Примеры гемангиобластомы сетчатки у пожилых и при синдроме Маршалла-Стиклера
Добавил пользователь Алексей Ф. Обновлено: 08.01.2026
Гемангиобластома - это опухоль, образующаяся в слизистой оболочке кровеносных сосудов в нижней части головного и спинного мозга. Гемангиобластомы составляют около 2% от всех первичных опухолей головного мозга и около 3% от всех опухолей позвоночника. Для получения своевременного лечения, необходимо обращать внимание на характерные симптомы гемангиобластомы.
- Что такое гемангиобластома?
- Какие симптомы наблюдаются при гемангиобластоме?
- Ключевые характеристики гемангиобластомы
- Факторы риска и причины возникновения гемангиобластомы
- Меры профилактики гемангиобластомы
- Какие прогнозы при гемангиобластоме
Что такое гемангиобластома?
Гемангиобластома - это редкое и медленно растущее доброкачественное новообразование. Образуется преимущественно в клетках, выстилающих сосуды, снабжающих кровью головной мозг, гораздо реже поражаются спинной мозг и другие органы. Несмотря на то, что этот вид опухоли не является раковым, он может вызывать серьезные неврологические нарушения и осложнения. Наиболее часто гемангиобластома возникает в возрасте до 40 лет.
В онкологических клиниках за границей существуют специализированные отделения, врачи которых работают над изучением и лечением редких опухолей, в том числе и гемангиобластом. В таких клиниках лечение проходит гораздо эффективнее благодаря обширному опыту специалистов международного уровня.
Какие симптомы наблюдаются при гемангиобластоме?
Основные симптомы, зафиксированные при лечении гемангиобластомы за рубежом, в некоторой степени зависят от месторасположения опухоли и имеют тенденцию развиваться постепенно. При этом симптоматика может появляться и исчезать, она связана с размерами опухоли.
Различают 3 подтипа симптомов гемангиобластомы:
- общемозговой;
- мозжечковый;
- отдаленный.
Первые общемозговые симптомы основываются на повышенном внутричерепном давлении и проявляются в форме:
- головной боли (в 70% случаев);
- гидроцефалии (в 50% случаев);
- измененном психическом состоянии (в 10% случаев);
- потере координации и дисбалансе;
- тошноте;
- рвоте;
- неконтролируемом (необычном) и резком движении глаз.
Если опухоль образуется в спинном мозге, она проявляется отдаленными симптомами:
- мышечной слабостью или онемением конечностей;
- болью в спине либо в шее;
- запорами;
- утратой контроля над кишечником или мочевым пузырем (недержание кала или мочи).
Также следует учитывать, что присутствие опухоли влияет на снабжение мозга цереброваскулярной жидкостью (CSF). Именно изменение уровня CSF дает толчок к внезапному проявлению первых симптомов, особенно головных болей.
При подборе тактики лечения рака мозга за рубежом учитываются симптомы, проявляющиеся из-за особенностей локализации гемангиобластомы:
- Гемангиобластома мозжечка часто демонстрирует отклонения, связанные с увеличением размеров новообразования, которое оказывает давление на мозжечок или вызывает избыток спинномозговой жидкости (гидроцефалию). Характерные проявления: расстройства координации движений; изменение привычного почерка (он становится более крупным и размашистым), появление некоординированных движений с широким охватом. Могут возникать затруднения при выполнении элементарного неврологического теста: закрыв глаза, прикоснуться пальцем к кончику носа. Мозжечковые гемангиобластомы стремительно растут, что приводит к отклонениям и двустороннему поражению обоих полушарий.
- Гемангиобластомы, локализующиеся в стволе спинного мозга, могут вести к утрате двигательной функции, появлению мышечной слабости, потере чувствительности конечностей либо сбою естественных функций кишечника, мочевого пузыря.
Ключевые характеристики гемангиобластомы
В отличие от раковых новообразований, гемангиобластомы не проникают в близлежащие ткани и в большинстве своем характеризуются медленным ростом, поэтому оставляют возможность провести лечение до возникновения каких-либо серьезных осложнений.
Согласно системе Всемирной организации здравоохранения (ВОЗ) опухолей нервной системы, гемангиобластомы классифицируются как менингеальные новообразования неопределенного происхождения. С момента своего начального изучения гемангиобластомы были обнаружены во многих областях центральной нервной системы. Отмечалось преимущественное вовлечение мозжечка и спинного мозга, но истинная частота этой опухоли не была обнаружена до тех пор, пока не стали доступны неинвазивные методы диагностической визуализации, в частности магнитно-резонансная томография. Вместе со значительным улучшением хирургических подходов и микрохирургической техники это делало гемангиобластому хоть и опасным, но потенциально излечимым заболеванием.
Гемангиобластома имеет 1 степень агрессивности по классификации ВОЗ и относится к опухолям, которые могут возникать в центральной нервной системе или в других частях тела (в почках, печени и поджелудочной железе). Эти опухоли, как правило, визуализируются в виде резко разграниченных однородных масс.
Наиболее распространенные места локализации в процентных показателях
Внутричерепная локализация составляет 87-97% всех случаев гемангиобластом:
- 95% - в задней ямке;
- 85% - в полушарии мозжечка;
- 5% - в продолговатом мозге;
- лишь в редких случаях патология выходит за пределы мозжечка и церебеллопонтинного угла;
- 5% - в супратенториальном (обычно в зрительных лучах);
- церебральные гемангиобластомы наблюдаются только у пациентов с болезнью фон Гиппеля-Линдау.
Локализация в позвоночнике составляет лишь 3-13%.
Факторы риска и причины возникновения гемангиобластомы
Большинство гемангиобластом возникает случайно и без установленных причин (75-80% случаев). В процессе лечения рака за границей было выявлено, что около 20-25% всех случаев гемангиобластом связано с наличием у пациента генетического заболевания, известного как синдром фон Гиппель-Линдау (VHL). Этот синдром является наследственным и характеризуется аномальным ростом опухолей в определенных частях тела. Специфические опухоли, которые связаны с синдромом VHL, включают:
- гемангиобластомы головного и спинного мозга, а также сетчатки;
- почечные кисты и почечно-клеточный рак;
- феохромоцитомы;
- опухоли эндолимфатического мешка.
Люди с генетическим заболеванием Гиппеля-Линдау имеют более высокий риск возникновения множественных гемангиобластом в мозге и позвоночнике в течение всей жизни. В остальных случаях новообразование носит, как правило, единичные проявления.
К тому же прослеживается:
- незначительная мужская преимущественность заболевания у взрослых;
- пик заболеваемости приходится на возраст около 30-60 лет, но развитие патологии возможно и ранее, если у пациента имеется синдром фон Гиппель-Линдау.
В качестве факторов риска следует упомянуть общий список возможных причин, провоцирующих развитие онкологических процессов в организме:
- работа на вредных производствах, контакт с опасными канцерогенными веществами и радиоактивным излучением;
- проживание в районах с неблагополучной экологической обстановкой;
- курение, алкоголизм и наркомания;
- наличие в анамнезе инфекций, поражающих центральную нервную систему.
Каковы бы ни были причины возникновения заболевания, его необходимо лечить. Стоимость лечения рака за границей будет зависеть не только от выбранной страны, но и от опыта и статуса врача, репутации клиники и т.д.
Меры профилактики гемангиобластомы
Для этого вида опухолей на сегодня еще не разработано четкой программы профилактики. Это прежде всего связано с неустановленными причинами и случайным характером возникновения опухоли. В качестве профилактических мер специалисты предлагают придерживаться общих рекомендаций:
- сбалансированное здоровое питание;
- отказ от вредных привычек;
- посещение регулярных профилактических осмотров;
- внимание к негативным симптомам, возникающим в организме и своевременное обращение к специалистам.
Какие прогнозы при гемангиобластоме
Прогнозы всегда составляются индивидуально и зависят от множества факторов:
- размера и местоположения опухоли;
- возраста пациента;
- используемого лечения.
Хирургическая резекция опухоли - это ключевой способ, позволяющий полностью излечиться. При успешном удалении гемангиобластомы вероятность осложнений невелика. Все симптомы после лечения обычно пропадают, позволяя человеку вернуться к привычной жизни.
При обширных опухолевых областях хирургия используется для предоперационной эмболизации. Профилактическая лучевая терапия рекомендована пациентам с неполным удалением гемангиобластомы, так как вероятность рецидива остается довольно высокой (25%).
Если гемангиобластома имеет большие размеры и в процессе роста повредила нервы в головном мозге, могут возникнуть длительные осложнения. Врач учитывает эти аспекты и рекомендует дополнительные лекарства, позволяющие свести к минимуму негативное влияние на качество жизни больного. После окончания курса лечения гемангиобластомы важно регулярно проходить профилактические обследования, в ходе которых лечащий врач сможет наблюдать за возможным образованием новых опухолей или проявлением признаков рецидива.
Онкологические клиники Израиля демонстрируют положительные результаты излечения гемангиобластомы. Это связано с внушительным опытом здешних специалистов в преодолении онкологических процессов центральной нервной системы.
Синдром Стиклера ( Врожденная артроофтальмопатия )
Синдром Стиклера — это редкий вариант наследственных коллагенопатий, при котором происходит нарушение структуры коллагена II, IX, XI типов. Наследуется по аутосомно-доминантному и аутосомно-рецессивному механизмах. Заболевание проявляется прогрессирующей миопией с отслойкой сетчатки, поражением костно-суставного аппарата, характерными фенотипическими особенностями. Диагностика синдрома проводится при помощи рентгенографии скелета, офтальмоскопии, биохимического и молекулярно-генетического тестирования. Лечение поддерживающее: реабилитационные программы, психологическая поддержка, хирургическая коррекция осложнений.
МКБ-10
Общие сведения
Синдром Стиклера назван в честь американского педиатра Г.Б. Стиклера, который в 1965 г. выпустил статью с анализом своей многолетней работы с пациентами, имевшими типичный комплекс симптомов: прогрессирующую миопию, дегенерацию суставов, расширение эпифизов. Генетические основы патологии были открыты только в 1980-1990-х гг. Заболевание имеет второе название «врожденная артроофтальмопатия». Распространенность синдрома Стиклера составляет 1 случай на 7,5-10 тыс. взрослого населения, половых различий в структуре заболеваемости нет.
Причины
При синдроме Стиклера выделено 5 вариантов мутаций в генах, кодирующих синтез коллагеновых волокон: COL2A1, COL11A1, COL11A2, COL9A1, COL9A2. Заболевание 1, 2, 3 подтипов характеризуется аутосомно-доминантным вариантом наследования, то есть вероятность рождения больного ребенка составляет 50%, если один из родителей — носитель мутантного гена. Синдром 4 и 5 подтипов передается аутосомно-рецессивно, проявляется только если оба родителя имеют мутантный ген.
Патогенез
Болезнь возникает при нарушении структуры коллагеновых волокон 2-го, 9-го и 10-го типов, которые входят в состав костной и хрящевой ткани, стекловидного тела глазного яблока. Дефектный коллаген изменяет нормальную структуру соединительной ткани. При этом у пациентов нарушается формирование суставов, происходят дегенеративные процессы, деформируется костный скелет и, как следствие, становятся заметными характерные фенотипические признаки.
Классификация
В клинической практике систематизация заболевания на отдельные подтипы используется редко, что обусловлено схожестью их симптоматики, общностью подходов лечения. По результатам молекулярно-генетической диагностики были выделены 5 разновидностей синдрома Стиклера, которые отличаются локализацией мутации, видом дефектного коллагена. В классификации наследственной коллагенопатии выделяют такие варианты:
- I — мутация COL2A1, кодирующего альфа-1 цепь коллагена 2 типа.
- II — аномалия COL11A1, который отвечает за синтез альфа-1 цепи коллагена 11 типа.
- III — патология COL11A2, отвечающего за образование α-2 цепи коллагеновых волокон 11 типа.
- IV — изменение последовательности COL9A1, регулирующего синтез α-1 коллагеновых цепей 9 типа.
- V — точечная мутация COL9A2, который кодирует альфа-2 цепь коллагеновых волокон 9 типа.
Симптомы
Дети с врожденной артроофтальмопатией имеют специфические изменения черт лица, которые заметны уже в неонатальном периоде. У больных проявляется аномалия Робина: плоское лицо с недоразвитой нижней челюстью, расщелины неба, западение языка. Также наблюдается выпирание верхней губы, деформация скуловых дуг, западение переносицы. Изредка пациенты имеют типичный марфаноидный фенотип, что затрудняет постановку диагноза.
Суставные поражения вначале проявляются гипермобильностью, чрезмерной гибкостью и подвижностью. Дети и подростки часто страдают искривлениями позвоночника в виде кифоза, лордоза, сколиоза. По мере прогрессирования синдрома дегенеративные изменения провоцируют контрактуры, боль, увеличение и припухлость суставов. Пациенты после 30 лет обычно сталкиваются с артропатией тазобедренных, коленных, голеностопных суставов.
Повреждение зрительного аппарата состоят в прогрессирующем ухудшении зрения, появлении катаракты, дегенеративных изменений стекловидного тела. Возникают нарушения слуха по типу кондуктивной тугоухости. У части больных отмечается задержка психомоторного развития, снижение интеллектуальных способностей легкой или средней степени тяжести.
Осложнения
Наиболее опасным последствием синдрома является отслойка сетчатки, которая может начаться в детском возрасте при отсутствии лечения прогрессирующей миопии. В результате наступает слепота на один или оба глаза. Поражение суставов со временем приводит к ограничению подвижности и без коррекции заканчивается инвалидностью больных. Неэстетичные изменения лицевого скелета становятся причинами комплексов, проблем с социализацией.
Вовлечение в процесс слухового анализатора чревато частыми отитами, а при неблагоприятном течении есть риск снижения слуха вплоть до глухоты. Изредка патология соединительной ткани затрагивает клапанный аппарат сердца, что вызывает пролапс митрального клапана, нарушения кровообращения, сердечную недостаточность. Аномалии строения ротовой полости сопровождаются проблемами с употреблением пищи, высоким риском аспирации, затруднениями при дыхании.
Диагностика
Синдром Стиклера отличается полиморфностью клинической картины, схожестью симптоматики с другими наследственными нарушениями соединительной ткани, что усложняет диагностический поиск. При первичном осмотре у педиатра определяются фенотипические проявления коллагенопатий, изучается семейный анамнез. Для подтверждения врожденной артроофтальмопатии назначаются следующие методы исследования:
- Рентгенография скелета. Патогномоничными симптомами синдрома Стиклера являются расширенные эпифизы костей, измененная структура диафизов, дегенерация суставов. Для более детальной диагностики делаются компьютерная томография суставов, КТ лицевого черепа.
- Молекулярно-генетическое тестирование. Исследование генома рекомендовано для 100% подтверждения диагноза у страдающих болезнью Стиклера. В генетических лабораториях используют методики флуоресцентной гибридизации, секвенирования генома, чтобы выявить поврежденный ген, уточнить подтип синдрома.
- Лабораторные методы. С помощью тонкослойной хроматографии выявляют повышение уровня гликозаминогликанов в крови, их усиленное выделение с мочой. Состояние коллагеновых волокон оценивают иммуногистохимическим способом с определением специфических антител, а также путем анализа свободного гидроксипролина в моче.
- Консультации специалистов. Осмотр офтальмолога с проведением офтальмоскопии необходим, чтобы оценить состояние зрительного аппарата, осмотреть глазное дно, выявить признаки начинающейся отслойки сетчатки. Прием отоларинголога с выполнением аудиометрии применяется при проблемах со слухом.
Лечение синдрома Стиклера
Консервативная терапия
Этиопатогенетическое лечение болезни Стиклера отсутствует. Усилия врачей направлены на коррекцию существующих проблем со здоровьем, повышение качества жизни пациентов. Комплексный подход к терапии таких больных включает несколько направлений:
- ЛФК. Лечебная физкультура необходима для устранения тугоподвижности суставов, возникающей при прогрессировании синдрома, коррекции искривлений позвоночника и сопутствующего им мышечно-тонического синдрома. ЛФК, как правило, дополняется массажем, кинезиотейпированием.
- Абилитация. Детям со сниженным интеллектом требуется обучение в специальных школах, занятия с дефектологами, логопедами, олигофренопедагогами. Таким образом удается повысить обучаемость пациентов, подготовить их к самостоятельной жизни.
- Психологическая помощь. Дефекты внешности в сочетании со снижением когнитивных возможностей затрудняют социализацию детей, провоцируют замкнутость, депрессии. Для коррекции этих проблем требуется регулярные визиты к детскому психологу.
Хирургическое лечение
Больным зачастую требуется помощь офтальмохирургов, которые проводят лазерную коагуляцию или криотерапию для лечения отслоения сетчатки, сохранения зрения. При критическом ухудшении слуха рассматривается вопрос о постановке слухового аппарата. Аномалии развития лицевого черепа требуют поэтапной пластической коррекции, чтобы устранить проблемы с дыханием и приемом пищи, улучшить эстетику внешности.
Прогноз и профилактика
Состояние пациентов зависит от своевременности постановки диагноза, полноты терапевтических, хирургических, реабилитационных мероприятий. При должном лечении удается поддерживать хорошее качество жизни больных, поэтому прогноз относительно благоприятный. Меры первичной профилактики синдрома не разработаны. Семейным парам с отягощенной наследственностью рекомендована консультация генетика при планировании зачатия.
1. Современные подходы к диагностике наследственных нарушений соединительной ткани/ А.В. Клеменов, А.С. Суслов// Лечащий врач. — 2014. — №3.
3. Синдром Стиклера I типа у детей/ А.Н. Семячкина, А.В Поляков, П.В. Новиков, Е.А. Каменец// Российский вестник перинатологии и педиатрии. — 2009. — №3.
4. Синдром Марфана/ В.М. Делягин, Ж.С. Жакулова, И.А. Нарычева, М.Б. Мельникова// Практическая медицина. — 2008. — №28.
Синдром Маршалла ( PFAPA-синдром )
Синдром Маршалла (PFAPA-синдром) - это заболевание преимущественно детского возраста, включающее периодическую лихорадку, афтозный стоматит, фарингит, шейную лимфаденопатию. Симптомами являются регулярно повторяющиеся эпизоды повышения температуры выше 39˚С, боли в горле, язвенные поражение слизистой рта, увеличение шейных лимфоузлов. Диагноз устанавливается на основании клиники, анализов крови, посевов отделяемого из зева, исключения других возможных причин рецидивирующей лихорадки. Лечение ограничено глюкокортикоидами или жаропонижающими препаратами, так как антибиотики и противовирусные средства неэффективны. В редких случаях проводится тонзиллэктомия.
В 1987 году американским педиатром Г.Маршаллом и соавторами описана ранее неизвестная периодическая лихорадка, которая первоначально была названа синдромом Маршалла. В современных источниках используется название PFAPA-синдром как аббревиатура симптомов, включающих периодическую лихорадку (periodic fever), афтозный стоматит (aphthous stomatitis), фарингит (pharingitis), шейный лимфаденит (cervical adenitis). Распространенность и заболеваемость точно неизвестны. Дебют патологии отмечается в возрасте от 1 до 5 лет и, как правило, к подростковому периоду симптоматика разрешается. В последнее время появились данные о возможности развития PFAPA-синдрома у взрослых. Среди заболевших преобладают мужчины (55-70%).
Причины синдрома Маршалла
Этиология остается неизвестной. Сегодня PFAPA-синдром рассматривается как полигенное или мультифакториальное заболевание, при котором модифицирующую роль играют генетические, средовые факторы, возможные особенности реагирования организма на инфекцию. У 7-10% пациентов выявляются мутации гена MEFV, который участвует в образовании белка пирина гранулоцитами, моноцитами, дендритными клетками, фибробластами кожи, брюшины и синовиальной оболочки. Его предполагаемая функция состоит в снижении воспалительного ответа путем ингибирования активации и хемотаксиса нейтрофилов. Выявляются мутации TNFRSF1A, МВК, гена, кодирующего белок NLRP3.
Цитокиновый профиль при данной патологии позволяет отметить повышение сывороточных уровней интерлейкина-1β (ИЛ-1β), ТNFα, ИЛ-6, ИЛ12р70, в том числе в период между атаками, что указывает на постоянно текущее субклиническое воспаление. Определенный инфекционный агент не выделен, некоторые исследования указывают на возможное участие вирусов Эпштейна-Барр, простого герпеса 1, 2 типов и цитомегаловируса, а также редких бактериальных агентов: Муcobacterium chelonae (микобактерии хелона), Plasmodium (плазмодии), Borrelia (боррелии), Brucella (бруцеллы). У части исследуемых больных выявляется дефицит витамина D.
Точный патогенез не установлен. Современные исследователи относят PFAPA-синдром к системным аутовоспалительным заболеваниям. Их отличием от аутоиммунных являются генетически обусловленные особенности протекания воспалительной реакции и реагирования иммунитета, а не механизмы синтеза антител и активации Т-лимфоцитов при контакте с антигеном. Наличие мутации гена MEFV ведет к синтезу дефектного пирина, который в норме ослабляет и ингибирует чрезмерную восприимчивость организма, а в измененном виде приводит к дефициту ингибитора хемотаксического фактора С5а, что нарушает функцию контроля процесса воспаления.
Ген NLRP3 кодирует белок криопирин, при его мутации моноциты под влиянием разнообразных триггеров начинают синтезировать огромное количество ИЛ-1β. В норме иммунная система способна защитить себя от избытка данного цитокина. При PFAPA-синдроме эта регуляция нарушена, вследствие чего развивается клиническая картина, так как интерлейкин-1β ответственен не только за гипертермию, но и за повреждение, ремоделирование тканей, повышенный уровень маркеров системного воспаления.
Симптомы синдрома Маршалла
Клиническая картина представляет собой лихорадочные эпизоды, которые повторяются каждые 2-12 недель (средний цикл 28 дней). Температура чаще повышается внезапно, лихорадка достигает высоких цифр (от 40 до 41°C). Иногда за сутки перед повышением температуры появляется общая слабость, астения, снижение аппетита. Затем присоединяется афтозный стоматит, при котором появляются мелкие (до 5 мм) язвенные поражения слизистой полости рта - афты. Фарингит характеризуется болью в горле, гиперемией слизистой глотки. В типичных случаях развивается шейный лимфаденит - лимфоузлы в области шеи увеличиваются, становятся болезненными при пальпации.
В 43-48% наблюдений симптомы возникают все вместе, чаще всего встречается стоматит (55%). Крайне редко пациентов беспокоит головная боль, тошнота, рвота, вздутие живота. На 4-5 сутки температура тела нормализуется, воспалительные явления разрешаются. Интервалы между атаками составляют от 2 до 7 недель. Со временем межприступные промежутки могут удлиняться. Особенностью протекания лихорадки является то, что при температуре 40°C общее самочувствие детей остается относительно удовлетворительным. Между эпизодами восстанавливается аппетит, набирается потерянная масса тела. Рост, общее психоэмоциональное развитие не страдает. Течение заболевания доброкачественное, атаки обычно прекращаются к подростковому возрасту. Описание дебютов синдрома Маршалла во взрослом возрасте делает его актуальным не только для педиатрии.
Осложнений синдрома Маршалла описано не было. Долгосрочных исследований у пациентов не проводилось. Однако на фоне афтозного стоматита, фарингита возможно присоединение вторичной инфекции, что может привести к развитию тонзиллита, заглоточного абсцесса, отита, гнойного медиастинита. Длительно текущий воспалительный процесс повышает риск возникновения амилоидоза. Кроме того, рецидивирующие эпизоды лихорадки оказывают изнуряющее действие, вынуждают ребенка пропускать школьные занятия, могут привести к неуспеваемости.
Для постановки диагноза PFAPA-синдрома используют диагностические критерии, предложенные Маршаллом (1987 г.): регулярно повторяющиеся лихорадки с раннего возраста (начало 2-5 лет); присутствие одного из следующих клинических признаков: афтозный стоматит, шейный лимфаденит, фарингит; полностью бессимптомный интервал между эпизодами лихорадки; нормальное физическое и нервно-психическое развитие ребенка; отсутствие циклической нейтропении. На данный момент нет специфических анализов для установления синдрома Маршалла. Диагностический поиск включает:
- Консультацию педиатра, ревматолога. Производится детальный сбор анамнеза пациента: история течения беременности и родов у матери, наследственность, особенности питания, роста, развития ребенка, перенесенные заболевания, информация о вакцинации, наличие или отсутствие контакта с инфекционными больными. Осматриваются слизистые оболочки щёк, глотки, миндалин; проводится аускультация сердца, легких, измерение АД, пульса; пальпация живота, лимфоузлов.
- Клинико-биохимические анализы. В период лихорадки общий анализ крови выявляет лейкоцитоз с повышением нейтрофилов, ускорение СОЭ. В периоды между приступами все воспалительные параметры нормализуются. Также во время атак повышается уровень С-реактивного белка, печеночные ферменты не изменены. Анализ крови на 25-OH может выявлять дефицит витамина Д3- холекальциферола. IgG, IgA, IgM, IgD, прокальцитонин, антинуклеарные антитела, ревматоидный фактор даже при повышении температуры тела остаются нормальными.
- Дополнительные исследования. Посев отделяемого из верхних дыхательных путей на микрофлору и чувствительность к антибиотикам, посев мочи, крови на стерильность (на высоте лихорадки), рентгенография органов грудной клетки, придаточных пазух носа выполняются для исключения инфекционной природы лихорадки. При синдроме Маршалла патологических изменений при данных исследованиях не выявляется.
Дифференциальный диагноз синдрома Маршалла проводят со следующими нозологиями: возвратный тонзиллит, инфекционные заболевания, ювенильный идиопатический артрит, циклическая нейтропения, семейная средиземноморская лихорадка (FMF), синдром гиперглобулинемии D, болезнь Бехчета.
Лечение синдрома Маршалла
Методы лечения все еще являются предметом споров. Антибиотикотерапия, применение противовирусных, антигистаминных препаратов не имеют эффективности; НПВС обладают только кратковременным жаропонижающим эффектом. На сегодняшний день для терапии PFAPA-синдрома успешно используются:
- Кортикостероиды. Одна или две дозы преднизолона (1-2 мг/кг), бетаметазона (0,1-0,2 мг/кг) могут резко прекратить приступы лихорадки в течение нескольких часов. Другие сопутствующие симптомы требуют больше времени для разрешения. Стероиды применяются только во время приступов, указанные дозировки не вызывают токсических эффектов. Глюкокортикоидная терапия способна сократить интервал между приступами, но не предотвращает рецидивы.
- Колхицин. Может быть эффективным для предотвращения частых эпизодов лихорадки, на течение лихорадочного периода он не влияет. Побочным эффектом являются желудочно-кишечные расстройства (в 20% случаев). Было проведено несколько исследований данного препарата, причем большинство из них - в Израиле, где большая доля пациентов несут патогенные варианты MEFV.
- Циметидин. В исследованиях на небольших группах примерно четверть пациентов (24-27%) имели полное разрешение эпизодов лихорадки при его приеме, а еще 24-32% сообщили о частичной эффективности с уменьшением частоты или тяжести приступов.
- Анакинра. В настоящее время в качестве экспериментальной терапии рассматриваются рекомбинантные антагонисты рецептора интерлейкина-1β (анакинра), проводятся исследования данной группы препаратов. Все пациенты продемонстрировали клиническое улучшение, снижение уровня цитокинов в крови.
- Тонзиллэктомия. Является радикальным методом, который приводит к полному излечению. Операция должна выполняться только в случае непереносимости или неэффективности стандартной лекарственной терапии в связи с определенными рисками инвазивного вмешательства (кровотечение, осложнения анестезии).
Все проявления синдрома Маршалла обычно самостоятельно разрешаются до подросткового возраста. Фатальные последствия и серьезные осложнения в литературе не описаны. Несмотря на благоприятный прогноз, при появлении схожих жалоб необходимо обратиться к детским специалистам (педиатру, отоларингологу, стоматологу) для обследования, установления диагноза и подбора адекватной терапии. Специфических методов профилактики не существует. Пациентам, страдающим данным синдромом, рекомендовано применять витамин Д3 в дозе 400 МЕ в зимнее время.
1. Клинико-лабораторная характеристика детей с синдромомм Маршалла/ Жданова Л.В., Бимбаев А. Б-Ж.// Вестник Бурятского государственного университета. - 2017.
2. Случай трудной диагностики больной с синдромом Маршалла/ Сафина А.И., Лутфуллин И.Я., Закиров К.З, Шапиро В.Ю.// Казанский медицинский журнал. - 2001.
3. Синдром периодической лихорадки, афтозного стоматита, фарингита и шейного лимфаденита (синдром Маршалла)/ Таточенко В.К. и соавт.// Вопросы современной педиатрии. - 2003.
Болезнь Гиппеля-Линдау: гемангиобластома сетчатки
Информация только для специалистов в сфере медицины, фармации и здравоохранения!
Содержание
- Гемангиобластома центральной нервной системы
- Гемангиобластома сетчатки
- Светлоклеточный почечно-клеточный рак
- Феохромоцитома
- Опухоли эндолимфатического мешка среднего уха
- Серозная цистаденома и нейроэндокринные опухоли поджелудочной железы
- Папиллярная цистаденома придатка яичка и широкой связки матки
Молекулярная биология болезни Гиппеля-Линдау

Ген фон Гиппеля-Линдау (VHL) был картирован на хромосоме 3p25 и клонирован в начале 1990-х, а дальнейшие исследования в понимании функции гена VHL продолжались в течение следующих 20 лет. Продукт этого гена, белок VHL, действует как супрессор опухолей. Как и в случае с патогенными вариантами некоторых других генов-супрессоров опухолей (например, гена ретинобластомы 1, RB1), модель «двух ударов» была подтверждена для БГЛ, при которой вариант потери функции зародышевой линии инактивирует одну копию VHL-гена во всех клетках. Для развития опухолей, связанных с VHL, должна произойти потеря экспрессии второго нормального аллеля либо из-за соматических изменений, либо из-за делеции второго аллеля, либо из-за гиперметилирования его промотора. При спорадическом почечно-клеточном раке инактивация VHL за счет соматических изменений обоих аллелей очень распространена.
За последние два десятилетия были достигнуты значительные успехи в понимании биологии, лежащей в основе формирования опухолей, связанных с VHL. Белок VHL образует стабильный комплекс с несколькими другими белками, включая элонгины В и С, а также куллин 2. Этот комплекс нацелен на несколько белков для протеасомной деградации, тем самым регулируя их уровни внутри клетки. Компонент VHL этого комплекса функционирует как убиквитинлигаза E3 для молекул-мишеней. После связывания с комплексом VHL молекулы-мишени ковалентно связываются с убиквитином, облегчая деградацию протеасомой.
В дополнение к своей функции убиквитинлигазы E3, VHL выполняет несколько других важных клеточных функций, включая поддержание первичной реснички, регуляцию цитокинеза, контроль функции микротрубочек, целостность внеклеточного матрикса и регуляцию клеточного цикла. Патогенные варианты гена VHL также связаны с врожденными формами истинной полицитемии.
Клиническая картина капиллярной гемангиобластомы сетчатки

Капиллярные гемангиобластомы сетчатки обычно локализуются либо в периферической части сетчатки, либо в юкстапапиллярной области. Потеря зрения при капиллярных гемангиобластомах сетчатки обычно вызывается экссудацией из опухоли, вызывающей отек сетчатки, или тракционными эффектами, при которых глиальная пролиферация на поверхности опухоли вызывает стрии и деформацию сетчатки. Кроме того, капиллярные гемангиобластомы сетчатки могут кровоизлиять, что приводит к отслойке сетчатки, глаукоме и потере зрения.
Капиллярные гемангиобластомы сетчатки обнаруживаются у 70% пациентов с БГЛ к возрасту 60 лет; они часто многоочаговые и двусторонние. По сравнению с пациентами со спорадическими гемангиобластомами сетчатки пациенты с БГЛ намного моложе и чаще имеют множественные поражения. В одной серии из 31 пациента с БГЛ и 37 пациентов без неё пациенты с БГЛ были моложе (18 лет против 36 лет соответственно), имели в среднем четыре опухоли и были более склонны к развитию новых опухолей, чем пациенты без заболевания. заболевание.
В исследовании 890 пациентов с БГЛ у 335 пациентов была капиллярная гемангиобластома сетчатки как минимум в одном глазу. Поражения были обнаружены односторонне у 42% и двусторонне у 58% пострадавших пациентов. Корреляции между возрастом, полом и латеральностью поражения выявлено не было. Из вовлеченных глаз 87% имели опухоли, которые можно было визуализировать индивидуально; из них опухоли обычно обнаруживались только в периферической части сетчатки (85%) и реже в юкстапапиллярной области (15%). Количество опухолей на периферии в среднем составляло 2,5+/-1,8 на глаз, при этом 25% глаз имели поражение более одного квадранта сетчатки. Оценка взаимосвязи генотип-фенотип при капиллярной гемангиобластоме сетчатки показала, что у 15% людей с вариантами, которые приводят к полной потере белка VHL, развилась гемангиобластома по сравнению с общей распространенностью в популяции пациентов 37%. Было обнаружено, что риск потери зрения увеличивается с возрастом, хотя количество опухолей существенно не увеличивается в зависимости от возраста.
Как и в случае с кажущимися спорадическими гемангиобластомами центральной нервной системы (ЦНС), любой пациент с капиллярной гемангиобластомой сетчатки (особенно в возрасте до 40 лет) должен пройти генетическое тестирование зародышевой линии на наличие патогенных вариантов гена VHL.
Рутинное наблюдение за капиллярной гемангиобластомой сетчатки рекомендуется для пациентов с заболеванием VHL из-за его высокой частоты. Частое появление таких поражений в детстве делает важным инициировать офтальмологическое наблюдение в педиатрической популяции после постановки диагноза, и это является одной из причин, по которой рекомендуется генетическое тестирование на патогенные варианты гена VHL у детей раннего возраста.
Диагностика капиллярной гемангиобластомы сетчатки
Диагноз болезни (фон) Гиппеля-Линдау (БГЛ) обычно устанавливается путем обнаружения зародышевого патогенного (обычно с потерей функции) варианта гена VHL. Это чаще всего наблюдается у пациентов, которые проходят генетическое тестирование после того, как у них диагностировано одно проявление БГЛ, или у тех, кто проходит тестирование, потому что у их близких родственников диагностирована эта болезнь. Диагноз также может возникнуть, когда генетическое тестирование проводится по другой причине и неожиданно обнаруживает вторичный патогенный вариант VHL. В редких случаях у пациентов, у которых нет доступа к генетическому тестированию, диагноз болезни может быть основан на клинических критериях (например, у пациентов с одним поражением, связанным с БГЛ, и соответствующим семейным анамнезом, или у пациентов с множественными поражениями, связанными с БГЛ).
Пациентов с подозрением на БГЛ следует направлять в специализированные центры для оценки, генетического консультирования и окончательного диагноза с помощью генетического тестирования, даже если в семейном анамнезе нет этой болезни.
Пациентов следует направлять на соответствующую генетическую консультацию в сочетании с генетическим тестированием на патогенные варианты гена VHL. Болезнь наследуется по аутосомно-доминантному типу, и больные люди имеют 50-процентную вероятность передачи ассоциированного с болезнью варианта гена VHL каждому потомку. Учитывая разный возраст появления опухоли, большинство людей с БГЛ доживают до зрелого возраста и рожают детей в основном до постановки диагноза. Таким образом, нет ничего необычного в том, чтобы увидеть многопоколенческие родословные БГЛ со многими пораженными людьми, каждый из которых имеет несколько разные модели диагнозов опухолей и разный возраст начала заболевания.
Среди редких пациентов с соматическим мозаицизмом риск для потомства зависит от того, несет ли зародышевая ткань патогенный вариант, хотя это обычно не определяется клинически. Таким образом, пациенты с документально подтвержденным мозаицизмом должны быть проинформированы о том, что риск рождения больного ребенка может достигать 50% и что любой больной ребенок унаследует патогенный вариант в 100% своих клеток и потенциально будет иметь более тяжелые проявления болезни, чем мозаичный родитель.
Диагноз БГЛ у ребенка здоровых родителей может быть очень тревожным, и следует тщательно объяснить концепцию патогенных вариантов de novo или переменной экспрессивности (например, когда у родителя еще не была диагностирована опухоль, связанная с БГЛ). Никогда не следует предполагать, что здоровые родители отрицательны на патологический вариант гена VHL без прямого генетического тестирования. Родителей следует успокоить, а возможную вину смягчить, объяснив, что патогенный вариант de novo вряд ли может быть результатом какого-либо действия, имевшего место непосредственно перед беременностью или во время нее.
Растет обеспокоенность родителей относительно того, когда предоставлять информацию о диагнозе детям с положительным генетическим тестом на VHL. В общем, лучше всего передавать эту информацию в различных условиях по мере взросления ребенка, и родители могут получить помощь от медицинского работника при инициировании этих дискуссий.
Лечение капиллярной гемангиобластомы сетчатки
Лечение капиллярной гемангиобластомы сетчатки требует, чтобы преимущества лечения были сбалансированы с потенциальными осложнениями, связанными с лечением. Данные о том, можно ли внимательно наблюдать за небольшими поражениями без специфического лечения, до тех пор, пока не появятся какие-либо признаки роста или симптомы, противоречивы. Некоторые группы рекомендуют лечить капиллярную гемангиобластому сетчатки сразу после обнаружения (чтобы предотвратить рост и осложнения), в то время как другие ждут некоторого изменения в размере, прежде чем начинать лечение. Для тех, кто начинает лечение, мы предлагаем лазерную фотокоагуляцию, а не другие методы лечения. Другие альтернативные варианты включают фотодинамическую терапию или лучевую терапию. Системная терапия белзутифаном является приемлемым вариантом для пациентов, которым противопоказана местная терапия из-за близости опухоли к зрительному нерву или множественных прогрессирующих поражений. Клинические испытания приветствуются, если таковые имеются.
Местная терапия. Лазерная фотокоагуляция эффективна более чем в 70% случаев, как правило, при однократном лечении и является предпочтительным методом лечения. Исключением является то, что гемангиобластомы зрительного нерва не следует лечить этими методами из-за вредных побочных эффектов на нормальную сетчатку. Фотодинамическая терапия также может рассматриваться как вариант лечения капиллярной гемангиобластомы сетчатки, хотя данные о ее эффективности ограничены. Внешняя лучевая терапия может быть использована в качестве терапии спасения, если другие методы оказались неэффективными.
Белзутифан, ингибитор индуцируемого гипоксией фактора-2альфа (HIF-2альфа), является вариантом лечения пациентов с капиллярными гемангиобластомами сетчатки, расположенными близко к зрительному нерву, или пациентов с множественными прогрессирующими гемангиобластомами; такие пациенты, как правило, не подходят для местной терапии. В исследовании фазы II белзутифан улучшил состояние во всех 16 глазах (100%) у 12 пациентов с подлежащими оценке гемангиобластомами сетчатки.
Экспериментальная терапия (антиангиогенные средства) - необходимы дальнейшие исследовательские исследования для лучшего понимания биологии исходной гемангиобластомной клетки и ее эндотелия, а также для разработки активной системной терапии. Несколько ингибиторов рецепторов фактора роста эндотелия сосудов (VEGF), которые препятствуют ангиогенезу, такие как сунитиниб, пазопаниб, бевацизумаб и ранибизумаб, продемонстрировали ограниченную эффективность при гемангиобластоме сетчатки.
Наблюдение за пациентами с капиллярной гемангиобластомы сетчатки

Заболеваемость и смертность у пациентов с болезнью (фон) Гиппеля-Линдау (БГЛ) значительно снизились благодаря лучшему пониманию естественного течения серьезных клинических проявлений заболевания, более совершенным методам визуализации и улучшениям в терапии. Наблюдение важно не только для выявления новых поражений на ранней стадии, но и для наблюдения за небольшими бессимптомными поражениями на предмет признаков прогрессирования.
Протоколы наблюдения сосредоточены на гемангиобластомах (включая капиллярные гемангиобластомы сетчатки), почечно-клеточном раке (ПКР), феохромоцитомах и аудиологическом исследовании, учитывая повышенный риск опухолей эндолимфатического мешка (ЭЛСТ) у пациентов с БГЛ. Рекомендации по наблюдению могут быть адаптированы к каждому пациенту с учетом текущего или предшествующего диагноза опухоли. Тем не менее, все люди с БГЛ, даже если в настоящее время они бессимптомны, должны понимать, что у них могут развиться проявления болезни, и им будет полезно следовать рекомендациям по эпиднадзору.
Несколько организаций предоставляют обновленные руководства по эпиднадзору, учитывающие современные методы визуализации и лабораторной диагностики. Международная группа клиницистов, занимающихся лечением детей с БГЛ, была созвана в 2016 году в рамках семинара Американской ассоциации исследований рака (AACR) по детской предрасположенности к раку. Группа рассмотрела как американские, так и европейские схемы по БГЛ и опубликовала рекомендации по эпиднадзору, которые предусматривали повышение интенсивности и более раннее начало скрининга. Эти рекомендации впоследствии были оценены консенсусной комиссией, сформированной Альянсом БГЛ, состоящей из клиницистов, занимающихся всеми областями знаний, связанных с лечением болезни, и представителей семинара AACR. Ниже приводится сводка этих рекомендаций.
Макулодистрофия (возрастная макулярная дегенерация) - симптомы и лечение
Что такое макулодистрофия (возрастная макулярная дегенерация)? Причины возникновения, диагностику и методы лечения разберем в статье доктора Казанцевой Екатерины Игоревны, офтальмолога со стажем в 6 лет.
Над статьей доктора Казанцевой Екатерины Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Макулодистрофия (макулярная дегенерация, или макулопатия) — это обширная группа хронических прогрессирующих заболеваний, при которых постепенно поражается центральная зона нервной ткани глаза — макулярная область сетчатки. Сопровождается снижением центрального зрения. Как правило, поражает оба глаза.
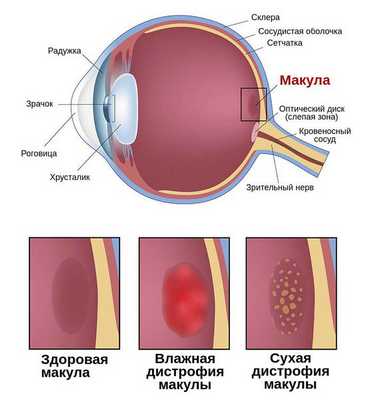
Заболевания, разрушающие макулярную область, входят в пятёрку главных причин снижения остроты зрения у взрослого населения России [1] . Одной из распространённых форм макулодистрофии является возрастная макулярная дегенерация, или, как её ещё называют, сенильная (старческая) макулодистрофия. Этой форме и посвящена данная статья.
Возрастная макулодистрофия — одна из основных причин необратимого ухудшения и потери зрения у людей старше 50 лет во всём мире [1] .
В основном данное заболевание поражает женщин, причём после 75 лет оно встречается в два раза чаще. Причина такой закономерности пока неизвестна.
В основе возрастной макулодистрофии лежит нарушение питания центральной зоны сетчатки. Причин такого нарушения множество. Прежде всего оно возникает в результате старения, о чём говорит связь частоты появления болезни с возрастом. Отдельная роль отводится наследственности [5] .
Другим доказанным фактором риска является курение. Отказ от этой пагубной привычки может снизить риск развития макулодистрофии в несколько раз [4] .
Также риск появления возрастной макулодистрофии повышают сердечно-сосудистые заболевания, особенно атеросклероз, атеросклеротические бляшки на сонной артерии и гипертоническая болезнь [6] .
Вероятность развития дистрофии повышается у людей, которые употребляют больше насыщенных жиров и холестерина, а также при высоком индексе массы тела. Ниже риск разрушения макулы у людей, потребляющих достаточное количество омега-3 полиненасыщенных жирных кислот — содержатся в морской рыбе, мясе диких животных, морских водорослях и др.
Одной из важных причин появления макулодистрофии является избыточная инсоляция — облучение солнечным светом. При попадании на сетчатку ультрафиолета коротковолнового или синего спектра видимого света в ходе фотохимических реакций формируются свободные радикалы. Они повреждают мембраны клеток, т. е. фоторецепторов [19] . В норме для защиты сетчатки от действия свободных радикалов в тканях присутствует антиоксидантная защита. При макулодистрофии в системе формирования свободных радикалов и антиоксидантной защиты возникает дисбаланс [8] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы макулодистрофии
При возрастной макулодистрофии зрение снижается постепенно и безболезненно в течение нескольких лет. Пациенты замечают снижение остроты зрения, которое не получается скорректировать подбором очков. Также появляются жалобы на затуманивание зрения, трудности при чтении, снижение контрастной чувствительности, особенно в помещении с плохим освещением или в сумерках.
Иногда при запущенных стадиях центральное зрение снижается до такой степени, что пациенты видят лишь боковым зрением или эксцентрично, а в центре — только чёрное или серое пятно.

На начальных стадиях развития возрастной макулодистрофии жалоб у пациентов может и не быть, поэтому заболевание становится находкой во время офтальмологического осмотра, проводимого по другому поводу [15] .
Патогенез макулодистрофии
Несмотря на многочисленные исследования возрастной макулодистрофии, патогенез этого заболевания до сих пор остаётся невыясненным. Но часть основных звеньев патогенеза всё же изучена.
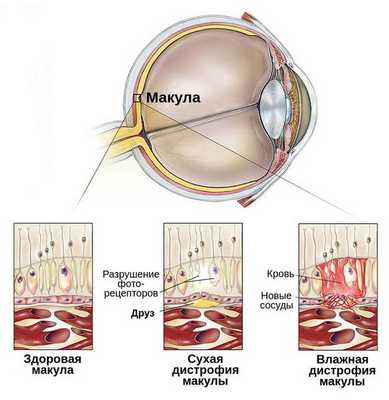
C возрастом между слоями сетчатки откладываются продукты жизнедеятельности фоторецепторов — липофусцин, или пигмент "старости". Их называют друзами. Они возникают из-за ухудшения питания тканей и замедленного выведения из них продуктов обмена.
![Друзы [13]](/pimg3/primeri-gemangioblastomi-setchatki-687E0A.jpg)
Механизм формирования друз похож на процесс образования атеросклеротических бляшек артерий. Избыточное отложение липофусцина способствует повреждению пигментного эпителия и фоторецепторов, так как это вещество способно под воздействием света производить активные формы кислорода, включая перекисное окисление липидов. Этот процесс ещё больше усугубляется при большом количестве ультрафиолета [19] . В итоге повреждаются окружающие клетки и ткани.
Далее патогенез может пойти по одному из путей:
- возникают обширные участки атрофии сетчатки — формируется сухая форма заболевания;
- высвобождаются факторы роста сосудов или VEGF с развитием неоваскуляризации — формируется влажная форма макулодистрофии.
Все эти процессы протекают с выраженной дисфункцией макулы и резким ухудшением центрального зрения.
Классификация и стадии развития макулодистрофии
Разные подходы и взгляды на патогенез макулодистрофии, развитие методов диагностики стали причиной появления нескольких вариантов классификаций возрастной макулодистрофии.
Одна из международных классификаций была принята в 1995 году. В ней разделяли обширное понятие "возрастная макулодистрофия" следующим образом:
- ранние формы заболевания, или возрастная макулопатия — образование друз, диспигментация;
- поздние формы заболевания, или, собственно, сама возрастная макулярная дегенерация — географическая атрофия и хориоидальная неоваскуляризация — разрастание сосудов.
Но большинство офтальмологов в России используют в своей практике классификацию макулодистрофии, основанную на этапах развития дистрофического процесса. Кацнельсон Л. А. с соавторами выделяют три формы (стадии) заболевания [9] :
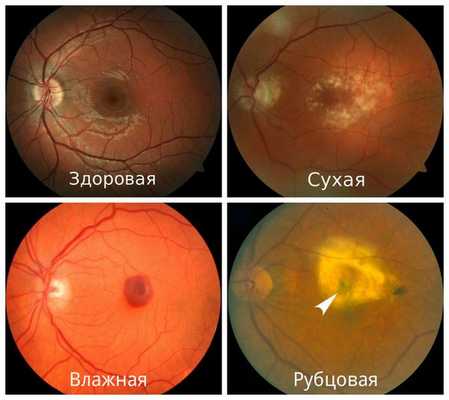
- Сухая форма (неэкссудативная). На начальном этапе может никак себя не клинически проявлять либо сопровождаться снижением центрального зрения при плохом освещении или в сумерках. Далее нарушение центрального зрения прогрессирует. Это проявляется наличием друз, дефектов пигментного эпителия, перераспределением пигмента, атрофией пигментного эпителия. На данном этапе заболевание может остановиться в своём развитии и не переходить в другие формы.
- Влажная форма (экссудативная). Она отличается процессами, которые возникают в ответ на прогрессивную отёк и ишемию сетчатки в виде неоваскуляризации сетчатки — разрастания сосудов. Является более агрессивной формой, чем сухая, и влечет за собой более быстрое и прогрессивное ухудшение центрального зрения. Проявляется быстрым снижением центрального зрения, искривлением предметов, появлением серого пятна перед глазом.
- Рубцовая форма. Является финалом заболевания. Проявляется стойким снижением остроты зрения вплоть до потери центрального зрения. Пациенты зачастую видят периферией или эксцентрично. Под сетчаткой формируется грубая соединительная ткань на фоне ранее новообразованных сосудов. Атрофируется пигментный эпителий и слой фоторецепторов, в результате чего макулярная зона становится нефункциональной или малофункциональной.
Осложнения макулодистрофии
Так как заболевание на начальных стадиях протекает практически бессимптомно, очень высок риск того, что впервые макулодистрофия обнаружится только тогда, когда сетчатка уже будет достаточно повреждена. Это состояние будет сопровождаться стойким снижением остроты и качества центрального зрения.
К тому же при нерегулярных осмотрах врача-офтальмолога можно пропустить переход возрастной макулодистрофии из сухой формы во влажную, из-за чего пациент не получит своевременное и эффективное лечение.
Осложняет ситуацию то, что сетчатка очень чувствительна к ишемии, которая усугубляется из-за прогрессирующей болезни. Если не провести эффективное лечение, то время до полного ухудшения макулодистрофии может исчисляться в месяцах. Поэтому при несвоевременном обращении за специализированной помощью пациент необратимо теряет центральное зрение на один глаз, и получает высокий риск развития данного заболевания и аналогичного исхода на втором глазу. В результате человек становится инвалидом [16] .
Диагностика макулодистрофии
Первым этапом диагностики является самодиагностика с помощью теста Амслера. Для этого пациент располагает тест на расстоянии 15-20 см от лица, сосредотачивает свой взгляд на центральной точке, прикрывает ладонью один глаз и оценивает квадраты и линии вокруг точки: есть ли искривления, искажения, все ли квадраты одинаковое или появились серые пятна. Потом тоже самое он повторяет и для второго глаза.
Такой периодический самоконтроль должен проводить каждый пациент с сухой формой возрастной макулодистрофии. При первых признаках искажений линий требуется немедленное обратиться к специалисту.
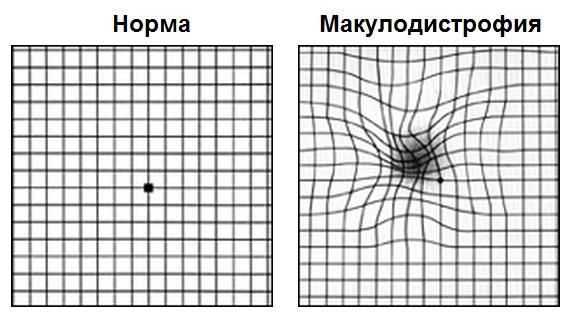
На картинке справа проиллюстрировано то, как видит тест Амслера пациент с сухой макулодистрофией развитой стадии или с рисками перехода из сухой формы во влажную; слева — зрение пациента без макулодистрофии или на начальных стадиях сухой формы болезни.
В целом диагностика возрастной макулодистрофии базируется на основных и дополнительных методах исследования.
Основные данные врач получает при стандартном офтальмологическом осмотре:
- сбор жалоб;
- уточнение истории болезни;
- оценка зрительных функций и данных офтальмоскопии, а лучше — биомикроофтальмоскопии (осмотр глазного дна).
К дополнительным методам диагностики относятся:
- флуоресцентная ангиография;
- ангиография с индоцианином зелёным;
- оптическая когерентная томография сетчатки;
- электроретинография.
Наиболее информативными методами выявления патологии сетчатки являются флуоресцентная ангиография и ангиография с индоцианином зелёным. Они визуализируют новообразованные сосуды, состояние пигментного эпителия и кровеносного русла глазного дна в целом. Для проведения этих двух методик использую различные модели фотокамер, а также флуоресцеин или индоцианин зелёный, которые вводят внутривенно перед исследованием. Однако при планировании и проведении данных манипуляций стоит помнить, что эти препараты могут вызывать аллергические реакции и другие побочные эффекты. Поэтому отбор пациентов для таких видов исследований должен быть тщательным.
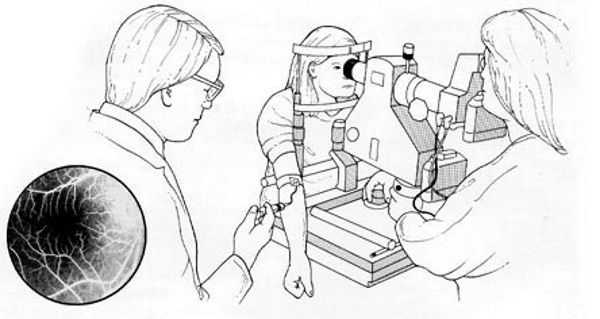
Противопоказания к ангиографии:
- а ллергические реакции в анамнезе; в стадии обострения;
- заболевания сердечно-сосудистой системы в стадии обострения;
- острый и хронический гломерулонефрит ; в стадии обострения;
- хроническая почечная недостаточность в стадии обострения;
- беременность и кормление грудью.
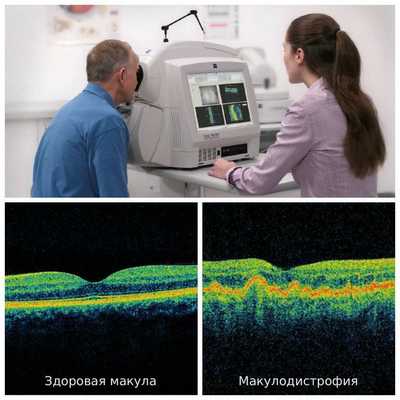
Наиболее информативным и безопасным методом диагностики для пациента является оптическая когерентная томография (ОКТ). Она используется для оценки морфологической и топографической оценки макулярной зоны, показывает структурные изменения сетчатки. Этот способ позволяет не только выявить начальные проявления возрастной макулодистрофии, такие как друзы и дефекты пигментного эпителия, но и уже явные признаки влажной формы болезни — отслойку пигментного эпителия и нейроэпителия, появление новых сосудов под сетчаткой. Эти данные являются основными показателями эффективности лечения, с помощью который оценивается дальнейшая тактика ведения пациента [2] .
Для определения степени функционального поражения сетчатки центральной зоны используется электроретинография (ЭРГ). Она может проводиться на начальных этапах болезни, когда морфологические изменения ещё не заметны, но уже присутствуют симптомы. Хотя, как показывает практика, функциональные нарушения в слоях сетчатки при возрастной макулодистрофии в явной степени появляются только на развитых и далеко зашедших стадиях заболевания [10] [11] .
Лечение макулодистрофии
Несмотря на большие успехи в диагностике возрастной макулодистрофии на ранних этапах развития, лечение этой патологии остаётся проблемой. Ни один из существующих вариантов лечения не способен полностью предупредить развитие возрастной макулодистрофии. FDA (управление по санитарному надзору за качеством пищевых продуктов и медикаментов) не одобрило ни одного лекарства от сухой макулодистрофии [14] .
При высоких рисках развития и начальных стадиях сухой макулодистрофии в первую очередь рекомендуется проводить курсы антиоксидантной терапии [18] . Они позволяют нормализовать обменные процессы и увеличить антиоксидантную защиту сетчатки. Согласно результатам исследований, лечение антиоксидантами и микроэлементами (цинком и медью) при продолжительных курсах снижают частоту развития поздних стадий возрастной макулодистрофии почти на четверть [12] .
Среди веществ с антиоксидантным эффектом, защищающих сетчатку от окислительного стресса и развития макулодистрофии, главное место занимают каротиноиды. Организм человека не способен синтезировать данные вещества, поэтому их количество в клетках сетчатки напрямую зависит от качества пищи. Каротиноиды содержатся в моркови, цитрусах, томатах, шпинате, кукурузе. Из поступаемой пищи только зеаксантин и лютеин транспортируются белками плазмы крови в фоторецепторы сетчатки, где они уже формируют жёлтый пигмент — ксантофилл. Он как раз и является главной защитой при окислительном стрессе, блокируя ультрафиолет [9] [13] . Также антиоксидантным воздействием на сетчатку обладают антоцианозиды, которые содержатся в экстракте плодов черники.
Для того, чтобы данные витаминно-минеральные комплексы работали, необходимо, чтобы все компоненты находились в правильных пропорциях: лютеин — минимум 2 мг, антоцианы — минимум 10 мг, цинк — более 10 мг, медь — менее 1 мг.
Однозначного ответа о длительности курсового лечения нет. По мнению некоторых авторов, витаминно-минеральные комплексы идут как заместительная терапия, их следует принимать постоянно [4] .
Как показали результаты всемирных клинических исследований, анти-VEGF препараты эффективны в отношении остроты зрения по сравнению с другими методами лечения — использования глюкокортикостероидов, лазерного лечения и др. Поэтому они заслуженно стали препаратами первой линии выбора при лечения влажной макулодистрофии, причиной которой является образование новых кровеносных сосудов.
Прогноз. Профилактика
Пациентам с сухой возрастной макулодистрофией рекомендуется принимать добавки с антиоксидантами — каротиноиды, витамины, микроэлементы. При лечении влажной формы болезни одним из главных и решающих факторов является время, так как перспектива лечения будет напрямую зависеть от своевременно начатой эффективной терапии, т. е. от начала постановки ингибиторов VEGF. Чем раньше начато правильное лечение, тем лучше результат. Терапевтическим окном считают один год от начала заболевания. Важен ежемесячный мониторинг показателей остроты зрения, данных ОКТ и биомикроофтальмоскопии для оценки необходимости повторных инъекций после стандартной "загрузочной дозы".
Стандартное ведение сухой возрастной макулодистрофии предполагает самоконтроль пациента с помощью теста Амслера, регулярные осмотры один раз в 6-12 месяцев с контролем остроты зрения и проведением биомикроофтальмоскопии, а также приём антиоксидантов. Регулярные осмотры позволят как можно раньше выявить признаки перехода болезни из сухой формы во влажную.
Не смотря на довольно простые меры профилактики влажной формы заболевания, меньше половины пациентов в России получают регулярное лечение сухой макулодистрофии, остальные либо получают его нерегулярно, либо оставляют заболевание вовсе бесконтрольным (18,2 %) [14] .
Курсовое лечение сухой макулодистрофии в условиях стационара не имеет смысла, так как весь объём наблюдений и необходимой замещающей терапии можно выполнять на уровне поликлиники.
Для профилактики возрастной макулодистрофии требуется ещё в молодом возрасте исключить факторы риска: курение, наличие в рационе большого количества насыщенных жирных кислот, избыточную инсоляцию. Если факторы риска не удалось исключить, то после 50-ти лет нужно начинать профилактический приём витаминно-минеральных комплексов с каротиноидами.
Читайте также:
- Борозды и шероховатости ногтей после травмы
- Примеры эффективности фотодинамической терапии при метастазе в хориоидею
- Оценка строения синуса. Оценка капсулы лимфатического узла.
- Свободолюбивый ребенок Стрелец и его характер. Воспитание ребенка Стрельца
- Краткое изложение процесса фотосинтеза. Метаболизм фосфоглицерата и триозофосфата.