Результаты лечения генетических болезней. Исходы
Добавил пользователь Евгений Кузнецов Обновлено: 01.02.2026
Описание наследственных заболеваний
Всего известно около 7000 генетических, или наследственных, заболеваний. Они вызваны нарушениями наследственного аппарата клеток: мутациями ядерной или митохондриальной ДНК. Слово «наследственный» при описании таких патологий означает способность передаваться по наследству. Иногда применяют термин «врожденный» к таким болезням, подразумевая присутствие их явной или скрытой форме при рождении.
Генетическая патология чаще всего проявляется рано, когда ребенок новорожденный или младенец. Она может нарушать работу органов и приводить к преждевременной смерти. Описаны наследственные патологии, проявляющиеся только у взрослых. Изучением наследственных нарушений занимается медицинская генетика.
Передача генетических болезней связана с типом наследования: аутосомный (доминантный, рецессивный) и сцепленный с половыми хромосомами (X, Y). Аутосомное нарушение находится в неполовых хромосомах (22 пары аутосом). Доминантность или рецессивность определяется тем, достаточно для проявления болезни мутации на одной хромосоме из пары (доминантный тип) или обязательно мутация должна быть на обеих хромосомах (рецессивный тип). При аутосомно-рецессивном наследовании носитель мутации на одной хромосоме обычно здоров, но может передать нарушение детям. Отдельную группу составляют митохондриальные болезни: мутация присутствует в ДНК энергетических станций клетки (митохондрий). Митохондрии человек получает только от матери (клеточные структуры сперматозоида при оплодотворении не проникают в яйцеклетку, только ДНК), поэтому наследование таких патологий происходит по материнской линии: от матери ребенку любого пола.
Частота встречаемости генетических болезней
Среди разных стран отличается список наиболее распространенных и редких генетических болезней. В России можно назвать самыми частыми муковисцидоз, галактоземию, спинальную мышечную атрофию (СМА). Среди генетических есть редкие болезни. В России такими считаются заболевания с частотой встречаемости не более 10 случаев на 100 тысяч человек. Например, перечень редких болезней России включает из генетических такие патологии как фенилкетонурию (ФКУ), глазной альбинизм, липофусциноз.
Причины наследственных заболеваний
Наследственное заболевание возникает при наличии у человека изменения генетического материала (мутация). Мутации подразделяют на генные (нарушение наблюдается внутри одного гена), хромосомные (нарушение наблюдается внутри одной хромосомы), геномные (изменяется число хромосом). Генные мутации, например, замена, добавление, исчезновение одного или нескольких нуклеотидов, нарушают генетический код организма: систему кодирования трех нуклеотидов в одну аминокислоту белка. Мутации, затрагивающие большие участки ДНК, например, когда появляется одна лишняя хромосома, чаще всего не совместимы с жизнью, поэтому приводят к самопроизвольному прерыванию беременности на ранних сроках. Например, эмбрионы с лишней хромосомой (трисомией) во всех парах (кроме 13, 18, 21 пары и половых хромосом) не жизнеспособны.
Мутация может быть унаследована от родителей или возникнуть в клетках зародышевой линии, зиготе (оплодотворенной яйцеклетке), на ранних этапах развития эмбриона (в таком случае говорят о возникновении мутации de novo). Некоторые мутации проявляются только, когда присутствуют на двух хромосомах (аутосомно-рецессивные). Здоровый человек может быть носителем мутации в одной хромосоме (гетерозиготная форма). Если у второго родителя такой же ген содержит мутацию, то у ребенка может произойти объединение двух нарушений и развиться заболевание. Это верно, если вариант генный. Если вариант геномный, то есть происходит увеличение или уменьшение количества хромосом в геноме, то он всегда приводит к нарушению развития органов.
Неонатальный скрининг
В России бесплатно проводят обследование новорожденных на 5 наследственных заболеваний: фенилкетонурия (нарушение обмена аминокислот), муковисцидоз (поражает органы дыхания и пищеварения), врожденный гипотиреоз (заболевание щитовидной железы), адреногенитальный синдром (избыточная выработка гормонов надпочечников), галактоземия (нарушение обмена углеводов). Это неонатальный скрининг, целью которого является ранняя диагностика болезней у детей. Неофициально его называют "пяточным тестом", так как для него врач берет кровь из пяточек новорожденного, когда младенец еще не покинул роддом. Пальчики ребенка слишком малы, чтобы брать периферическую кровь, поэтому используется пятка. Для анализа требуется несколько капель крови, которые наносят на специальный тест-бланк. Обычно анализ проводится на 4 (доношенный ребенок) или 7 (недоношенный ребенок) день жизни. Если тест не успевают сделать в роддоме, то анализ проводит патронажная медсестра в первые дни после выписки малыша.
Если результат этого анализа отклоняется от нормы, то родитель узнает об этом. В таком случае проводят повторное исследование. Если повторный результат оказался таким же, как и первый, то врач начинает разрабатывать программу лечения. Несмотря на небольшое число случаев в год заболеваний, входящих в неонатальный скрининг, их раннее выявление и начало лечения крайне важно, так как позволяет снизить проявления болезни или даже спасти жизнь. Ранняя постановка диагноза также важна для определения того, как лечить ребенка, построения схемы терапии. Заниматься лечением наследственных болезней будут несколько врачей разных специальностей, так как нарушения развиваются в разных системах органов.
Молекулярная диагностика у новорожденных является основным способом предположить болезнь, так как у маленьких детей симптомы либо еще не проявились, либо проявились не полностью, что может не позволять провести диагностику по клиническим признакам. В дальнейшем развитие симптомов приведет к постановке диагноза, но лечение уже может оказаться неэффективным. Примером служит фенилкетонурия: без раннего назначения диеты, исключающей аминокислоту фенилаланин, у ребенка развиваются необратимые поражения головного мозга.
Генетический тест на заболевания ребенка
Ребенку может быть проведено расширенное генетическое тестирование для того, чтобы определить риск развития других наследственных болезней, помимо тех пяти, что выявляет при рождении неонатальный скрининг. Медико-генетический центр Genotek проводит тест «Риски заболеваний» для того, чтобы определить предрасположенность к более чем 180 болезней. А будущие родители могут пройти тест «Заболевания ребенка», который поможет определить риск развития патологий до того, как наступила беременность. Врач генетик после проведения тестирования расскажет о рисках. Поэтому рекомендуется консультация генетика после анализа. Это тестирование можно проводить на этапе, когда супруги осуществляют планирование беременности. Такое обследование является важной частью подготовки. Чтобы осуществить рождение здорового ребенка, делать его необходимо обоим супругам.
Неинвазивный пренатальный тест
Также может проводиться исследование генетических болезней плода неинвазивным способом. Метод проведения такого анализа называется НИПТ (неинвазивное пренатальное тестирование). Он позволяет выявлять крупные нарушения 21, 18, 13, половых хромосом в циркулирующей ДНК плода. НИПТ предполагает взятие венозной крови матери, перед процедурой не требуется специальной подготовки. Проводить анализ можно, начиная с 10 недели беременности. Тест не исследует весь геном, а только крупные перестройки определенных хромосом. Это метод скрининга, выявления группы риска, а не постановки диагноза. Поэтому при положительном результате такого исследования рекомендуется проведение инвазивного анализа (амниоцентеза или кордоцентеза) для подтверждения того, что плод действительно имеет нарушение. Проведение инвазивной процедуры может сопровождаться риском прерывания беременности (1-2%), так как беременный организм воспринимает вмешательство как стрессовый фактор. Поэтому женщина может отказаться сдавать инвазивный тест после НИПТ, но пройти консультацию врача-генетика и обсудить различные варианты дальнейшего ведения беременности.
Результаты лечения генетических болезней. Исходы
Ревматоидный артрит (РА) — воспалительное ревматическое заболевание неизвестной этиологии, характеризующееся симметричным хроническим эрозивным артритом (синовитом) периферических суставов и системным иммуновоспалительным поражением внутренних органов. Клиническая картина РА крайне многообразна и во многом зависит от преимущественной локализации воспалительных изменений в соединительной ткани различных органов. Согласно данным ВОЗ, частота встречаемости РА в популяции составляет от 0,6 до 1,3%, при этом у близких родственников она достигает 3-5%, что свидетельствует о генетической детерминированности патологии. Женщины болеют в 2,5-3 раза чаще мужчин, преимущественно в возрасте 35-50 лет, в более поздние возрастные периоды отмечается увеличение частоты заболевания.
РА по-прежнему относится к заболеваниям с неизвестной этиологией, в литературе широко дискутируется вопрос о его мультифакторной природе, в развитии РА активное участие принимают генетические, внешнесредовые, иммунологические, гормональные, инфекционные и другие факторы. В отличие от классических генетических болезней, при которых множество различных генов и их комбинаций предрасполагают к развитию заболевания, РА представляет собой генетически гетерогенное заболевание, в первую очередь обусловленное генетическим несовершенством иммунорегуляторных процессов.
По данным многочисленных исследований, риск развития РА ассоциирован с носительством антигена главного комплекса гистосовместимости класса II HLA-DR4 и HLA-DR1, который включает более 20 аллелей. Активно обсуждаются роли и других генетических факторов, непосредственно не связанных с HLA-DR. К ним относят полиморфизм генов пептидиларгинин дезаминазы, белка тирозин фосфатазы N22 (protein tyrosine phosphatase N22 (PTPN22 C1858T), цитотоксичный Т-лимфоцитсвязанный антиген (CTLA-4 A49G), ген хемокиновых рецепторов 5 CCR5-∆32, ген NO-синтетазы ENOS 4 a/b, ген матриксных металлопротеиназ (MMР) ММР9-1562 C/T. Эти гены являются наименее изученными с точки зрения предрасположенности к РА. Рассмотрим их взаимосвязь непосредственно с РА и другими заболеваниями.
Ген PTPN22 расположен на коротком плече первой хромосомы в позиции 13.2. Он кодирует синтез тирозин фосфатазы - фермента, который регулирует порог активации Т-клеточных рецепторов, участвует в регулировании сигнальной трансдукции, т. е. ретранслирует сигналы извне в ядро клетки. Эти сигналы помогают клетке расти и делиться, выполнять специализированные функции. Белок, синтезируемый под влиянием гена PTPN22, участвует в сигнализации, которая помогает контролировать активность Т-клеток. Т-клетки идентифицируют инородные вещества и защищают организм от инфекции [15, 26, 36].
В 2012 г. учеными G. Song, S. Bae, S. Kim и Y. Li проведено исследование взаимосвязи гена PTPN22, полиморфизмов C1858T с РА в популяциях разных национальностей. Был проведен метаанализ полиморфизма C1858T гена и РА с использованием аллельного контраста и доминантной модели. Сравнивались 17 961 больных РА и 18 611 здоровых людей. Метаанализ показал связь между аллелем Т и РА. При распределении по этническому принципу анализ показал, что T-аллель был в значительной степени связан с РА у европейцев и неевропейцев. Кроме того, прямое сравнение РФ-положительных и РФ-отрицательных субъектов (РФ - ревматоидный фактор) выявило взаимосвязь аллеля Т у пациентов с РФ-положительным РА. Эти исследования подтверждают, что C1858T полиморфизм гена PTPN22 связан с восприимчивостью к РА в различных этнических группах, особенно у европейцев, и T-аллель значительно более распространен у РФ-положительных больных, чем у РФ-отрицательных [32, 41].
Также полиморфизм C1858T гена PTPN22 рассматривался как фактор риска развития РА и системной красной волчанки (СКВ) среди населения Колумбии. В исследование были включены 413 пациентов с РА, 94 пациента с СКВ и 434 здоровых. Результаты исследования доказывают ассоциацию между аллелями С1858T и РА, а также между С1858T и СКВ [31].
Выявлена связь полиморфизма гена у АЦЦП-позитивных (АЦЦП - антитела к циклическому цитруллинированному пептиду) больных РА в Турции. Обследовано 323 пациента с РА и 426 здоровых пациентов, генотипированных методом полимеразной цепной реакции (ПЦР) по полиморфизму C1858T гена. Частота гетерозиготного генотипа (СТ) составила 8,4% у пациентов с РА и 5,4% у здоровых соответственно. Гомозиготный генотип (TT) отсутствовал в обеих группах РА. Таким образом, определено, что полиморфизм C1858T гена PTPN22 является геном восприимчивости АЦЦП-положительного РА в Турции [14].
Была доказана связь полиморфизма C1858T гена PTPN22 и с другими системными заболеваниями, например, с системной склеродермией (СС). Проведенный метаанализ показывает, что полиморфизм C1858T гена PTPN22 связан с восприимчивостью к СС у европейцев, и его существование зависит от этнической принадлежности. Кроме того, у афроамериканцев оказалась намного ниже распространенность аллеля T, чем в других исследуемых группах населения, а европейцы имели самый высокий показатель распространенности [40, 42].
У венгерских больных РА и здоровых людей также был генотипирован полиморфизм С1858T гена PTPN22, результат показал увеличение распространенности аллеля Т у пациентов с РА по сравнению с контрольной группой. Ассоциация была обнаружена как у РФ-серопозитивных, так и у АЦЦП-положительных пациентов. В TT гомозиготном генотипе восприимчивость к РА более чем в 2 раза больше, чем в СТ [39].
Была доказана связь гена PTPN22 у больных с РФ-положительным РА, который был независим от HLA-DRB1 генотипа у больных кавказского происхождения, живущих в Великобритании [22].
Многочисленными исследованиями ученых показано участие полиморфизма C1858T гена PTPN22 в развитии таких заболеваний, как туберкулез [13], сахарный диабет 1-го типа [45], аутоиммунный тиреоидит (АИТ) [28, 46], витилиго [30, 44].
Наименее изученным в отношении предрасположенности к РА является A49G ген цитотоксического Т-лимфоцит-связанного иммуноглобулина 4 (CTLA4). Он расположен на длинном плече второй хромосомы в 33-й позиции. Этот ген является членом надсемейства иммуноглобулинов и кодирует белок, который передает ингибирующий сигнал Т-клеткам. Белок содержит V домен, трансмембранный домен и цитоплазматический хвост. Мутации в этом гене связаны с развитием таких заболеваний, как инсулинзависимый сахарный диабет [1, 10], болезнь Грейвса [36], тиреоидит Хашимото [8, 21, 33], рассеянный склероз [47], гепатит С [20].
Связь наличия этого гена и аутоиммунных заболеваний была выявлена среди жителей Словакии в результате проводимых исследований. Аутоиммунные заболевания щитовидной железы нередко сочетаются с РА [21]. Целью данного исследования было изучение частоты аллелей и генотипов полиморфизма А496G, в 1-м экзоне гена CTLA4 у словацких пациентов с РА, АИТ, как у больных с РА + АИT, так и у здоровых. Обследованы 57 пациентов с РА, 57 - с АИT, 34 - с обеими патологиями (РА + АИT) и 51 здоровый человек. Все были этнически однородны (словаки), проживали в одном географическом районе. А49G генотип и частота аллеля G гена CTLA4 в группе с РА не была существенно выше по сравнению с контролем. Частота GG-генотипа гена CTLA4 была незначительно выше у больных с АИT, чем в контрольной группе. Однако частота GG-генотипа и аллеля G у пациентов с РА и АИT была значительно выше, чем в контрольной группе. Частота GG-генотипа гена CTLA4 у словацких пациентов с РА была ненамного выше по сравнению с контрольной группой [21]. Полиморфизм А49G гена CTLA4 также связан с развитием сахарного диабета 1-го типа [10], АИТ Хашимото [21, 27], ювенильным идиопатическим артритом [34], рассеянным склерозом [47].
Роль гена ММР и полиморфизмов СТ в предрасположенности к РА активно обсуждается в настоящее время. Ген MMP9 расположен на длинном плече 20-й хромосомы между позициями 11,2 и 13,1. Семейство ММР представляет собой цинк- и кальций-зависимые эндопептидазы, способные специфически гидролизовать основные компоненты внеклеточного матрикса. Протеиназы присутствуют во всех без исключения клетках, внеклеточном матриксе и различных биологических жидкостях организма. Физиологически представители семейства ММР синтезируются как препробелки и секретируются как проферменты в очень незначительных количествах. В основном ММР секретируются под действием провоспалительных цитокинов, а главными источниками их продукции считаются активированные макрофаги, нейтрофилы, фибробласты [5].
При РА формируется особый тип воспаления, в т. ч. с повреждающим действием семейства ММР на соединительную ткань. Среди ферментов системы протеолиза наибольшее значение принадлежит семейству ММР, которые, имея особенности доменных структур и функций, действуют на коллаген и протеогликановый матрикс, разрушая основное внеклеточное вещество соединительной ткани. Предполагается, что семейство ММР проявляет более выраженный деструктивный эффект в присутствии оксида азота, выработку которого усиливает индуцибельная NO-синтетаза. Совместное действие медиаторов интерлейкина-1 и фактора некроза опухоли-α вносит значительный вклад в развитие периартикулярного и системного разрушения хрящевой ткани, свойственного РА. Так, при исследовании плазменной активности ММР-3 у пациентов с различными формами РА, остеоартрозом и подагрой была установлена ее значительная активность у больных РА.
Исследование активности ММР-1, -3 и тканевых ингибиторов ММР-1 в сочетании с уровнями С-реактивного белка (CРБ) и цитокинов у пациентов с эрозивными и неэрозивными ревматическими заболеваниями выявило значительное увеличение активности протеиназ в сыворотке крови больных с эрозивным артритом. При этом установлена прямая корреляция между уровнем СРБ и активностью ММР-3, которые лучше всего коррелировали с клиническими проявлениями РА. Следовательно, можно утверждать, что диагностически значимым является определение активности ММР-3 и уровня СРБ в сыворотке крови больных РА. Результаты исследования свидетельствуют о том, что активность ММР-3 в большей степени, чем цитокины, отражает степень воспаления при РА. Указанный функциональный потенциал позволяет рассматривать ММР-3 как одну из основных протеиназ, участвующих в процессах деструкции соединительной ткани при РА, что дает основание рекомендовать ее в качестве маркера указанной деструкции [9].
Согласно современным данным, можно выделить 2 протеиназы, представляющие соответственно подсемейство стромелизинов - ММР-3 (стромелизин 1) и подсемейство желатиназ - ММР-9 (желатиназа В), с максимальной активностью участвующих в нарушении структуры соединительной ткани и отвечающих на аутоиммунное воспаление и эрозирование суставов при РА.
Также наличие гена ММР связано с такими заболеваниями, как болезнь Кавасаки [27], колоректальный рак [35], эндометриоз и аденомиоз [37]. Установлена значимость полиморфных локусов генов MMP-3, MMP-9, ADAM33 и TIMP3 в качестве маркеров риска развития хронической обструктивной болезни легких [3]. Определенную роль играет наличие функционального полиморфизма промоторного региона генов ММР-2 и ММР-9 в развитии острых коронарных осложнений [2].
Еще одним геном предрасположенности к РА является ген CCR5 - это ген хемокиновых рецепторов 5, кодирующий β-хемокиновые рецепторы. Ген находится на коротком плече 3-й хромосомы в позиции 21,31. Белок, кодируемый этим геном, - важный рецептор для макрофагов вирусов, включая ВИЧ. Дефектные аллели этого гена были связаны с сопротивлением ВИЧ-инфекции [4, 43]. Экспрессия гена CCR5 была также обнаружена в линии промиелобластных лейкозных клеток. Предполагается, что этот белок может играть роль в гранулоцитарной пролиферации и дифференцировке. Ученый S. Han установил, что полиморфизм гена CCR5 является генетическим фактором риска для развития радиографического поражения суставов при РА [19]. Ген CCR5 выявляется в различных иммунных клетках и влияет на патогенез РА. При его исследовании определялась связь 4-х полиморфизмов гена CCR5 - 1118 и 303A, 927-С и 4833G и их гаплотипов с восприимчивостью к РА. Были обследованы 157 пациентов с РА и 383 здоровых индивидуумов. Между здоровыми людьми и больными РА не было отмечено статистически значимых отличий в генотипах, аллелях и гаплотипах по выбранным полиморфизмам [19]. Полиморфизмы 1118 и 303A гена CCR5 показали существенную связь с тяжестью радиографических рецессивных моделей и в результате анализа многомерной логистической регрессии были признаны независимыми предикторами радиографической тяжести РА [19, 38]. Также полиморфизмы 1118 и 303A гена CCR5 показали существенную связь с сужением суставной щели пораженных суставов при РА. Эти результаты доказывают, что генетические полиморфизмы гена CCR5 являются независимым фактором риска для установления радиографической тяжести, особенно с развитием эрозий при РА [48]. Также выявлена взаимосвязь гена ССR5 с развитием ювенильного РА [12].
Таким образом, согласно данным литературы, мы проанализировали непосредственную связь этих генов с предрасположенностью к РА. Можно сделать выводы, что дальнейшее изучение наличия у пациентов с РА таких генов является очень актуальным в настоящий момент для того, чтобы оценить их влияние на возможность развития РА, варианты клинического течения, лечения и прогноз этого заболевания.
Управление по контролю качества пищевых продуктов и медикаментов США (FDA) 24 мая 2019 года одобрило использование генно-терапевтического препарата Zolgensma для лечения спинальной мышечной атрофии 1-го типа у детей в возрасте до двух лет. Рассказываем, что такое генная терапия и почему она может стать лекарством будущего.
Генно-терапевтический подход к лечению наследственных заболеваний разрабатывается уже 40 лет. Основная технология генной терапии основана на замещении гена с мутацией правильно функционирующей копией этого гена. Но есть еще две стратегии: выключение неправильно работающего гена и введение нового гена, который поможет организму победить заболевание.
Надо подчеркнуть принципиальное отличие генной терапии от редактирования генома, которое сейчас тоже активно разрабатывается для лечения наследственных заболеваний. Генная терапия доставляет ген в клетки, чтобы компенсировать дефектный ген. Но при этом не происходит удаление дефектной ДНК из клеток. При редактировании генома происходит удаление или изменение дефектной ДНК в клетках пациента.
Для доставки генная терапия использует различные вирусы, которые транспортируют ген специфично в определённый орган. Вирусы - внутриклеточные паразиты, они встраивают свою генетическую информацию в ДНК клетки и таким образом заставляют клетку делать копии вирусной ДНК. Это оказалось очень полезным свойством для генной терапии. Перед использованием вирусы делают безвредными, чтобы они не могли вызывать заболевание, но могли доставить ген в клетки.
В зависимости от цели генная терапия бывает соматической и фетальной. В первом случае вирус с геном вводят в клетки тела, во втором - в эмбрион на ранней стадии развития. В результате фетальной генной терапии генетический материал попадает во все клетки и может быть передан детям.
В ходе клинических испытаний препарат Zolgensma показал хорошие результаты у пациентов со спинальной мышечной атрофией: из 21 пациентов 19 смогли начать двигать головой и самостоятельно сидеть.
Спинальная мышечная атрофия - наследственное заболевание, к развитию которого приводят мутации в гене SMN1. При этом заболевании из-за нарушения работы нервных клеток спинного мозга развивается слабость мышц и их атрофия. Дети теряют способность ходить, а по мере развития заболевания - самостоятельно дышать. Более 90% случаев заболевания заканчиваются смертью детей до 2-х лет.
Препараты генной терапии могут стать эффективным средством лечения многих наследственных заболеваний, для которых не существовало лечения до этого. Так, в ближайшее время ожидает одобрения международного регулятора еще один новый генно-терапевтический препарат для лечения талассемии и серповидноклеточной анемии. Оба этих заболевания связаны с мутациями, результатом которых является синтез неправильно работающего гемоглобина - белка, переносящего кислород в крови.
Прежде всего, препараты генной терапии разрабатываются для заболеваний, причиной которых является одна в мутация в одном гене. Таких болезней 10 000. В связи с большими затратами на разработку генно-терапевтических препаратов, они очень дорогие. Но постепенно отработка технологии позволит значительно снизить стоимость таких лекарств и сделать их доступными большинству пациентов.
Наследственные опухолевые синдромы

Понятие наследственной предрасположенности к различным патологиям широко известно. На сегодняшний день наследственная предрасположенность особенно интересна в контексте формирования опухолей наследственного типа. Зная о наличии такой предрасположенности, можно реализовывать возможности предиктивной медицины и не допустить развития опухоли у пациента в группе риска. Что такое наследственные опухолевые синдромы и как наиболее эффективно диагностировать их в нашей стране, расскажем в этой статье.
По условиям возникновения все виды злокачественных новообразований можно разделить на спорадические и наследственные. Случаи спорадических опухолей составляют подавляющее число во всемирной популяции. По различным данным, от 5 до 10 % составляют случаи наследственных новообразований или наследственные опухолевые синдромы.
Наследственный опухолевый синдром — это состояние, связанное с формированием опухоли в результате наследования мутаций в генах, ассоциированных с онкогенезом. Мутировавшие гены чаще играют контролирующую роль и ответственны за течение клеточного цикла или репарацию ДНК. При этом, в отличие от спорадических опухолей, при наследственных опухолевых синдромах мутации обнаруживаются в генах клеток всего организма с момента формирования зиготы. Опухоль при наследственном синдроме развивается в более раннем возрасте (на 10-15 лет раньше спорадических опухолей), когда наследственный дефект репарации ДНК или течения клеточного цикла будет сочетаться с накопившимися за жизнь соматическими мутациями . Необходимо учитывать, что понятие наследственных новообразований не тождественно понятию семейных опухолей. В первом случае опухоль имеет известную молекулярно-генетическую причину, семейные же опухоли могут ей не обладать, но демонстрировать высокую частоту встречаемости в одной семейной линии.
Широкое распространение наследственных опухолевых синдромов связано прежде всего с постоянным накоплением генетических мутаций в популяции. Большинство опухолей наследственного типа можно рассматривать как патологии с аутосомно-доминантным типом наследования. Носительство мутаций не влияет на фертильность, что позволяет мутациям широко распространяться в популяции.
Еще одной особенностью наследственных опухолевых синдромов является более частое возникновение первично-множественных новообразований.Так, у женщин билатеральные опухоли молочных желез чаще развиваются именно в контексте наследственного рака молочной железы и яичников. По сравнению с опухолями спорадического типа, лечение наследственных опухолевых синдромов носит более длительный характер и требует больших усилий со стороны онкологов.
Одним из наиболее распространенных наследственных опухолевых синдромов является синдром Линча, или наследственный колоректальный рак без полипоза. Частота встречаемости такого синдрома в общей популяции составляет 1:500, а пенетрантность патологии — до 80 %. Согласно Амстердамским критериям II, подозревать у пациента синдром Линча можно по совокупности признаков:
— наличие как минимум трех членов семьи с гистологически подтвержденным колоректальным раком или раком эндометрия, тонкой кишки, мочеточника или лоханки почки, при этом один из членов семьи является родственником первого порядка для двух других;
— исключен семейный аденоматозный полипоз;
— заболевание имеется у представителей как минимум двух последующих поколений;
— как минимум у одного члена семьи заболевание было диагностировано в возрасте до 50 лет.
В нашей стране большое значение имеет высокая распространенность наследственного рака молочной железы и яичников. Среди всех случаев рака молочной железы наследственные составляют 5-10 %, среди случаев рака яичников — 10-15 %. Критерии, определяющие подозрение на наследственный рак молочной железы и яичников, не являются общепринятыми и варьируют в разных странах. Так, критериями NCCN (Национальной онкологической сети США) являются:
— рак молочной железы в возрасте до 50 лет;
— трижды негативный рак молочной железы в возрасте до 60 лет;
— рак яичников или рак поджелудочной железы в любом возрасте;
— рак молочной железы в любом возрасте при наличии в семье случаев рака молочной железы, рака яичников, рака простаты или рака поджелудочной железы;
— рак молочной железы в любом возрасте при наличии еврейских корней в родословной.
Диагностика наследственного опухолевого синдрома у пациентов с уже имеющейся патологией имеет высокую значимость для подбора средств таргетной терапии. Так, например, при выявлении дефектов BRCA1 и BRCA2 возможно назначение препарата Линпарза (олапариб) — ингибитора белка PARP, ответственного за репарацию ДНК.
Наследственным опухолевым синдромом с наиболее широким спектром возможных новообразований является синдром Ли-Фраумени. Этот синдром характеризуется высоким риском формирования злокачественных новообразований в детском и подростковом возрасте. Часто обнаруживаются опухоли молочной железы, головного мозга, надпочечников, лейкозы и другие новообразования. Синдром Ли-Фраумени связан с мутацией в гене TP53. Белок, экспрессируемый им, играет центральную роль в регуляции клеточного цикла. По этой причине синдром Ли-Фраумени ассоциирован с повышением риска возникновения большого количества различных новообразований.
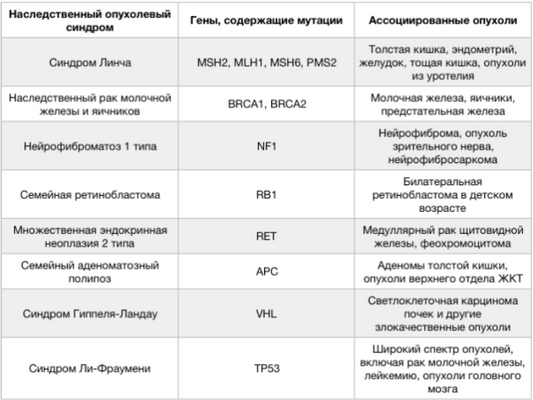
Рисунок 1. Характеристика основных наследственных опухолевых синдромов.
При подозрении на наследственный опухолевый синдром показано проведение генетического тестирования. Среди возможных методов определения генных мутаций одним из простых и наименее затратных является полимеразная цепная реакция (ПЦР). ПЦР подразумевает амплификацию, то есть образование дополнительных копий участков нуклеиновых кислот, в том числе мутировавших, что и используется в формате генетических тестирований. Метод ПЦР позволяет выявить определенный набор — «панель» — мутаций в исследуемых генах. Низкая чувствительность метода для российских пациенток с подозрением на наследственный рак молочной железы и яичников определяется тем, что ПЦР-панель на 8 мутаций в генах BRCA1 и BRCA2, используемая в России, позволяет выявить лишь половину пациенток с мутациями в этих генах. Остальные пациентки получают отрицательные результаты.
В настоящее время все более широкое распространение в диагностике наследственных опухолевых синдромов получают методы секвенирования. Секвенирование — это процесс определения нуклеотидных последовательностей ДНК и РНК. Методы секвенирования нового поколения (next generation sequencing, NGS) — это целая группа технологий определения нуклеотидных последовательностей ДНК и РНК как в формате отдельных участков генов, так и в объеме целого генома. Определение генетических мутаций методом NGS высокоэффективно: метод позволяет обрабатывать большие объемы данных за короткое время, выявлять и анализировать весь спектр имеющихся мутаций, что снижает число индивидуальных ошибок, связанных с особенностями генома пациента.
В нашей стране генетические исследования методом секвенирования нового поколения проводятся в крупных федеральных центрах, куда можно попасть по направлению врача-онколога. Кроме того, на сегодняшний день любой желающий при подозрении на наследственный опухолевый синдром может пройти генетическое тестирование методом NGS в лаборатории yRisk. Компания работает с использованием современных платформ секвенирования, позволяющих выявлять мутации, ассоциированные с наследственными опухолевыми синдромами. Чувствительность такого анализа значительно превышает данные ПЦР.
Положительный результат исследования не означает наличие у пациента опухоли на данный момент. Обнаружение опухоль-ассоциированных мутаций позволяет включать пациентов в группы риска и регулярно проводить диспансеризацию внутри этих групп. Профилактические мероприятия среди групп пациентов с наследственными опухолевыми синдромами дают возможность предотвратить появление опухоли в раннем возрасте и развитие первично-множественных опухолей, что крайне важно для сохранения высокого качества жизни. Как уже было сказано, генетическое тестирование методом NGS показано и пациентам, уже имеющим опухоль, для подбора наиболее эффективных средств таргетной терапии.
Выявление носительства опухоль-ассоциированных мутаций также позволяет планировать профилактические операции, проведение которых предотвращает развитие опухоли в дальнейшем. Так, при наследственном раке молочной железы и яичников высок превентивный эффект профилактической подкожной мастэктомии. Сохранить качество жизни после этой операции позволяет последующее эндопротезирование молочных желез. Женщинам старше 40 лет с такими мутациями рекомендуется профилактическая овариоэктомия. Профилактические операции рекомендованы и при некоторых других наследственных синдромах.
Диагностика наследственных опухолевых синдромов в современном мире имеет особую значимость. К сожалению, пока не был создан метод, позволяющий эффективно предотвращать развитие спорадических опухолей. Выявление же групп людей с наследственной предрасположенностью позволяет существенно снизить частоту злокачественных новообразований, а в конечном итоге — спасти жизнь не одной тысяче пациентов.
Источники
Parkes A., Arun B. K., Litton J. K. Systemic treatment strategies for patients with hereditary breast cancer syndromes //The oncologist. - 2017. - Т. 22. - №. 6. - С. 655.
Mork M. E. et al. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer //Journal of Clinical Oncology. - 2015. - Т. 33. - №. 31. - С. 3544.
Rahner N., Steinke V. Hereditary cancer syndromes //Deutsches Ärzteblatt International. - 2008. - Т. 105. - №. 41. - С. 706.
Молекулярные связи свидетельствуют о наследственной природе болезни Крона

Мутации гена NOD2 являются факторами риска болезни Крона. Многие аспекты того, как они влияют на состояние человека при данной болезни, неизвестны. Обнаружение вовлеченных в патогенез заболевания популяций клеток выдвигает новые терапевтические возможности.
Болезнь Крона — хроническое воспалительное заболевание кишечника, поражающее многих людей. Например, более 0,3 % населения Канады и Германии страдают от этого заболевания, а частота выявления случаев возрастает во всем мире [1]. Существует острая потребность в новых методах лечения, однако их эволюции препятствует отсутствие четкого понимания того, как возникает болезнь. Своей статьей в «Naturе» Nayar с соавт. [2] проливают свет на давнюю загадку одного фактора риска болезни Крона; выводы ученых имеют важное клиническое значение.
Болезнь Крона может поражать любой отдел кишечника. Чаще всего это подвздошная кишка, где развивается воспалительный процесс, который часто приводит к фиброзу (отложению волокнистой соединительной ткани как реакция на повреждение). Это приводит к сужению (стриктуре) просвета подвздошной кишки, что требует хирургического вмешательства [3]. Болезнь Крона представляет собой удобную для изучения модель, отображающую заболевания, развитие которых опосредовано комбинацией генетической предрасположенности и влияния окружающей среды. В данном случае, генетическая предрасположенность лежит в основе развития воспалительных реакций на воздействие микроорганизмов кишечника, что в итоге приводит к заболеванию.
Генетические вариации гена NOD2 (полиморфизм) являются самым сильным фактором риска болезни Крона; примерно 20 % от этого всего риска развития болезни связано с тремя однонуклеотидными полиморфизмами данного гена [4]. Кроме того, мутации NOD2 являются сильными прогностическими факторами для развития стриктур подвздошной кишки и, как следствие, необходимости хирургического вмешательства при болезни Крона. Это свидетельствует о сильной взаимосвязи между генетической основой этого состояния и проявлениями болезни [3].
Однако взаимосвязь гена NOD2 и восприимчивость к болезням представляют собой интересный парадокс. NOD2 — это внутриклеточный рецептор (см. рис. 1), который распознает молекулу мурамилдипептида (MDP) — распространенного компонента стенок бактериальных клеток. До того, как NOD2 был описан как ген риска развития болезни Крона, функция его была лучше всего изучена на примере иммунных клеток, обеспечивающих функционирование врожденного звена иммунитета. Активация NOD2 в этих клетках приводит к экспрессии воспалительных молекул, называемых цитокинами, а аномально интенсивный воспалительный ответ может опосредовать повреждение тканей кишечника при болезни Крона [5, 6]. Следовательно, можно предполагать, что мутации NOD2, известные как мутации, приводящие к потере функции (которые не приводят к образованию полноценного кодируемого геном белка), будут защищать от болезни Крона. Тем не менее, такие мутации NOD2, приводящие к потере функции, идентифицируются как факторы риска заболевания. Поэтому последующие исследования были сосредоточены на другом аспекте функции и метаболизма NOD2 в кишечнике — поддержании гомеостаза в кишечнике и роли NOD2 в этом процессе. Ведь именно здесь самая большая биомасса иммунокомпетентных клеток организма постоянно подвергается воздействию MDP кишечных микроорганизмов, а мутации NOD2 нарушают равновесие между микробиомом и иммунной системой, что ведет к болезни [5].
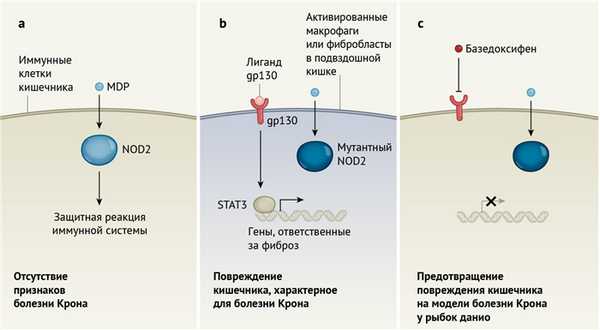
Роль мутаций NOD2 в возникновении фиброза подвздошной кишки до настоящего исследования была неизвестна. Авторы стремились понять, что запускает воспаление и фиброз при болезни Крона. Пытаясь обнаружить взаимосвязь между этими патологическими процессами и NOD2, они применили для исследования человеческие клетки, ткани кишечника человека и рыбок данио.
Во-первых, авторы использовали секвенирование РНК одиночных клеток, взятых из образцов воспаленной ткани подвздошной кишки во время операций у людей с болезнью Крона. В этих клетках обнаружилась сигнатура экспрессии генов, связанных с функциями активированных макрофагов и фибробластов. Авторам также удалось идентифицировать ключевую популяцию клеток, которая экспрессирует маркеры как миелоидных клеток, так и фибробластов. В соответствии с данными сведениями, популяция воспалительных макрофагов в подвздошной кишке дифференцируется для превращения в активированные фибробласты во время болезни.
Поразительно, но авторам удалось обнаружить факт эволюционной консервации этих клеточных популяций на экспериментальной модели воспаления тканей кишечника — на рыбках данио, обработанных декстраном сульфата натрия (DSS). Это вещество уже давно используется для индукции повреждения и воспаления кишечника на стандартных моделях грызунов. При моделировании воспалительных заболеваний кишечника человека in vivo предпочтение отдается именно мышиным моделям. Однако, как демонстрируют Nayar с соавт., рыбки данио представляют собой удобную альтернативу для исследований, от которых ожидается высокая степень результативности и быстрая оценка корреляции с заболеваниями человека. Действительно, кишечник рыбок данио и млекопитающих имеет сходную морфологию. Более того, как и у людей, у рыбок данио имеются звенья врожденного и адаптивного иммунитета, а воспаление тканей кишечника у рыбок данио также зависит от кишечного микробиома [7]. Методики редактирования генов, такие как CRISPR, помогают быстро модифицировать интересующие гены у рыбок данио.
Авторы изучили воспаление кишечника у рыбок данио, генетически модифицированных с дефицитом nod2. У этих рыб, обработанных DSS, было обнаружено повышенное содержание лейкоцитов в кишечнике, что является признаком воспаления, по сравнению с рыбками данио с нормальным количеством nod2. Но модель рыбок данио актуальна только в том случае, если можно найти корреляцию с организмом человека. Соответственно, используя сведения, полученные от детей с впервые диагностированной болезнью Крона, авторы демонстрируют, что увеличение количества копий мутации NOD2 (что связывается с риском развития болезни Крона) действительно коррелировало с сигнатурой экспрессии генов активированных макрофагов и фибробластов в тканях подвздошной кишки.
Чтобы понять функцию NOD2 в клетках человека, дифференцирующихся in vitro, авторы использовали моноциты периферической крови здоровых добровольцев и определили, есть или нет в клетках одна или две копии мутации NOD2, связанные с восприимчивостью к болезни Крона. Затем клетки дифференцировались in vitro в присутствии MDP и без него. Результатом стало большее количество активированных фибробластов в пропорции к клеткам с двумя копиями мутаций NOD2 по сравнению с клетками с нативным NOD2. Кроме того, увеличение количества мутаций NOD2 было связано с соответствующим увеличением количества фибробластов с сигнатурой экспрессии генов, характерной для активированных клеток. Интересно, что рыбки данио с дефицитом nod2, которые испытали воздействие MDP, обладали характеристикой экспрессии генов, характерной для активированных фибробластов, которая сохранялась даже во время восстановления после повреждения, опосредованного DSS. Рыбки данио с нативным типом nod2 таковой характеристикой не обладали. Исходя из этого, можно предположить, что дефицит nod2 препятствует эффективному разрешению фиброза и воспаления.
Для дальнейшего выяснения молекулярных основ связанной с риском мутаций NOD2 сигнатуры экспрессии генов, ответственных за развитие фиброза, авторы решили найти регуляторы транскрипции этого пути, стоящие выше на ступени контроля. Ученым удалось определить, что ген, кодирующий белок STAT3, в большей степени экспрессируется в активированных фибробластах и макрофагах. STAT3 является регулятором транскрипции ключевых компонентов воспалительных реакций и фиброза при воспалительных заболеваниях кишечника и действует через цитокиновый рецептор gp130. Анализ клинических данных, полученных от людей с болезнью Крона (у которых обнаружилось отсутствие реакции на антиФНО-терапию), выявил усиленную экспрессию генов, регулируемых gp130 и кодирующих белки IL-6, онкостатин M и IL-11 (терапия, основанная на антиФНО-антителах [ФНО — фактор некроза опухоли] — наиболее распространенный способ лечения болезни Крона). Это открытие подтверждает роль сигнализации посредством gp130 в этой группе лиц, устойчивых к упомянутой форме терапии.
Авторы предположили, что блокада gp130 может снизить частоту аномалий, возникающих при мутации NOD2. Они проверили эту гипотезу посредством использования базедоксифена (ингибитор gp130) на обработанных MDP клетках человека с мутациями NOD2. Базедоксифен действительно привел к снижению сигнатуры экспрессии генов, связанных с фибротическими процессами и обратил вспять изменения конфигурации клеток, являющихся характерными для активированных фибробластов. Этот препарат также снижал степень повреждения тканей кишечника, что было обнаружено у мутантных по отношению к nod2 рыбок данио, подвергшихся обработке DSS.
Начиная с описания клинических характеристик фиброза при болезни Крона, в данной работе описан молекулярный путь, связанный с мутациями NOD2, характерными для этого заболевания. Работа завершается предложением потенциальных подходов терапии для разрешения настоящих проблем лечения фиброза и лекарственной устойчивости к антиФНО-препаратам. Опираясь на генетику и клинические результаты этого внутриклеточного и молекулярного пути, исследование представляет собой «дорожную карту» для осуществления настоящих и будущих методов лечения.
Не у всех лиц с болезнью Крона существуют мутации NOD2, связанные с риском развития заболевания. Действительно, у людей определенных этнических групп (китайцев, малайцев, индусов) поражение подвздошной кишки является ведущим клиническим проявлением болезни Крона, однако NOD2 не связан с риском развития заболевания в этой популяции [5, 9]. Возможно, молекулярная сигнатура экспрессии генов активированных макрофагов и фибробластов является актуальной ключевой особенностью для людей с поражением подвздошной кишки при болезни Крона. Вероятно, различный генетический ландшафт может привести к сходным клиническим и молекулярным исходам. Следовательно, результаты Nayar с соавт. продвигают эту область на шаг ближе к молекулярной классификации болезни Крона, что может прояснить аспекты этого тяжелого состояния, насчитывающего около 200 генетических областей, связанных с риском развития заболевания [10] и его различными клиническими проявлениями.
Читайте также: