Синдром Aicardi у ребенка
Добавил пользователь Morpheus Обновлено: 01.02.2026
Российская медицинская академия последипломного образования;
Тушинская детская городская больница, Москва
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2011;111(7): 95‑97
Милованова О.А. Синдром Айкарди. Журнал неврологии и психиатрии им. С.С. Корсакова. 2011;111(7):95‑97.
Milovanova OA. Aicardi syndrome. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2011;111(7):95‑97. (In Russ.).
Синдром Айкарди - врожденное мультисистемное заболевание неясной этиологии, характеризующееся тетрадой симптомов: эпилептическими приступами по типу инфантильных спазмов, частичным или полным отсутствием мозолистого тела, хориоретинальными лакунарными очагами, умственной отсталостью [1, 15, 21, 31, 32, 34]. Впервые заболевание было описано J. Aicardi в 1958 г. [4]. Всего в мире отечественными и зарубежными авторами описано 200 случаев синдрома Айкарди [1, 4, 11]. Считается, что синдром Айкарди - Х-сцепленное доминантное заболевание, приводящее к летальному исходу у лиц мужского пола [22, 26]. J. Holmes и соавт. [16] описали единственного мальчика с XXY-кариотипом. Некоторые исследователи предполагали, что все известные случаи заболевания являются результатами хромосомных мутаций, однако сведения о пораженных сибсах отсутствовали [11]. Не исключено, что эмбриональный мозаицизм при мутации генов может быть наследственно обусловленным [20]. H. Ropers и соавт. [26] выявили микроделецию в Хр22.3 у девочки с синдромом Айкарди. Предполагается, что патогенез данного заболевания связан с генетически детерминированными нарушениями на этапе нейрональной миграции. Морфологическое исследование головного мозга подтверждает множественные аномалии его развития, включающие полную или частичную агенезию мозолистого тела, гетеротопию коркового вещества мозга, аномалии строения извилин (чаще по типу микрогирии и гетеротопии), нарушения клеточной архитектоники.
Наиболее яркими клиническими проявлениями синдрома Айкарди являются неврологические расстройства. Эпилептические приступы в виде инфантильных спазмов доминируют у детей 1-го года жизни. По классификации О. Dulac и соавт. [12] различают флексорные, экстензорные, флексорно-экстензорные, тонические или миоклонические, симметричные или асимметричные, синхронные или асинхронные, с фокальным компонентом, серийные или единичные инфантильные спазмы. У детей с синдромом Айкарди чаще выявляются латерализованные флексорные инфантильные спазмы, сочетающиеся у части больных с другими видами эпилептических приступов (чаще фокальными). Установлено, что чем меньше возраст дебюта приступов при данном заболевании, тем тяжелее степень нарушения психомоторного развития [6, 8, 10].
У детей с синдромом Айкарди часто встречаются черепно-лицевые дисморфии (микро-, гидроцефалия, аномалии ушной раковины), реже - вертебральные и реберные мальформации.
Практически у всех детей с синдромом Айкарди выявляются нарушения психомоторного развития различной степени. В случаях с задержкой психомоторного развития, предшествующей дебюту эпилептических приступов, неврологический регресс в дальнейшем усугубляется [23]. Отсутствует интерес к окружающему миру, потерян контакт с ребенком, снижается дифференцировка эмоциональных реакций, практически прекращается предречевое развитие. Постепенно развивается двигательный дефект, в ряде случаев до степени тетрапареза центрального генеза с полным отсутствием развития двигательных навыков и установочных рефлексов. В 90% наблюдений нарушаются познавательные функции [25, 35].
В ряде случаев метод ультразвукового сканирования плода позволяет выявить наличие синдрома Айкарди на ранних стадиях гестационного развития.
При визуализационных методах - нейросонографии (НСГ), рентгеновской компьютерной томографии (КТ), магнитно-резонансной томографии (МРТ) головного мозга - патогномоничным признаком синдрома Айкарди является агенезия мозолистого тела. Выделяют полную агенезию мозолистого тела, при которой отсутствуют все комиссуральные структуры вместе с фрагментами прозрачной перегородки, и частичную агенезию, которая в свою очередь подразделяется на агенезию ростральных и каудальных отделов мозолистого тела [29]. Чаще встречается агенезия каудальных отделов мозолистого тела, которая может сочетаться с изолированной дилатацией задних отделов боковых желудочков - кольпоцефалией. КТ-признаки агенезии мозолистого тела включают визуализацию межполушарной кисты, смещение вверх расширенного III желудочка и специфическое изменение формы тел боковых желудочков с увеличением расстояния между ними в виде симптома «ухвата» [7, 31]. Определяется расширение задних рогов боковых желудочков, своеобразный U-образный характер передних (фронтальных) рогов, удлиненная форма отверстия Монро. Кроме того, часто выявляются признаки атрофии вещества головного мозга в виде углубления и расширения конвекситальных борозд, расширения межполушарной и латеральной щелей, множественные пороки развития головного мозга.
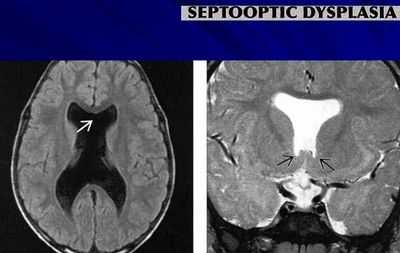
S. Carney и соавт. [9] описали девочку с сочетанием признаков двух синдромов: Айкарди и де Морсье (септооптической дисплазии).
При записи паттерн-реверсивных зрительных вызванных потенциалов (ЗВП) у детей регистрируются ответы как на низко-, так и высокочастотные стимулы [1].
У детей раннего возраста с синдромом Айкарди, не имеющих нарушений со стороны оптических сред, макулы и диска зрительного нерва, острота зрения, определенная при помощи паттерн-реверсивных ЗВП или методом предпочтительного взора, может соответствовать возрастной норме [1, 19].
Синдром Айкарди необходимо дифференцировать от других мультисистемных заболеваний, характеризующихся церебральными аномалиями, изменениями диска зрительного нерва и сетчатки: туберозный склероз, септооптическая дисплазия, папилло-ренальный синдром, врожденные TORH-инфекции, перинатальные гипоксически-ишемические поражения, изолированная агенезия мозолистого тела, сочетанные и изолированные церебральные аномалии (полимикрогирия, гетеротопия, лиссэнцефалия) [1, 2, 9, 13].
В настоящее время лечение синдрома Айкарди носит исключительно паллиативный характер. Основная стратегия терапии - купирование эпилептических приступов у детей грудного возраста, т.е. инфантильных спазмов, которые зачастую резистентны к противосудорожной терапии. Применяют различные комбинации антиконвульсантов в максимально переносимых дозах. Для монотерапии применяют противоэпилептические препараты - вальпроат (конвулекс-капли) 30-100 мг/кг в сутки в 2-3 приема или вигабатрин (сабрил) 50-150 мг/кг в сутки (средняя суточная доза 100 мг/кг) в 2 приема. При неэффективности монотерапии переходят к политерапии: 1) вальпроат 30-80 мг/кг в сутки в 2-3 приема + вигабатрин (сабрил) 50-150 мг/кг в сутки в 2 приема; 2) вальпроат 30-80 мг/кг в сутки в 2-3 приема + этосуксимид 20-35 мг/кг в сутки в 2-3 приема; 3) вальпроат 30-80 мг/кг в сутки в 2-3 приема + фризиум 0,5-1,0 мг/кг в сутки (максимально 1 мг/кг в сутки) в 2 приема. Высокоэффективны кортикостероиды: синактен-депо, начиная с 0,1 мг внутримышечно 1 раз в сутки, с постепенным наращиванием по 0,1 мг 1 раз в 2-5 дней до дозы 1,0 мг в сутки. Продолжительность терапии синактеном-депо составляет 1-3 мес. Преднизолон (2-10 мг/кг в сутки), гидрокортизон (5-20 мг/кг в сутки) применяют перорально. Продолжительность лечения варьирует от 3 до 8 нед, возможна длительная терапия - 4-6 мес. Гормональную терапию целесообразно назначать в сочетании с базовыми антиконвульсантами [3].
Таким образом, практически у всех детей с синдромом Айкарди присутствует нарушение психомоторного развития различной степени. От возраста дебюта эпилептических приступов зависит степень тяжести когнитивного дефицита.
На ЭЭГ при инфантильных спазмах в ряде случаев регистрируются типичные изменения в виде гипсаритмии. Методы прижизненной нейровизуализации - НСГ, КТ, МРТ головного мозга - окончательно подтверждают агенезию мозолистого тела у детей с синдромом Айкарди. Хориоретинальные лакунарные очаги, увеличение экскавации диска зрительного нерва, сочетающиеся с поражениями постгеникулярных зрительных путей, в частности зрительной лучистости, определяемыми при радиологических исследованиях, являются офтальмоскопическими проявлениями данного заболевания.
Ранняя диагностика синдрома Айкарди, включающая невро-, радио- и офтальмологические исследования, позволяет достаточно полно представить диагностический профиль этого тяжелого и прогностически неблагоприятного заболевания.
Неревматические кардиты у детей
Неревматические кардиты у детей - воспалительные поражения одной или нескольких оболочек сердца, не связанные с ревматической или другой системной патологией. Течение неревматического кардита у детей сопровождается тахикардией, одышкой, цианозом, аритмией, сердечной недостаточностью, отставанием в физическом развитии. При диагностике неревматического кардита у детей учитываются клинические, лабораторные, электрокардиографические, рентгенологические данные. В терапии неревматического кардита у детей используются сердечные гликозиды, НВПС, гормоны, мочегонные, метаболические, противовирусные и антимикробные препараты.

Общие сведения
Неревматические кардиты у детей - группа воспалительных заболеваний сердца, преимущественно инфекционно-аллергической этиологии. Целесообразность выделения неревматических кардитов в педиатрии обусловлена не только изолированным, но и часто сочетанным поражением 2-х и 3-х оболочек сердца у детей. Среди неревматических кардитов в детской кардиологии встречаются миокардиты, перикардиты, эндокардиты, а также миоперикардиты и панкардиты. Истинная распространенность неревматических кардитов в детской популяции неизвестна; по данным аутопсии, патология обнаруживается у 3-9% детей. Неревматическими кардитами болеют дети различных возрастных групп, однако среди них преобладают дети раннего возраста, преимущественно мальчики.

Причины неревматических кардитов у детей
Неревматический кардит у ребенка может быть обусловлен инфекционными или аллергоиммунологическими факторами. Среди инфекционных агентов преобладают вирусы (ECHO, Коксаки А и В, аденовирусы, вирусы гриппа типа А или В), встречаются бактерии (стрептококки, стафилококки), риккетсии, грибы, ассоциированная флора. Причиной врожденного кардита у ребенка выступают внутриутробные инфекции, воздействующие на плод. Бактериальные неревматические кардиты у детей нередко являются осложнением назофарингеальной инфекции, сепсиса, гематогенного остеомиелита, дифтерии, сальмонеллеза.
Кардиты аллергоиммунологической этиологии могут развиваться как следствие вакцинации, введения сывороток, приема лекарственных препаратов. Довольно часто прослеживается инфекционно-аллергическая природа поражения сердца. Примерно у 10% детей этиология неревматического кардита так и остается невыясненной.
Предрасполагающими факторами, на фоне которых активизируется вирусно-бактериальная микрофлора, повышается восприимчивость к токсинам и аллергенам, изменяется иммунологическая реактивность, могут выступать интоксикации, перенесенные ребенком инфекции, переохлаждения, психоэмоциональные и физические перегрузки, предшествующие хирургические манипуляции на сердце и сосудах, тимомегалия. У части детей с неревматическими кардитами обнаруживаются наследственные нарушения иммунной толерантности.
Классификация неревматических кардитов у детей
Таким образом, в зависимости от этиологии различают вирусные, бактериальные, паразитарные, грибковые, аллергические, идиопатические неревматические кардиты у детей. Разновидностью инфекционно-аллергического кардита служит миокардит Абрамова-Фидлера.
С учетом фактора времени кардиты делятся на врожденные (ранние и поздние) и приобретенные. По длительности течение кардита может быть острым (до 3-х месяцев), подострым (до 18 месяцев), хроническим (более 18 месяцев); по степени тяжести - легким, среднетяжелым и тяжелым.
Исходом и осложнениями неревматических кардитов у детей могут являться выздоровление, сердечная недостаточность (левожелудочковая, правожелудочковая, тотальная), гипертрофия миокарда, кардиосклероз, нарушение ритма и проводимости, тромбоэмболия, легочная гипертензия, констриктивный перикардит и пр.
Симптомы неревматических кардитов у детей
Врожденные кардиты
Ранний врожденный неревматический кардит обычно манифестирует сразу после рождения или в первом полугодии жизни. Ребенок рождается с умеренной гипотрофией; с первых дней жизни у него отмечается вялость и быстрая утомляемость при кормлении, бледность кожных покровов и периоральный цианоз, беспричинное беспокойство, потливость. Тахикардия и одышка, выраженные в покое, еще более усиливаются при сосании, плаче, дефекации, купании, пеленании. Дети с врожденными неревматическими кардитами рано и заметно отстают в наборе веса и физическом развитии. Уже в первые месяцы жизни у детей выявляется кардиомегалия, сердечный горб, гепатомегалия, отеки, рефрактерная к терапии сердечная недостаточность.
Клиника позднего врожденного неревматического кардита у детей развивается на 2-3 году жизни. Часто протекает с поражением 2-х или 3-х оболочек сердца. Признаки кардиомегалии и сердечной недостаточности выражены в меньшей степени, по сравнению с ранним кардитом, однако в клинической картине преобладают явления нарушения ритма и проводимости (трепетания предсердий, полная атриовентрикулярная блокада сердца и др.). Наличие у ребенка судорожного синдрома указывает на инфекционное поражение ЦНС.
Приобретенные кардиты
Острый неревматический кардит чаще развивается у детей раннего возраста на фоне перенесенного инфекционного процесса. Неспецифическая симптоматика характеризуется слабостью, раздражительностью, навязчивым кашлем, приступами цианоза, диспепсическими и энцефалитическими реакциями. Остро или постепенно возникает левожелудочковая недостаточность, характеризующаяся одышкой и застойными хрипами в легких. Клиническую картину неревматического кардита у детей обычно определяют различные нарушения ритма и проводимости (синусовая тахикардия или брадикардия, экстрасистолия, внутрижелудочковые и атриовентрикулярные блокады).
Для подострого кардита характерна повышенная утомляемость, бледность, аритмии, сердечная недостаточность. Хронический неревматический кардит обычно свойственен детям школьного возраста; протекает малосимптомно, преимущественно с экстракардиальными проявлениями (слабостью, утомляемостью, потливостью, отставанием в физическом развитии, навязчивым сухим кашлем, тошнотой, болями в животе). Распознавание хронического кардита затруднительно; дети нередко длительно и безрезультатно лечатся у педиатра с диагнозами «хронический бронхит», «пневмония», «гепатит» и др.
Диагностика
Распознавание неревматического кардита у детей должно проходить при обязательном участии детского кардиолога. При сборе анамнеза важно установить связь манифестации заболевания с предшествующей инфекцией или другими возможными факторами.
Постановке диагноза неревматического кардита у детей помогает совокупность клинико-инструментальных данных. Электрокардиография при кардитах не демонстрирует каких-либо патогномоничных признаков; обычно у детей выявляются длительно сохраняющиеся нарушения ритма сердца, АВ-блокады, блокады ножек пучка Гиса, признаки гипертрофии левых отделов сердца.
При рентгенографии органов грудной клетки выявляется кардиомегалия, изменение формы сердечной тени, усиление легочного рисунка за счет венозного застоя, признаки интерстициального отека легких. Результаты УЗИ сердца у ребенка демонстрируют дилатацию полостей сердца, снижение сократительной активности миокарда левого желудочка и фракции выброса.
При проведении иммунологического анализа крови отмечается повышение иммуноглобулинов (IgM и IgG), нарастание титров вирусных антител. Наиболее точные диагностические сведения возможно получить при эндомиокардиальной биопсии сердечной мышцы.
Врожденные неревматические кардиты у детей необходимо дифференцировать от врожденных пороков сердца (прежде всего, открытого атривентрикулярного канала, аномалии Эбштейна, синдрома Бланда-Уайта-Гарланда), перинатальной гипоксии. Приобретенные неревматические кардиты требуют разграничения с ревматизмом, кардиомиопатией, аритмиями другого генеза, констриктивным перикардитом, пролапсом митрального клапана у детей, опухолями сердца.
Лечение неревматических кардитов у детей
Терапия неревматического кардита у детей предусматривает стационарное и реабилитационное амбулаторное лечение. В период госпитализации ограничивается двигательная активность ребенка - постельный режим соблюдается в течение 2-4 недель. Основу питания составляет диета с повышенным содержанием солей калия и витаминов. Ребенку показаны занятия ЛФК под контролем инструктора.
Медикаментозную терапию неревматического кардита у детей составляют НПВС, глюкокортикостероиды, сердечные гликозиды, диуретики, препараты метаболического действия, антиагреганты, антикоагулянты, антиаритмические средства, ингибиторы АПФ и др. Если известен этиологический фактор неревматического кардита, ребенку назначается соответствующее этиотропное лечение (иммуноглобулины, интерфероны, антибиотики).
На амбулаторном этапе показаны реабилитационные мероприятия в условиях санатория кардиоревматологического профиля. Диспансерное наблюдение детей, перенесших острый и подострый неревматический кардит, проводится в течение 2-3 лет; врожденный и хронический варианты требуют пожизненного наблюдения. Профилактические прививки детям, перенесшим неревматический кардит, проводятся после снятия с диспансерного учета; хронический кардит является противопоказанием к вакцинации.
Прогноз и профилактика неревматических кардитов у детей
При благоприятном развитии событий постепенно регрессируют симптомы сердечной недостаточности, уменьшаются размеры сердца, нормализуется сердечный ритм. Легкие формы неревматического кардита у детей обычно заканчиваются выздоровлением; при тяжелых летальность достигает 80%. Факторами, отягощающими прогноз, служат прогрессирующая сердечная недостаточность, кардиосклероз, легочная гипертензия, стойкие нарушения ритма и проводимости.
Профилактика врожденного неревматического кардита у детей заключается в предупреждении внутриутробного инфицирования плода. Исключить развитие приобретенного кардита позволяет закаливание ребенка, лечение очаговых инфекций, предупреждение поствакцинальных осложнений.
Синдром Оллгрова ( Синдром ААА , Тройной синдром )
Синдром Оллгрова - это редкая генетическая болезнь, проявляющаяся алакримией, ахалазией и аддисонизмом. Заболевание вызвано мутацией AAAS, которая локализована 12-й хромосоме, и наследуется аутосомно-рецессивным путем. Патология характеризуется синдромом «сухого глаза», нарушениями глотания и продвижения пищи в желудок, дефицитом глюкокортикоидов и неврологическими симптомами. Для диагностики синдрома Оллгрова назначают генетическое тестирование, гормональные исследования крови и мочи, инструментальную визуализацию надпочечников, пищевода, головного мозга. Лечение симптоматическое с учетом ведущих клинических проявлений.
МКБ-10



Характерная триада симптомов была впервые описана в 1978 году английским педиатром и эндокринологом Дж. Оллгровом и позже названа в его честь. Генетические основы заболевания выявлены только спустя 18 лет. Болезнь Оллгрова также называют синдромом «трех А». В практической генетике распространенность заболевания составляет 1 случай на 1 млн. населения. Учитывая полиморфизм симптоматики и низкую настороженность врачей относительно этого синдрома, во многих случаях он диагностируется с запозданием.

Причины
Этиологическая структура синдрома Оллгрова была окончательно расшифрована в 1996 году. Заболевание развивается вследствие мутации в гене AAAS. Он расположен на длинном плече 12 хромосомы в локусе 12q13.13. Ген состоит из 16 экзонов и кодирует более 500 аминокислот в составе белка ALADIN, который имеет регуляторные функции. Этот протеин в больших количествах экспрессируется в надпочечниках, желудочно-кишечном тракте и нервной ткани.
Патология имеет аутосомно-рецессивный путь наследования. Синдром наблюдается у новорожденных мужского и женского пола с одинаковой вероятностью, если родители являются носителями мутантного гена. Вероятность повторения болезни при следующей беременности составляет 25%. Семейные случаи патологии в основном прослеживаются у жителей Африки и Азии, тогда как среди европейцев регистрируются единичные случаи.
Патогенез
Механизм развития заболевания досконально не изучен. Хотя ученые связывают мультисистемность поражения с патологиями протеина ALADIN, пока не найдено общего поражающего агента для 3-х типичных органов-мишеней: пищевода, слезных желез и надпочечников. Сложность определения патогенеза обусловлена вариативностью фенотипа у пациентов с синдромом Оллгрова, поскольку при одинаковом характере мутации характер течения варьирует от бессимптомного до крайне тяжелого.
Симптомы
Первым признаком синдрома Оллгрова является алакримия - снижение или полное отсутствие слез, которое обусловлено гипоплазией слезных желез. Она проявляется у детей раннего возраста, в некоторых случаях родители замечают настораживающий симптом у младенцев 5-6 месяцев. Из-за недостаточного увлажнения глаз дети старшего возраста жалуются на жжение, рези, ощущение инородного тела. Зачастую у них снижается зрение, возникают рецидивирующие кератиты.
Второй патогномоничный признак - ахалазия кардии, которая встречается у 75% пациентов в возрастном периоде до 15-16 лет. Дискоординация кардиального сфинктера и нарушение перистальтики пищевода проявляется расстройствами глотания. Пациенты давятся и поперхиваются пищей, могут случайно вдохнуть частички еды. Со временем развивается функциональная обструкция нижней трети пищевода, которая нарушает энтеральное питание.
Хроническая недостаточность надпочечников, которая входит в триаду синдрома Оллгрова, развивается на втором-третьем десятилетии жизни. У некоторых людей клинические проявления гормонального дефицита возникают только в пожилом возрасте. Внешне патология проявляется пигментацией кожи и слизистых, которые приобретают легкий бронзовый оттенок. Наблюдается снижение массы тела, немотивированная слабость, низкое артериальное давление.
Клиническая триада зачастую дополняется неврологическими симптомами. Для синдрома Оллгрова характерны двигательные нарушения, которые вызваны бульбоспинальной амиотрофией, мотосенсорной полинейропатией, нижним спастическим парапарезом. Возникают вегетативные расстройства: ортостатическая гипотония, нарушение зрачковых реакций, снижение потоотделения и выработки слюны.
Осложнения
Надпочечниковая недостаточность при отсутствии лечения манифестирует аддисоническим кризом. Развивается гипогликемия, шок, нарушение сознания. Алакримия осложняется кератопатией и прогрессирующей потерей зрения. Неврологические расстройства становятся причиной умственной отсталости, деменции, паркинсонизма. Ахалазия сопровождается аспирацией пищевых масс и пневмониями с затяжным течением. При вдыхании крупной порции пищи есть риск удушья.
Синдром Оллгрова нередко сопровождается комплексом врожденных аномалий внешности. Дети имеют вытянутое лицо, редкие или отсутствующие ресницы, большое расстояние от ноздрей до края верхней губы. Поражение опорно-двигательного аппарата проявляется сколиозом, остеопорозом, низкорослостью. Серьезную угрозу представляет синдром удлиненного интервала QT, который сопряжен с риском жизнеугрожающих аритмий.
Первичное обследование пациентов проводит врач-педиатр или терапевт, в зависимости от сроков манифестации и характера симптоматики. При физикальном осмотре обращает на себя внимание краснота и сухость глаз, «загорелый» оттенок кожи, типичные аномалии лица. Для постановки диагноза при синдроме Оллгрова требуется комплекс лабораторно-инструментальных исследований, таких как:
- Инструментальные методы.УЗИ надпочечников назначается для визуализации признаков атрофии и кальцинации органа, объемных новообразований. Наличие ахалазии подтверждают данные рентгеноскопии пищевода с контрастированием, эзофагеальной манометрии. Исключить первичное поражение нервной системы позволяет нейросонография, КТ или МРТ головного мозга.
- Проба Ширмера. Исследование проводится для объективной диагностики нарушений слезоотделения. Интенсивность выработки слезной жидкости оценивают по длине смоченного участка бумаги, помещенной на край нижнего века. Алакримию диагностируют при длине мокрого пятна менее 5 мм.
- ДНК-диагностика. Выявление мутации AAAS - единственный метод подтверждения диагноза, который назначается всем больным с подозрением на синдром Оллгрова. Однако около 5% пациентов имеют атипичные мутации, которые пока не удается идентифицировать рутинными методами.
- Гормональный профиль. Для недостаточности надпочечников характерно снижение суточного выделения 17-КС и кортизола с мочой, повышение концентрации АКТГ в крови. Дополнительно проводят стимуляционный тест с АКТГ, пробу инсулиновой гипогликемии.
- Консультации специалистов. Пациентам требуется осмотр у офтальмолога с проведением биомикроскопии глаза, визометрии, исследования глазного дна. Двигательные и интеллектуальные нарушения - показания к осмотру у невролога. При подтвержденном гормональном дефиците к обследованию привлекают взрослого или детского эндокринолога.
Дифференциальная диагностика
При постановке диагноза исключают болезнь Аддисона, семейный изолированный дефицит глюкокортикоидов (СИДГ), гипоплазию надпочечников. Патологии слезоотделения при болезни Оллгрова необходимо дифференцировать с синдромом Шегрена. Сочетание патогномоничной триады с неврологическими расстройствами требует дифференциальной диагностики с полинейропатиями, адренолейкодистрофией, врожденными патологиями головного и спинного мозга.
Лечение синдрома Оллгрова
Консервативная терапия
Как и при других генетических синдромах, схема этиотропного лечения заболевания пока не разработана. На первый план выходит заместительная гормонотерапия глюкокортикоидами. Пожизненный прием гормонов стабилизирует самочувствие пациента и предупреждает развитие аддисонического криза. При сочетанном дефиците минерало- и глюкокортикоидов лечение проводится комбинированными препаратами. Для коррекции алакримии назначают капли «искусственная слеза».
Возможности медикаментозного лечения неврологических симптомов ограниченны нейропротекторами, витаминами, антиоксидантами и метаболической терапией. В практической педиатрии значимую роль занимает реабилитация пациентов с привлечением логопеда, детского психолога, коррекционного педагога. Учитывая легкую степень интеллектуальных нарушений, при соответствующем лечении возможна социализация и профессиональная реализация пациентов.
Хирургическое лечение
Для коррекции эзофагеальной патологии требуется помощь хирургов. Щадящим методом лечения признана эндоскопическая баллонная эзофагодилатация, которая улучшает проходимость пищи в желудок, однако имеет нестойкий эффект. «Золотым стандартом» при ахалазии считается эзофагокардиомиотомия по Геллеру, которая показывает хорошие отдаленные результаты без рецидивов.
Прогноз и профилактика
Заболевание отличается медленным прогрессированием, поэтому при своевременной диагностике и коррекции надпочечниковой недостаточности состояние пациентов остается стабильным. Отдаленный прогноз зависит от степени тяжести неврологических нарушений, наличия умственной отсталости. Профилактика болезни предполагает медико-генетическое консультирование семейных пар, имеющих высокий риск рождения ребенка с синдромом Оллгрова.
1. Синдром Оллгрова. Почему важны все симптомы/ И.А. Мацнева, Н.И. Волкова, И.Б. Решетников// Молодой ученый. - 2016. - №15.
2. Редкий генетический синдром в практике педиатра-эндокринолога/ Е.В. Тозлиян// Практика педиатра. - 2014. - №2.
4. Синдром Оллгрова: как заподозрить проблему? Опыт эндокринолога/ Н.И. Волкова, И.Ю. Давиденко, И.Б. Решетников, С.С. Бровкина// Проблемы эндокринологии. - 2020;66(1).
Синдром Айкарди
Синдром Айкарди — это Х-сцепленное генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела с формированием хориоретинальных лакун и вариабельными эмбриональными аномалиями скелета. Типичной клинической картиной выступает триада признаков: инфантильные спазмы, задержка психического развития, нарушения зрения. Диагностика осуществляется при помощи электроэнцефалографии, церебральной МРТ, офтальмоскопии, изучения зрительных потенциалов, рентгенографии. Генетические методы диагностики пока не найдены. Лечение паллиативное с назначением комбинаций антиконвульсантов для постоянного приема, периодических курсов кортикостероидов.



Синдром Айкарди относится к редким расстройствам эмбрионального развития нервной системы. Впервые описан в 1965 году французским детским неврологом Жаном Айкарди как заболевание, характеризующееся следующей триадой признаков: агенезия мозолистого тела, офтальмологические расстройства, мышечные спазмы. Название синдрому в честь Ж. Айкарди было дано в 1972 г. Дальнейшее его изучение расширило список характерных клинических признаков, включив в них лицевой дисморфизм, умственную отсталость, аномалии позвоночного столба. Развитие методов нейровизуализации и генетических исследований позволило сделать диагностику более точной. По данным мировой статистики, всего насчитывается около 500 заболевших. Синдром связан с патологией Х-хромосомы, встречается исключительно у девочек. У плодов мужского пола возникают аномалии, не совместимые с жизнью.

Заболевание является генетической патологией, связанной с нарушением участка на Х-хромосоме. Считается, что оно имеет доминантное сцепленное с Х-хромосомой наследование. Теоретически вероятность рождения больной девочки от матери, страдающей синдромом, составляет 50%. Наличие патологической Х-хромосомы у мальчиков приводит к внутриутробной смерти.
На практике генетическая передача не зафиксирована, поскольку заболевшие девочки не могут иметь потомства в силу выраженных психомоторных расстройств. Все исследованные случаи синдрома Айкарди возникали de novo. Встречаются преимущественно спорадические варианты заболевания. Исключение составляет единственный известный случай у двух сестер. В связи с малым количеством заболевших, проведение достоверного исследования мутагенных триггеров затруднительно.
Патогенетические механизмы развития заболевания находятся в стадии изучения. Предполагается, что мутации генов обуславливают расстройство нейрональной миграции, происходящей в период между 12-й и 24-й неделями эмбрионального развития. В эмбриогенезе миграция неокортикальных нейронов на периферию приводит к образованию коры и подкорковых зон. Ее нарушения обуславливают дисгенезию мозолистого тела, аномальное формирование извилин и архитектоники мозга.
В норме в мозолистом теле проходят межполушарные проводящие пути, поэтому его дисэмбриогенез влечет нарушение связей между полушариями. Макроскопически в головном мозге выявляется микрогирия, субатрофия церебральной коры, перивентрикулярные кисты, гетеротопия, агенезия мозолистого тела. Микроскопически в сетчатке отмечается истончение слоев, уменьшение количества сосудов, гиперплазия пигментного эпителия.
Дети с синдромом Айкарди рождаются без видимых патологий, обычно в срок. В большинстве случаев беременность и роды протекают без осложнений. Заболевание манифестирует инфантильными спазмами в первые месяцы жизни. Спазмы представляют собой фокальные эпиприступы, при патологии Айкарди отличаются большой вариабельностью.
Возможны миоклонии, тонические судороги, ретропульсии, пропульсии. Наблюдаются симметричные и асимметричные, единичные и серийные судорожные приступы, типична тенденция к серийности. Флексорные спазмы отмечаются у 97% пациенток. В 42% случаях заболевания инфантильные спазмы сочетаются с другими видами эпиприступов, как фокальными, так и генерализованными. Фокальные припадки происходят с вовлечением преимущественно лицевой мускулатуры.
Задержка психомоторного развития характерна для всех больных синдромом. Отмечается обедненность эмоциональных реакций, утрачен контакт с окружающими, останавливается развитие навыков и предречевое развитие. Позже присоединяются моторные расстройства: геми- и тетрапарезы с повышением или снижением тонуса мышц, возможна спастичность. В отдельных случаях описана умеренная задержка развития, легкая степень олигофрении.
Офтальмологические аномалии вариабельны, включают: атрофию зрительного нерва, микроофтальмию, колобому. Возможна катаракта, пигментный ретинит. Клинически наблюдается значительное снижение зрительной функции. Со стороны желудочно-кишечного тракта характерна неустойчивость стула, проблемы с кормлением, гастроэзофагальный рефлюкс.
Среди челюстно-лицевых дисморфий описаны асимметрия лица, гипертелоризм, аномалии ушной раковины, выступающие резцы. У 33% пациенток отмечаются дисэмбриогенетические изменения позвоночника и ребер в виде полупозвонков, расщепления позвоночника, агенезии ребра, аномального реберно-позвоночного сочленения. В возрасте после 7 лет типично замедление роста, в пубертате — задержка полового развития.
Гемангиомы, невусы и другие дерматологические проявления описаны у 20% больных. В 7% случаев синдром Айкарди протекает с аномалиями конечностей: гипоплазией пальцев, синдактилией, каптодактилией.
Выраженная олигофрения, двигательные расстройства с первых месяцев жизни обуславливают глубокую инвалидность ребенка. Девочки нуждаются в постоянном уходе, половина из них неспособны к элементарному самообслуживанию. Нарушение мышечной иннервации при отсутствии постоянных реабилитационных мероприятий сопровождается гипотрофиями, в случаях спастичности — развитием контрактур.
Судорожный синдром с генерализованными припадками и кластерными приступами усугубляет неврологический дефицит, олигофрению. Позвоночные аномалии приводят к развитию выраженного сколиоза. Больные с синдромом Айкарди подвержены рецидивирующей пневмонии, имеют больший риск возникновения новообразований.
Внешний вид ребенка с лицевыми аномалиями, уменьшенным кончиком носа, латерально расположенными бровями позволяет врачу предположить генетическое заболевание. В неврологическом статусе показательны мышечная гипотония или односторонняя спастичность, снижение мышечной силы в конечностях, оживление рефлексов, снижение психического развития. Диагностика синдрома осуществляется совместно с генетиками, однако специфический генетический анализ еще не разработан. Назначаются следующие исследования:
- Электроэнцефалография. Наиболее типичным ЭЭГ-признаком синдрома Айкарди выступает гипсаритмия, появляющаяся после манифестиции инфантильных спазмов. Типично различие ЭЭГ-паттерна двух полушарий, феномен «расщепленного мозга». У ряда пациенток энцефалографическая картина имеет стертый характер. Наличие вариабельных судорожных припадков обуславливает полиморфизм изменений ЭЭГ.
- Офтальмоскопия. Выявляет наличие хориоретинальных лакун, визуализирующихся как белесоватые депегментированные участки сетчатки. Двусторонние лакуны расположены асимметрично, в 10-20% случаев очаги диагностируются только в одном глазу. При офтальмоскопии пациенток более старшего возраста диагностируется катаракта, колобома и/или атрофия диска зрительного нерва.
- Зрительные вызванные потенциалы (ЗВП). Исследуются для определения остроты зрения в младенческом возрасте и проведения дифдиагностики с другими офтальмологическими заболеваниями. У маленьких пациенток без изменений на глазном дне острота зрения может соответствовать возрастной норме.
- МРТ головного мозга. Визуализирует полное или частичное недоразвитие мозолистого тела, признаки корковой атрофии, пахигирию, расширение борозд. III желудочек расширен, вместе с боковыми желудочками смещен вверх, вокруг него выявляются церебральные кисты. Отмечается изменение формы латеральных желудочков, множественные области гетеротопии с отсутствием дифференцировки слоев коры, гидроцефалия.
- Рентгенография скелета. Необходима для диагностики костных аномалий, сопровождающих синдром Айкарди.
Перинатальная ДНК-диагностика не разработана. Выявление патологии возможно в ходе ультразвукового исследования беременной. По данным УЗИ 1 триместра возможна диагностика агенезии мозолистого тела. Позже выявляются микроцефалия, интракраниальные кисти, позвоночные аномалии, прочие пороки развития.
Необходимо дифференцировать патологию Айкарди от прочих ранних мультисистемных поражений, включающих сочетание дисгенеза церебральных структур и нарушения строения сетчатки. Первым симптомом септооптической дисплазии выступает горизонтальный нистагм, отсутствующий у пациентов с синдромом Айкарди. Отличительной чертой туберозного склероза служит наличие дерматологических симптомов: гиперпигментаций, ангиофибром, участков «шагреневой» кожи. Папилло-ренальный синдром протекает с поражением почек. Дифференциальная диагностика с врожденными TORH-инфекциями проводится на основании данных лабораторных обследований.
Следует отличать синдром Айкарди от заболевания Айкарди-Гутиера с аутосомно-рецессивным типом наследования. Последнее относится к прогрессирующим лейкодистрофиям детского возраста, характеризуется потерей приобретенных навыков, лимфоцитозом цереброспинальной жидкости, повышенным уровнем альфа-интерферона, кальцификатами в подкорковых структурах.
Лечение синдрома Айкарди
В основе терапии заболевания лежит паллиативная медикаментозная помощь и реабилитационные мероприятия. Базовой схемой является сочетание противосудорожных и гормональных фармпрепаратов, что позволяет добиться снижения частоты эпиприступов. Основными компонентами лечения являются:
- Антиконвульсанты. На первом этапе проводится монотерапия в максимально переносимой дозировке. Поскольку инфантильные спазмы при синдроме Айкарди отличаются стойкостью к противосудорожным препаратам, то приходится переходить к комбинированной схеме терапии, включающей 2-3 препарата.
- Глюкокортикоиды. Показали свою эффективность при внутримышечном введении в течение 1-3 месяцев. В ряде случаев по назначению врача возможна более длительное глюкокортикоидное лечение.
- Реабилитационная терапия. Направлена на поддержание двигательной активности, предотвращение мышечной атрофии, формирования контрактур при спастичности. Лечебные мероприятия включают массаж и специальную гимнастику, разработанные индивидуально с учетом имеющихся аномалий. Детям со средней степенью когнитивного дефицита показаны занятия с психологом, логопедом для максимально возможного развития навыков.
Поскольку синдром Айкарди является генетическим заболеванием с неуточненными этиофакторами генных мутаций, то его специфическая профилактика затруднительна. Возможно проведение общих профилактических мероприятий, направленных на охрану женщины от вредоносных воздействий в период беременности.
1. Синдром Айкарди/ Милованова О.А.// Журнал неврологии и психиатрии им. С.С. Корсакова - 2011. - 111(7).
2. Синдром Айкарди/ Петухова Н.М., Павловец Л.П., Жакашева А.С.// Вестник Алматинского государственного института усовершенствования врачей. - 2010. - 1.
3. Aicardi syndrome: an epidemiologic and clinical study in Norway/ Lund, C., Bjørnvold, M., Tuft, M., Kostov, H., Rosby, O., Selmer, K. K.// Pediatric Neurology. - 2015. - 52(2).
4. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children/ Glasmacher, M. A., Sutton, V. R., Hopkins, B., Eble, T., Lewis, R. A., Park Parsons, D., & Van den Veyver, I. B. // Journal of child neurology. - 2007. - 22(2).
Голопрозэнцефалия — тяжелый порок развития головного мозга

Голопрозэнцефалия — это порок развития головного мозга (переднего), характеризующийся неполным разделением на полушария либо полным его отсутствием.
Данная патология довольно часто встречается в медицинской практике и в год количество пациентов составляет приблизительно 1250 человек. Таким образом, этот порок наиболее часто встречаемый среди других аномалий в развитии переднего мозга.
Что характерно, есть у голопрозэнцефалии и свои ареалы, где процент появления заболевания намного выше, чем у других.
Так, наиболее часто это порок развития мозга наблюдается у представителей дальневосточных стран Азии, филиппинцев, пакистанцев и жителей острова Гавайи. Что характерно, у женщин этих народов аномалия встречается гораздо чаще, чем у мужчин.
Тип наследования
Голопрозэнцефалия - порок, который может быть результатом заболеваний, характерных для Х-сцепленного, аутосомно-рецессивного и аутосомно-доминантного типа наследования.

Таким образом, для носителей аутосомно-доминантной голопрозэнцефалии процент рождения ребёнка со стёртой формой порока, составляет 14%, а с выраженной - 21%.
Данный порок весьма разнороден, поэтому его проявление у одного из членов семьи может быть обусловлено основным заболеванием: например, если мать больна сахарным диабетом, то есть риск родить ребёнка с голопрозэнцефалией и он составляет 1 %.
В случае, если у родителей присутствует синдромальная форма, то у детей может проявиться в случае повторения соответствующего синдрома.
Цитогенетическая аномалия может иметь повторение в случае наличия порока у одного из потенциальных родителей.
В случаях, если в роду не было ни одного случая проявления данного порока развития головного мозга, его появление может иметь место в 4-5% случаев. В некоторых из них возникновение аномалии объясняется дигенным наследованием.
Провоцирующие факторы и патогенез нарушения
Причины развития порока можно условно разделить на два вида: наследственные и экологические.
Наследственные являются хромосомными аномалиями, которые ведут к анеуплоидии (синдром триплоидии, трисомия 18, трисомия 13). По некоторым данным голопрозэнцефалия может наблюдаться у пациентов с синдромом Дауна, но это, скорее, совпадение.
Наиболее часто встречающиеся мутации, связанные с голопрозэнцефалией:
- синдром 13 q;
- синдром Генуя;
- хвостовой дисгенез;
- синдром Айкарди;
- псевдотрисомия 13;
- синдром Меккеля-Грубера.
К экологическим причинам относятся:
- наличие сахарного диабета у матери;
- снижение уровня холестерина;
- употребление алкогольных напитков;
- приём Аспирина;
- употребление ретиноевой кислоты;
- приём мизопростола;
- использование Метотрексата;
- употребление дифенилгидантоина.
Патогенез голопрозэнцефалии заключается в нарушениях формирования головного мозга по срединной линии. Зачастую, это происходит на 5-10 неделе беременности.
Алобарный порок — это последствие нарушений деления мозга в горизонтальной, вертикальной, сагиттальной плоскостях. В результате развития порока головного мозга параллельно могут проявляться и сопутствующие патологии.
Клинические виды с симптомами
В медицинской практике выделяют несколько видов голопрозэнцефалии:
- алобарная;
- семилобарная;
- лобарная;
- средний межполушарный вариант;
- септо-преотипический тип.
Голопрозэнцефалия алобарная
Алобарный вид порока является самым тяжёлым и характеризуется полным слиянием двух полушарий мозга. Проявляться он может в следующих симптомах:
- один или два глаза могут располагаться в одной орбите с хоботком над органом зрения — циклопия;
- циклопия без наличия хоботка;
- заячья губа двусторонняя;
- анофтальмия;
- заячья губа;
- очень близко посаженные глаза;
- глаза расположены очень близко друг к другу и между ними имеется нос с одной ноздрёй — цебоцефалия;
- глаза могут быть расположены чрезвычайно близко друг к другу, но разделяются хоботком и орбитами.
- у пациентов с мутациями в zic2 гене черты лица могут сохраняться нормальными.
Семилобарная форма нарушения
Семилобарная голопрозэнцефалия проявляется в слиянии лобных долей при наличии незначительной перегородки в задней части с присутствием серпа и межполушарной щели. Проявляться данная форма аномалии может по-разному:
- вид лица может быть нормальным;
- может отсутствовать носовая перегородка;
- анофтальмия;
- глаза часто посажены очень близко друг к другу;
- заячья губа двусторонняя;
- депрессии переносицы;
- может иметься срединная расщелина нёба и губы (нёба или губы).
Лобарная форма нарушения
Данный вид порока есть наименее серьёзным среди основных видов. Лобарная голопрозэнцефалия характеризуется слитыми лобными долями при разделённом мозге на левое и правое полушарие, в том числе присутствуют боковые желудочки.
Порок может проявляться как:
- депрессии переносицы;
- сравнительно нормальный вид лица;
- глаза, посаженные близко друг к другу;
- голова может иметь нехарактерно маленькие размеры;
- заячья губа двусторонняя;
- проблемное психомоторное развитие;
- судороги;
- тяжёлая умственная отсталость.
Слияние среднего межполушарного пространства
Среднее межполушарное слияние - это вид голопрозэнцефалии, обозначенный в 1993 году, когда уже были изучены и выделены три основных вида: алобарный, лобарный и семилобарный. Данному пороку присущи аномалии связей между теменной и лобной областями больших полушарий.
- отсутствие эндокринопатии;
- хорея, атетоз;
- узкий нос;
- глаза близко посажены;
- депрессии переносицы;
- черты лица в норме;
- проявляются эндокринные расстройства;
- часты приступы эпилепсии;
- умственная отсталость;
- атетоидные движения.
Септо-преоптический вид
Септо-преоптический тип встречается крайне редко. Характеризуется соединением преоптических и септальных участков мозга, при этом существенного слияния коры головного мозга (фронтальной) не наблюдается.
На фото МРТ головного мозга — септо-преоптический вид голопрозэнцефалии
- пороки развития, касающиеся черепа и лица незначительны;
- задержка речи;
- трудности обучения;
- расстройства поведения.
Диагностические критерии
В большинстве случаев голопрозэнцефалию можно диагностировать ещё во время внутриутробного развития плода при помощи ультразвукового исследования.
При визуализации головной мозг представляет собой одну область, также может быть в наличии киста верхних отделов мозга. Одним из характерных проявлений порока есть аномалии области лица.
Если во время беременности патология не была выявлена, её наличие может быть определено после появления малыша на свет. Так, если медицинский персонал определит по внешним признакам наличие отклонений от нормы, могут быть назначены МРТ и КТ. С помощью этих видов исследования можно определить подтип порока развития головного мозга.
В случаях, когда визуально определить проблему не удалось и никаких особенных проявлений не замечено, выявить аномалию можно будет только после достижения малышом одного года: в это время будут налицо задержки развития.
Молекулярно-генетическое тестирование может быть применимо в случаях, если в семье имели место быть случаи голопрозэнцефалии. Производится исследование путём изучения ДНК клеток эмбриона.
Оказание медицинской помощи и прогноз
Лечение голопрозэнцефалии, в основном, направлено на устранение симптомов. Также большую роль играют консультации и поддержка родителей, выявление причины порока.
Так, при наличии алобарного вида порока единственным лечением есть симптоматическое лечение: ни один из других методов не действует. В такой ситуации можно предложить родителям лишь один выход - улучшить качество жизни малыша. Хотя, согласно статистике, при определении данного порока женщинам предлагается аборт.
Симптоматическим есть лечение и при лобарном, семилобарном виде. Также в случае с лобарной голопрозэнцефалией применяется хирургической вмешательство для исправления черт лица (например, заячьей губы).
Такие операции можно проводить после достижения младенцем пяти месяцев. Устранение подобных проблем поможет улучшить качество жизни: например, упростить сосание молока.
Последствия во многом зависят от тяжести порока: если это алобарная голопрозэнцефалия, велик процент летального исхода. В большинстве случаев у пациентов имеются задержки развития, к которым присоединяются припадки эпилепсии, дисфункция ствола мозга, нарушения сна.
40% пациентов с алобарной голопрозэнцефалией не переживают пятимемячный возраст, 80% — один год. С лобарным видом порока младенец может прожить более года в 50% случаев.
Дородовая профилактика
Для того чтобы уберечь себя и малыша от развития патологий головного мозга, будущая мать должна быть предельно осторожна со своим здоровьем: регулярно следует посещать медицинское учреждение и лечащего врача.
В идеале, до зачатия необходимо сделать все необходимые анализы для минимизации процента проявления порока у малыша.
Кроме этого важно вести активный образ жизни, отказаться от вредных привычек и медикаментов, которые могут спровоцировать аномалию.
Читайте также: