбарные нарушения, пожилой возраст.
Читайте также: