Преждевременное заращение черепных швов у детей. Синдрома Апера, Карпентпера, Хотпцена, Пфейффера
Добавил пользователь Alex Обновлено: 22.01.2026
Практически любой врач курса общей анатомии помнит о существовании специфических форм черепа, таких, как скафоцефалический (вытянутый в передне-заднем направлении) и брахицефалический (увеличенный в ширину). Но редко кто вспоминает о том, что необычная форма черепа у ребенка во многих случаях является признаком преждевременного заращения черепных швов.
Конечно, все доктора знакомы с термином краниостеноз - преждевременное заращение швов черепа, приводящее к неспецифическому повреждению головного мозга вследствие недостаточного расширения полости черепа в период наиболее активного роста мозга. Когда возникает вопрос о том, как лечат краниостеноз немногие вспоминают о возможности хирургического иссечения преждевременно заросших швов и лишь единицы знают о существовании метода двухлоскутной краниотомии.
Между тем, по международной статистике, преждевременное закрытие одного из швов черепа (изолированный краниосиностоз) возникает примерной у одного из 1000 детей. Интересно отметить, что такая же частота характерна и у детей, имеющих расщелину губы. При этом ни у кого не вызывает трудностей диагностика расщелин губы, потому что таких пациентов, несомненно, видел каждый врач. Тогда как почти никто из врачей общей практики не может припомнить, видел ли он когда-нибудь ребенка с преждевременным заращением швов черепа.
Хождение по мукам
Краниосиностоз - преждевременное заращение одного или нескольких швов черепа, приводящее к формированию характерной деформации головы. Таким образом, уже в родильном доме ребенок с подозрением на краниосиностоз может быть выделен из общей массы новорожденных и направлен на дообследование. На практике, к сожалению, на этом этапе все деформации черепа, обнаруженные у детей, расцениваются врачами как особенности послеродовой конфигурации головы и им не уделяется должного внимания. В период новорожденности форме черепа также не придается большого значения. Обычный ответ педиатра на обеспокоенность родителей: «…Ничего страшного, хорошо прибавляет в весе, ушла желтуха, а голова такая оттого, что лежит на боку. Вот начнет ходить, и все исчезнет».
Психомоторное развитие детей проходит с отставанием, деформации черепа самопроизвольно не исчезают, некоторые деформации становятся менее заметными, скрываясь под волосами, другие ошибочно расцениваются врачами как иные заболевания, а третьи отступают на второй план при наличии более очевидных нарушений функции органов и систем. Зачастую малышей с краниосиностозами консультируют генетики, и нередко правильно устанавливается группа заболеваний и даже предполагается непосредственный генетический синдром. Несмотря на это, единицы таких пациентов поступают в специализированные клиники для проведения лечения. Подавляющее большинство детей, не получивших лечения, имеют сниженный интеллект и становятся инвалидами. Из-за необычной формы черепа нарушаются пропорции лица, и к периоду полового созревания у таких детей чаще, чем у других, возникают трудности в социальном общении и даже возможны суицидальные попытки.
Родители постепенно перестают обращать внимание на легкие деформации черепа, а при наличии выраженной у ребенка деформации лица детские хирурги разъясняют им, что исправление косметических дефектов проводится только в 16 лет.
Диагностика
Рисунок 1
Основными швами свода черепа являются сагиттальный, коронарный, лямбдовидный и метопический (рис. 1). При преждевременном заращении костного шва происходит компенсаторный рост костей перпендикулярно к его оси (закон Вирхова). В результате появляется характерная деформация. Опишем наиболее часто встречающиеся формы краниосиностозов.
Сагиттальный краниосиностоз
Преждевременное заращение сагиттального шва приводит к увеличению передне-заднего размера черепа с нависающими лобной и затылочной областями и к уменьшению его ширины с формированием узкого овального лица (рис. 2). Такой вид деформации называют скафоцефалией, или ладьевидным черепом. Это наиболее частое заболевание среди общего числа изолированных синостозов (50-60%). Характерная форма черепа видна уже с рождения. При осмотре головы сверху заметно втяжение теменных областей, это дает ощущение циркулярной перетяжки свода черепа на уровне или чуть кзади от ушных раковин. Четко определяется большой родничок, причем его размеры не отличаются от нормы. Характерным считается наличие костного гребня, пальпируемого в проекции сагиттального шва.
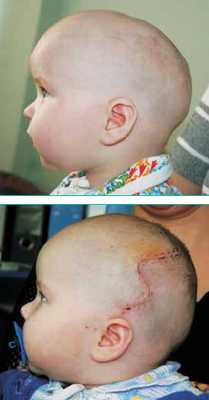
Рисунок 2: а - ребенок до и б - после устранения скафоцефалии.
Метопический краниосиностоз
Самым редким представителем группы изолированных краниосиностозов является метопический краниосиностоз, или тригоноцефалия, составляющая 5-10% от общего их количества. Несмотря на это, данное заболевание, пожалуй, чаще всего распознается как врожденная деформация черепа из-за характерной клинической картины.
При раннем замыкании метопического шва происходит формирование треугольной деформации лба с образованием костного киля, идущего от надпереносья до большого родничка. При взгляде на такой череп сверху видна четкая треугольная деформация с вершиной в области надпереносья. При этом верхние и латеральные края орбит смещаются кзади, что дает ощущение разворота плоскости орбит кнаружи и уменьшения межорбитального расстояния (гипотелоризм). Деформация лба настолько необычна, что дети с тригоноцефалией часто обследуются у генетиков и наблюдаются как носители наследственных синдромов, сопровождаемых снижением интеллекта. Действительно, тригоноцефалия рассматривается как неотъемлемая часть таких синдромов, как Opitz, Oro-facio-digital syndrom, и некоторых других. Верно и то, что многие синдромальные заболевания приводят к задержке интеллектуального развития, но частота их настолько низка, а клиническая картина настолько характерна, что не стоит всех детей, имеющих лишь метопический синостоз, причислять к группе риска по развитию умственной неполноценности.
Односторонний коронарный краниосиностоз
Коронарный шов расположен перпендикулярно срединной оси черепа и состоит из двух равноценных половин. Так что при преждевременном заращении одной из его половин формируется типичная асимметричная деформация, именуемая плагиоцефалией. Вид ребенка с плагиоцефалией характеризуется уплощением верхнеорбитального края орбиты и лобной кости на стороне поражения с компенсаторным нависанием противоположной половины лба (малыш как будто хмурится одной стороной лица). С возрастом более отчетливо начинает проявляться ипсилатеральное уплощение скуловой области и искривление носа в ту же сторону. В школьном возрасте присоединяется деформация прикуса, связанная с увеличением высоты верхней челюсти и как следствие - смещением нижней челюсти на стороне преждевременно закрывшегося шва. В тяжелых случаях имеется даже компенсаторное выбухание затылочной области со стороны синостоза. Нарушения со стороны органа зрения представлены чаще всего односторонним косоглазием. Плагиоцефалия чаще других расценивается как особенности послеродовой конфигурации головы. Но в отличие от последней она не исчезает в первые недели жизни, а, наоборот, с возрастом прогрессирует.
Таким образом, огромную роль в правильной постановке диагноза играет именно форма черепа.
Из инструментальных методов диагностики наилучшим является проведение компьютерной томографии с трехмерным ремоделированием изображения костей свода черепа и лица. Это обследование помогает выявить сопутствующую патологию головного мозга, подтвердить наличие синостоза в случае изолированного повреждения и установить все заинтересованные швы в случае полисиностоза.
Лечение
Самым активным периодом роста головного мозга считается возраст до двух лет. Таким образом, с функциональной точки зрения предотвратить краниостеноз можно ранним хирургическим лечением. Оптимальным возрастом для проведения операции по поводу краниосиностоза можно считать период с 3 до 9 месяцев. Преимуществами лечения в данном возрасте можно считать:
- легкость манипулирования с тонкими и мягкими костями черепа;
- облегчение окончательного ремоделирования формы черепа быстро растущим мозгом;
- более полное и быстрое заживление остаточных костных дефектов.
Если лечение выполняется после пяти лет, сомнительно, что оно приведет к значительному улучшению функции головного мозга. В большей степени операция будет направлена на устранение деформации головы.
Основной особенностью современного хирургического лечения является не только увеличение объема черепа, но и исправление его формы и сочетанной деформации лица в ходе одной операции.
Сведения об авторах:
Андрей Вячеславович Лопатин, зав. отделением челюстно-лицевой хирургии ГУ «Российская детская клиническая больница» Росздрава, профессор, д-р мед. наук
Сергей Александрович Ясонов, врач отделения челюстно-лицевой хирургии ГУ «Российская детская клиническая больница» Росздрава
Краниосиностоз
Краниосиностоз - это заболевание, основным симптомом которого является деформация мозгового отдела черепа, возникающая вследствие преждевременного зарастания костных швов. Клиника включает в себя деформации черепа, симптомы внутричерепной гипертензии, патологию зрительного нерва, отставание в психическом развитии. Редко заболевание сопровождается аномалиями костей лицевого черепа. Диагностика заключается в оценке степени зарастания черепных швов и определении костных дефектов путем физикального обследования, рентгенографии, КТ и МРТ. Основное лечение - ранняя хирургическая коррекция формы костей черепа.
Общие сведения
Краниосиностоз - это патологическое состояние в педиатрии, возникающее на фоне раннего зарастания черепных швов, характеризующееся деформацией черепной коробки и нарушением развития тканей головного мозга. В среднем распространенность разных форм заболевания в станах СНГ составляет 0,03-3,5% от всех новорожденных. Мужской пол более склонен к развитию данной патологии. Наиболее распространенный вариант - моносиностоз. Чаще всего наблюдается преждевременное зарастание сагиттального шва (скафоцефалия) - 50-65% от всех краниосиностозов. Самой редкой и прогностически неблагоприятной является синдромальная форма, при которой имеется высокий риск летального исхода на первом году жизни ребенка. При своевременной диагностике и адекватном лечении в первые 6-9 месяцев жизни дальнейшее развитие пациента проходит без отклонений.
Причины краниосиностоза
Точная этиология краниосиностоза не установлена. Согласно выдвинутым теориям, данное заболевание может развиваться в результате внутриутробного нарушения гормонального фона ребенка, перинатальных травм и сдавливания костей черепа в полости матки. Также данная патология возникает при наследственных патологиях - синдроме Апера, синдроме Крузона и синдроме Пфайффера. Доподлинно известна одна из ведущих причин развития краниосиностоза - аномалия гена, отвечающего за образование рецепторов фактора роста фибробластов (FGFR типы I, II и III).
Патогенетически краниосиностоз обусловлен преждевременным синостозированием одного или сразу нескольких черепных швов: коронарного, сагиттального, лямбдовидного или метопического. На фоне этого, согласно закону Вирхова, возникает компенсаторный рост костной ткани в перпендикулярном направлении, из-за чего формируется деформация черепа. Полисиностоз (а зачастую - и моносиностоз) часто сопровождается внутричерепной гипертензией, которая может проявляться неврологическими нарушениями вследствие сдавливания коры головного мозга, венозным застоем глазного дна, отеком диска зрительного нерва, а при длительном течении - полной атрофией зрительного нерва и потерей зрения.
Классификация краниосиностоза
Краниосиностоз, согласно этиологическим факторам, разделяют на две группы:
- Синдромальный. В данном случае патология сочетается с другими врожденными пороками. Сюда относятся сцепленные с Х-хромосомой, моногенные, хромосомные и другие краниосиностозы. Например - комбинация синостоза с дисплазией костей лицевого черепа, синдром Смита-Лемли-Опица или рото-пальце-лицевой синдром.
- Несиндромальный. Это изолированная форма, которая возникает самостоятельно и не имеет сопутствующих заболеваний.
В зависимости от количества заросших черепных швов выделяют:
- Моносиностоз. Характеризуется поражением только 1 шва. В случае с коронарным и лямбдовидным швом зарастание может быть одно- или двухсторонним. Наиболее распространенная форма.
- Полисиностоз. В патологический процесс втягиваются 2-3 шва.
- Пансиностоз. При этой форме наблюдается сращивание всех костных швов черепа ребенка. Встречается крайне редко.
Симптомы краниосиностоза
Клинически краниосиностоз проявляется с момента рождения ребенка. Для всех форм характерны плагиоцефалия и раннее закрытие большого родничка (в норме это происходит в 12-18 месяцев). Только при полисиностозе или сопутствующей гидроцефалии он может оставаться открытым до 3-х летнего возраста. Также при краниосиностозах зачастую наблюдается повышение внутричерепного давления, которое может проявляться неврологическими нарушениями: беспокойством, интенсивным плачем, тошнотой и рвотой, нарушением сна, снижением аппетита, позитивным симптомом Грефе, судорогами.
Каждая из форм заболевания имеет характерные клинические особенности. Краниосиностоз стреловидного шва (скафоцефалия или ладьевидный череп) характеризуется увеличением переднезаднего размера головы ребенка при недостаточности ее ширины. Визуально определяется вытягивание черепа, «вдавливание» височных областей, «нависание» лба и затылочной части, сужение лица и приобретение им овальной формы. Пальпаторно над местом прохождения стреловидного шва выявляется костный гребень. В раннем возрасте возможна задержка психического развития.
Зарастание лямбдовидного шва чаще всего носит односторонний характер и проявляется уплощением затылочной области. Является трудно диагностируемой формой, поскольку плагиоцефалия практически незаметна под волосами, а неврологические нарушения минимальны. При взрослении пациента динамика заболевания практически отсутствует.
Коронарный или венечный краниосиностоз может быть как одно-, так и двухсторонним. Зарастание только одной половины шва сопровождается типичной деформацией черепа ребенка - уплощением лобной кости и верхней части глазницы с пораженной стороны. При этом противоположная половина компенсаторно «нависает». Со временем развиваются искривление носа в противоположную сторону, уплощение скулы, нарушение прикуса и косоглазие. Двухсторонний коронарный краниосиностоз проявляется широким, плоским и высоким лбом с уплощенными глазничными краями лобной кости, редко - башенной деформацией черепа (акроцефалией). Неврологические нарушения неспецифичны и аналогичны другим формам.
Нетопический краниосиностоз или тригоноцефалия характеризуется развитием треугольного лба с костным килем, проходящим от глабеллы до большого родничка. Также наблюдается гипотелоризм - смещение глазниц кзади с уменьшением межглазничного промежутка. Со временем происходит некоторое сглаживание костного гребня и нормализация формы лба. В половине случаев возникают нарушения зрения и отставание в психическом развитии.
Синдромальный краниосиностоз является самой редкой и тяжелой формой. Помимо плагиоцефалии отмечается дисплазия костей лицевой части черепа, из-за чего возникают дыхательная недостаточность, нарушение приема пищи и патология зрения. Характеризуется синостозом венечного шва и, как результатом - брахицефалической формой головы ребенка. Также возникают гипоплазии костей верхней челюсти, выпячивание глазных яблок из орбит, гипертелоризм. Часто наблюдается значительное расширение родничка и расхождение стреловидного шва. Без лечения у детей развивается выраженное отставание в психическом развитии, зачастую они погибают на протяжении первых 12 месяцев жизни от ОРВИ, осложнившихся пневмонией.
Диагностика краниосиностоза
Диагностика краниосиностоза базируется на физикальном осмотре и инструментальных методах исследования. Анамнез нередко малоинформативен, но его данные позволяют педиатру проследить динамику клинической симптоматики, если таковая имеет место. Важным моментом становится визуальный осмотр ребенка, который дает возможность обнаружить характерные деформации черепа, аномалии костей и т. д. Лабораторные анализы специфических изменений не выявляют и могут использоваться с целью определения генетической патологии или диагностики осложнений.
Обязательными являются инструментальные методы, позволяющие визуализировать костные деформации и оценить степень поражения тканей головного мозга. Сюда относятся нейросонография, рентгенография, компьютерная и магнитно-резонансная томография. Нейросонография используется с целью оценить состояние тканей головного мозга и размеры желудочков, выявить внутричерепную гипертензию. На рентгенограмме удается определить нарушения структуры костей, окостенение черепных швов, а при повышенном внутричерепном давлении - усиление пальцевых вдавлений. КТ и МРТ применяются для получения более информативных результатов. При подозрении на поражение зрительной системы проводится офтальмоскопия, позволяющая обнаружить поражение диска зрительного нерва. Рекомендованы консультации нейрохирурга и офтальмолога.
Дифференциальная диагностика краниосиностоза осуществляется с позиционной плагиоцефалией, родовой травмой новорожденных (кефалогематомой, подапоневротическим кровоизлиянием, переломом костей черепа), кистами головного мозга, рахитом и микроцефалией.
Лечение краниосиностоза
Основное лечение краниосиностоза - хирургическая коррекция костной деформации черепа. Оптимальное время для проведения оперативного вмешательства - первые 6-9 месяцев жизни ребенка. Данные сроки обусловлены тем, что в этом периоде наблюдается наиболее интенсивное развитие тканей головного мозга, которому может препятствовать деформация черепной коробки. Кроме того, кости черепа в этом возрасте быстро восстанавливают свою структуру без развития осложнений. Объем и техника операции зависят от формы краниосиностоза и сопутствующих патологий. В 2-3-х летнем возрасте коррекция проводится исключительно с целью ликвидировать косметический дефект. Помимо хирургического лечения осуществляется изменение рациона ребенка в соответствии с возрастными требованиями. При развитии интеркуррентных заболеваний показана медикаментозная терапия.
Прогноз и профилактика краниосиностоза
Прогноз для детей с краниосиностозом напрямую зависит от формы заболевания, своевременности диагностики и эффективности оперативного вмешательства. При качественном проведении лечебных мероприятий исход заболевания, как правило, благоприятный. Прогностически неблагоприятной принято считать синдромальную форму краниосиностоза.
Специфической профилактики для данной патологии не существует. Неспецифические меры подразумевают медико-генетическую консультацию семьи и планирование беременности, охрану здоровья женщины при вынашивании ребенка, рациональное питание, отказ от вредных привычек и исключение всех потенциальных этиологических факторов развития краниосиностоза.
Синдром Апера
Синдром Апера - генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей. Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований. Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Синдром Апера (акроцефалосиндактилия 1 типа) - генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания. Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее - лишь в 1995 году. Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных. Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний - акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей. Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности - к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей - различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы синдрома Апера
Некоторые проявления синдрома Апера заметны с самого рождения - например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног. Среди неонатологов симптом иногда носит название «среднего пальца» - в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь. Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа. Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие. Деформации затрагивают и кости лицевого черепа - переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса. Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты. Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост. При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов. При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа - башенный череп или брахикефалия, характерный внешний вид лица.
С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов - стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности. Также возможно наличие иных пороков развития - аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения.
Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию. Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния. Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера - пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни. Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления. По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект. Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах. Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется. При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения. При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов. В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Синдром Пфайффера
Синдром Пфайффера - генетическое заболевание с аутосомно-доминантным механизмом наследования, характеризующееся нарушением формирования костей черепа и конечностей. Симптомами являются деформации черепа (в результате краниосиностоза), костей пальцев рук и ног. Некоторые формы заболевания характеризуются глухотой, нарушением интеллектуального развития, стоматологическими патологиями. Диагностика синдрома Пфайффера производится на основании данных настоящего статуса больного, рентгенологических исследований, молекулярно-генетических анализов. Специфическое лечение отсутствует, применяют паллиативные мероприятия для предотвращения тяжелых осложнений (в том числе и хирургического характера) и симптоматическую терапию.
Синдром Пфайффера - врожденное генетически обусловленное нарушение формирования черепа и конечностей, которое характеризуется краниосиностозом, деформацией лица, расширением и укорочением пальцев, частыми синдактилиями. Впервые это заболевание было описано в 1964 году немецким педиатром Рудольфом Пфайффером, в дальнейшем было выявлено несколько клинических форм патологии.
По данным современной генетики, синдром Пфайффера является аутосомно-доминантным состоянием с полной пенетрантностью, половое распределение больных не имеет особенностей - с одинаковой частотой болеют как мальчики, так и девочки. Встречаемость точно не определена, по некоторым данным она составляет примерно 1:100 000 новорожденных. Каких-либо национальных или расовых особенностей в распределении этого заболевания также не выявлено. В отношении синдрома Пфайффера крайне важна ранняя диагностика, так как от времени начала паллиативного лечения во многом зависит прогноз заболевания и качество дальнейшей жизни больного.
Причины синдрома Пфайффера
Несмотря на то, что синдром Пфайффера является наследственным заболеванием с аутосомно-доминантным механизмом передачи, большинство случаев этой патологии являются следствием спонтанных герминативных мутаций в половых клетках родителей. При этом за развитие состояния отвечают дефекты двух генов, кодируемые ими белки обладают сходными функциями.
По причине того, что вышеуказанные белки обладают очень похожими функциями, патогенез синдрома Пфайффера при дефектах гена FGFR1 и FGFR2 тоже весьма схож. В результате миссенс-мутаций изменяется структура экспрессируемого белка, из-за чего полученный рецептор становится неспособен полноценно связываться с веществом-стимулятором (фактором роста фибробластов) и передавать информацию внутрь клетки. Так как фибробласты активно участвуют в процессах остеогенеза в эмбриональный период и в первые годы жизни ребенка, такое изменение рецепторов ведет к разнообразным костным патологиям.
Доказательством этого является тот факт, что дефекты гена FGFR1, помимо синдрома Пфайффера, приводят к тригоноцефалии 1-го типа и ряду других нарушений, а FGFR2 - к неспецифическому краниосиностозу, синдрому Крузона и иным похожим патологиям. При синдроме Пфайффера происходит преждевременное сращение костей черепа, что становится причиной деформации головы и лица, повышения внутричерепного давления - это, в свою очередь, способно вызывать ряд вторичных нарушений от нейросенсорных расстройств до глубокой умственной отсталости.
Классификация и симптомы синдрома Пфайффера
На сегодняшний день выделяют как минимум три клинические формы синдрома Пфайффера, которые различаются между собой выраженностью нарушений, течением и прогнозом заболевания. Причина такого различия лежит в молекулярно-генетических механизмах развития патологии - 1-й тип обусловлен единственной мутацией гена FGFR1 (252Pro-Arg) и некоторыми нарушениями структуры FGFR2, тогда как 2-й и 3-й типы вызваны различными вариантами дефектов только гена FGFR2.
Более тонкие особенности патогенеза каждой формы заболевания в настоящее время находятся в стадии изучения. Благодаря характерной клинической картине, специалисты могут определить разновидность синдрома Пфайффера, основываясь только на симптомах патологии. Молекулярно-генетический анализ во многих случаях необходим лишь для окончательного подтверждения диагноза.
Синдром Пфайффера 1-го типа является наиболее распространенным и благоприятным в прогностическом отношении вариантом заболевания. При этом состоянии уже с самого рождения ребенка отмечаются аномалии развития лица - гипертелоризм, недоразвитие верхнечелюстных костей, расширенные плоские переносица и спинка носа. Иногда могут отмечаться расщепление твердого нёба и экзофтальм. При этой форме синдрома Пфайффера характерные изменения формы черепа сводятся к увеличению его высоты и длины, очень редко отмечается асимметрия. В дальнейшем могут возникать аномалии зубов - искривление зубного ряда, гипоплазия, склонность к кариесу. Деформации костей конечностей сводятся к расширению фаланг первых пальцев на руках и ногах, может отмечаться частичная синдактилия.
Синдром Пфайффера 2-го типа - более тяжелая форма заболевания, проявляющаяся значительной выраженностью сращения костей черепа между собой и рядом других аномалий. Голова из-за краниосиностоза приобретает характерную форму «трилистника» или «листка клевера», наблюдается значительная гипоплазия средней трети лица. По причине грубого нарушения формирования черепа данный тип синдрома Пфайффера характеризуется выраженной умственной отсталостью и рядом неврологических нарушений. Пороки развития конечностей сводятся к расширению первых пальцев, синдактилии, анкилозу локтевых суставов. Кроме того, синдром Пфайффера 2-го типа часто сопровождается различными пороками развития внутренних органов. Все вышеперечисленные нарушения могут стать причиной летального исхода в раннем возрасте.
Синдром Пфайффера 3-го типа во многом схож по своим проявлениям с предыдущим вариантом заболевания, но краниосиностоз при этом не приводит к формированию деформации черепа в виде «листка клевера». У больных отмечается увеличение головы в высоту, пороки развития конечностей (синдактилии, анкилозы суставов), экзофтальм, неврологические нарушения. Для этого варианта синдрома Пфайффера характерно раннее развитие зубов, нередко младенцы уже рождаются с ними (натальные зубы). Также возможны пороки внутренних органов и ранняя смерть из-за совокупности нарушений.
Такое различие в клинической картине и прогнозе различных вариантов синдрома Пфайффера накладывает свой отпечаток на наследование этого заболевания. Так как 1-й тип, особенно в случае своевременного выявления и правильного лечения, характеризуется сохранением нормального уровня интеллекта, фертильности и выживаемости больных, возможна аутосомно-доминантная передача патологии потомству. Более тяжелые 2-й и 3-й типы чаще всего приводят к ранней смерти или глубокой инвалидизации больных, эти варианты синдрома Пфайффера возникают только в результате спонтанных мутаций.
Своевременная диагностика синдрома Пфайффера крайне важна, так как она позволяет вовремя составить схему паллиативного лечения, тем самым избежать осложнений и значительно улучшить качество жизни больного. Но это справедливо лишь в отношении 1-го типа заболевания, так как при других вариантах патологии комплекс пороков развития настолько тяжел, что практически не оставляет шансов на сохранение интеллекта, а в дальнейшем - жизни больных.
Для выявления синдрома Пфайффера используют ультразвуковые методики (в том числе и в качестве пренатальной диагностики), рентгенографию, молекулярно-генетические анализы. На профилактических УЗИ во время беременности при наличии этого заболевания у ребенка во 2-3 триместре можно выявить нарушение формирования черепа, синдактилию, пороки развития внутренних органов.
Рентгенологические методы редко используют при ранней диагностике синдрома Пфайффера у детей - в основном, производят исследование черепа, которое обнаруживает наличие краниосиностоза различной степени выраженности, гипоплазию верхнечелюстных костей, изменение формы глазниц. Также на рентгене можно определить изменение формы костей больших пальцев рук и ног.
Молекулярно-генетическая диагностика производится врачом-генетиком - в этом случае при подозрении на наличие синдрома Пфайффера 1-го типа производят поиск точечной мутации 252Pro-Arg методом прямого секвенирования экзона 7а гена FGFR1. В других случаях выполняют секвенирование 7-го и 9-го экзонов гена FGFR2 - именно там возникает большинство дефектов, приводящих к развитию синдрома Пфайффера. Вспомогательную роль в диагностике заболевания могут играть методы медицинской визуализации, направленные на определение аномалий развития внутренних органов.
Лечение синдрома Пфайффера
Лечение синдрома Пфайффера только симптоматическое и паллиативное - специалисты стараются устранить повышенное внутричерепное давление, обеспечить нормальное развитие нервной системы, снизить выраженность нарушений со стороны внутренних органов. Для этого широко применяют хирургические методики - разделение пальцев, пластику черепа и ряд других. Также назначают препараты, направленные на улучшение питания нервной ткани - ноотропные средства, витамины. При своевременно начатом лечении синдрома Пфайффера 1-го типа выживаемость больных значительно увеличивается. К сожалению, объем медицинской помощи зачастую недостаточен для полноценного лечения больных синдромом Пфайффера 2-го и 3-го типов.
Прогноз синдрома Пфайффера определяется вариантом этого заболевания. Первый тип характеризуется относительно благоприятным прогнозом, который зависит от своевременности выявления патологии и правильности подобранного лечения. В благоприятных условиях больные сохраняют нормальный интеллект, доживают до преклонного возраста и способны иметь потомство - при этом риск передачи заболевания детям составляет минимум 50%.
Однако синдром Пфайффера 2-го и 3-го типов обладает крайне неблагоприятным прогнозом - совокупность неврологических и других нарушений часто становится причиной летального исхода в раннем детстве. Даже при выполнении паллиативных мероприятий у больных наблюдается выраженная олигофрения и глубокая инвалидизация. Профилактика синдрома Пфайффера, учитывая часто спонтанный механизм развития этого заболевания, возможна только в рамках пренатальной диагностики ультразвуковыми или генетическими методами.
Краниосинтостоз (краниостеноз) - раннее закрытие черепных швов, что способствует ограниченному объему черепа, его деформации и внутричерепной гипертензии. Заболевание встречается у одного новорожденного на 2000, чаще наблюдается у мальчиков.
БСФ - биосоциальные функции
ВИЧ-вирус иммунодефицита человека
ЖДА - железодефицитная анемия
ИФА - иммуноферментный анализ
КТ -компьютерная томография
МДК - мультидисциплинарная группа
МР - медицинская реабилитация
МРТ -магниторезонансная томография
ОАК - общий анализ крови
ОАМ - общий анализ мочи
СОЭ - скорость оседания эритроцитов
ТМО - твердая мозговая оболочка
УЗИ - ультразвуковое исследование
ЭКГ - электрокардиография
эхоКГ- эхокардиография

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
Анатомо-топографическая классификация по Tessier: Формы краниостеноза зависят от характера деформации черепа[2].
- Тригононоцефалия (раннее сращение метопических швов, характеризующееся треугольным выпячиванием черепа в области лба)
- Брахиоцефалия (раннее сращение венечного и ламбовидного швов, характеризующееся увеличение черепа в поперечном диаметре)
Е. Группа Crouzon (преждевременное заращение швов черепа, венечного и ламбдовидного, в сочетании с сохранившимся или увеличенным поперечным размером всех отделов мозгового черепа)
- Pfeiffer cиностоз коронарного и ламбдовидного швов. Иногда встречается необычная патология - череп в форме трилистника или листа клевера;
II. МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ [1]
Основные (обязательные) диагностические мероприятия, проводимые на стационарном уровне (при экстренной госпитализации проводятся диагностические обследования, непроведенные на амбулаторном уровне):
Дополнительные диагностические мероприятия (при экстренной госпитализации проводятся диагностические обследования непроведенные на амбулаторном уровне)
Диагностические мероприятия, проводимые на этапе скорой неотложной помощи: нет.
Лабораторные исследования:
Изменения в клинических, биохимических анализах при отсутствии сопутствующей патологии не специфичны.
КТ головного мозга: (маленькие желудочки мозга, уменьшение или отсутствие подпаутинных щелей, может сочетаться с субкомпенсированной внутренней гидроцефалией, внутричерепные гипертензии, подтвердить наличие синостоза в случае изолированного повреждения и установить все заинтересованные швы в случае полисиностоза);
Рентгенограмма черепа в 2-х проекциях: отсутствие одного или нескольких черепных швов и связанное с этим изменение формы черепа, кости свода черепа значительно истончены, пальцевые вдавления по всему свода черепа.
- осмотр офтальмолога - с целью осмотра глазного дна и выявления признаков внутричерепной гипертензии.
Дифференциальный диагноз
Лечение
Диета при отсутствии сопутствующей патологии - соответственно возрасту и потребностям организма.
Перечень дополнительных лекарственных средств (менее 100% вероятность применения):
- Карбамазепин 200 мг.
Медикаментозное лечение, оказываемое на этапе скорой неотложной помощи: нет.
Хирургическое лечение является основным методом лечения краниостеноза. Основной особенностью современного хирургического лечения заключается в ремоделировании костей свода черепа. Для этого кости деформированных участков снимаются и переставляются в правильное анатомическое положение. При этом полость черепа увеличивается для дальнейшего беспрепятственного роста головного мозга. Для надежной фиксации ремоделированных костей между собой используют рассасывающиеся фиксирующие материалы: титановые минишурупы и минипластины, что значительно облегчает лечение пациентов.
Оптимальные сроки оперативного вмешательства у ребенка - в период между шестью и девятью месяцами жизни. Именно в эти сроки оперативное вмешательство минимально, риск самый низкий, а эффект от операции оптимальный.
- избегать провоцирующих факторов развития риска судорожного синдрома (яркий свет, громкий звук и т.д.)
Первый этап (ранний) медицинской реабилитации- оказание МР в остром и подостромпериоде травмы или заболевания в стационарных условиях (отделение реанимации и интенсивной терапии или специализированное профильное отделение) с первых 12-48 часов при отсутствии противопоказаний. МР проводится специалистами МДК непосредственно у постели больного с использованием мобильного оборудования или в отделениях (кабинетах) МР стационара. Пребывание пациента на первом этапе завершается проведением оценки степени тяжести состояния пациента и нарушений в соответствии с международными критериями и назначением врачом-координатором следующего этапа МР.[6]
Индикаторы эффективности лечения и безопасности методов диагностики и лечения, описанных в протоколе:
Читайте также: