Синдром Бернута (Bernuth) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Синдром Жильбера - разновидность дистрофичных доброкачественных поражений печени. Для патологии, именуемой также простая семейная холемия, характерно хроническое течение. Конституциональная гипербилирубинемия (повышение билирубина) возникает у человека от рождения и наблюдается всю жизнь, однако заболевание не опасно в плане увеличения риска летального исхода. Чем опасен патологический недуг? Основное проявление болезни: периодическое возникновение негемолитической желтухи.
Классификация
Выделяют две формы негемолитической семейной желтухи.
- Для первого вида характерно снижение коэффициента очищения (клиренса) желчного пигмента - билирубина при отсутствии повреждения эритроцитов.
- Вторая форма синдрома Жильбера наблюдается при уменьшении скорости выведении билирубина на фоне скрытого разрушения эритроцитов (гемолиза).
Причины
Чем опасен синдром Жильбера? Заболевание передается по наследству, поэтому у детей, больных семейной желтухой, существует высокий риск развития гипербилирубинемии.
Патогенетическими факторами развития синдрома Жильбера являются:
- генетическая предрасположенность к сбоям в процессе захвата билирубина;
- нарушение транспорта этого желчного пигмента;
- врожденная неполноценность и дефицит уридиндифосфатглюкуронилтрансферазы - фермента печени, из-за чего наблюдается увеличение содержания «виновника» желтухи в крови.
Симптомы
Чем опасен синдром Жильбера? Хотя при негемолитической желтухе показатель билирубина в крови непостоянный, визуальные симптомы заметны только в стадии обострения недуга. Признаки заболевания более очевидны при беременности и у лиц с сопутствующими соматическими патологиями. Клинические проявления заболевания впервые эпизодически появляются у детей и в подростковом возрасте.
Наиболее распространенные симптомы:
- незначительный желтушный окрас кожного покрова;
- мышечная слабость;
- упадок сил;
- дискомфорт в брюшной полости;
- диспепсические явления: тошнота, диарея или запор, отрыжка, метеоризм.
Диагностика
Невзирая на то, что синдром Жильбера особо не опасен, необходимо выполнить диагностические исследования для исключения более серьезных патологий, в числе которых:
- развернутый анализ состава крови и мочи;
- исследование уровня желчного пигмента;
- проведение проб после воздержания от пищи, на фоне лечения барбитуратами, после употребления никотиновой кислоты;
- анализ ДНК на определение мутаций в гене UGT1A1.
Лечение
Несмотря на то, что в фазе ремиссии конституциональная гипербилирубинемия не опасна для жизни пациента, заболевание требует периодического проведения комплексного лечения в период расцвета болезни. Как лечить негемолитическую желтуху, определяет в каждой конкретной ситуации врач после проведения комплексного обследования. Фармакологическое лечение включает применение медикаментов различных групп:
- желчегонные средства растительного происхождения, например: гепабене (Hepabene);
- активаторы микросомальных ферментов, в числе которых - барбитураты, например: фенобарбитал (Phenobarbitalum);
- гепатопротекторы, например: эссенциале форте Н (Essentiale forte N);
- адсорбирующие препараты, например: полисорб (Polysorb).
Профилактика
Профилактические мероприятия включают:
- избегание умственных и физических перегрузок;
- исключение потребления алкогольных напитков;
- санация хронических очагов воспаления;
- соблюдение специальной диеты № 5;
- витаминотерапия.
Чтобы определить лечение, вам нужно обратиться к доктору. Выберите и запишитесь к доктору из нашего рейтинга ниже.
Бернута синдром
Синдром Бернута (F. Bernuth, нем. педиатр 20 в.) - болезнь неясной этиологии, по клиническим проявлениям напоминающая гемофилию, но не передающаяся по наследству.
Википедия
Статьи для врачей
- 12.02.2105 Восприятие марки настроено позитивно. Диктат потребителя
- 12.02.2105 Восприятие марки настроено позитивно. Диктат потребителя
- 12.02.2105 Восприятие марки настроено позитивно. Диктат потребителя
Статьи для пациентов

SPRINT: О целевом уровне артериального давления у пожилых

Антидепрессанты. Влияние на сон и когнитивные функции у пожилых
IDSA/ ATS. Рекомендации по лечению внутрибольничных пневмоний
Головная боль у взрослых. Виды головных болей.
Головная боль. Вопросы и ответы
Мигрень
Болезнь Альцгеймера и антидепрессанты
Артериальная гипертензия. Какое из имеющихся руководств лучше
Насыщенные жиры и риск сердечно-сосудистых заболеваний. Первые шаги революции?
Терпены
Терпены - группа природных ненасыщенных углеводородов, содержащихся гл. обр. в эфирных маслах (камфора, ментол и др.); применяются, напр., при изготовлении лекарственных средств и инсектицидов.
Тандлера схема
Тандлера схема (J. Tandler, 1869-1936, австрийский анатом) - схема расчета проекции долей и извилин большого мозга на кости черепа по наружным костным ориентирам.
Сустав локтевой
Сустав локтевой (a. cubiti, PNA, BNA, JNA) - сложный сустав, объединяющий плечелоктевой, плечелучевой и проксимальный лучелоктевой сустав, заключенные в одну суставную капсулу; в суставе локтевом возм.
Синдром Бернетта ( Диетическая гиперкальциемия , Молочно-щелочной синдром , Молочное отравление )
Синдром Бернетта - это дисметаболическое заболевание, развивающееся после приема реабсорбируемых щелочей: молока, карбонатов кальция, магния. Характеризуется гиперкальциемией и защелачиванием организма. Проявляется отвращением к молочной пище, приступами тошноты и рвоты, быстрой утомляемостью, апатией, зудом кожи. Нарушается работа почек, возникают суставные и мышечные боли. Диагностика включает выявление гиперкальциемии и алкалоза с помощью лабораторных методик, определение отложений кальция методом пальпации и рентгенографии. Терапия сводится к отмене препаратов кальция, коррекции молочной лечебной диеты и устранению дегидратации.
МКБ-10
Общие сведения
Распространенное название синдрома Бернетта - молочно-щелочной синдром. Реже используются такие синонимы как синдром алкалоза, диетическая гиперкальциемия, молочное отравление. Впервые негативное влияние молока и щелочей на метаболизм было отмечено еще древнегреческим целителем Гиппократом. Подробное описание и выделение самостоятельной нозологической единицы было подготовлено американским врачом Ч. Бернеттом в 1949 году. Распространенность синдрома в общей популяции составляет около 1%, среди людей с гастритом и язвой желудка, придерживающихся молочной диеты - 30%. Заболевание чаще диагностируется у мужчин. Эпидемиологические показатели заметно возросли за последние десятилетия, что связано с широким распространением БАДов, содержащих кальций.
Причины синдрома Бернетта
В группе риска по развитию синдрома находятся лица с нарушением регуляции абсорбции кальция через кишечник и со снижением функциональной способности почек. Усиление всасывания микроэлемента и его соединений может происходить при гипервитаминозе D, гиперпаратиреозе, тиреотоксикозе. Болезни мочевыводящей системы затрудняют выведение избытка Ca+. Синдром Бернетта, спровоцированный приемом лекарственных препаратов, чаще всего развивается при следующих патологиях:
- Остеопороз. При остеопорозе снижается плотность костей, разрушается их органический матрикс. Для восполнения минерального дефицита назначаются кальцийсодержащие средства.
- Гастрит. Воспаление слизистой оболочки желудка провоцируется повышенной кислотностью желудочного сока. Для снижения активности соляной кислоты применяют антациды - щелочные препараты.
- Язвенная болезнь желудка. Антацидные средства, назначаемые при язвенной болезни, уменьшают интенсивность болезненных ощущений, проявления изжоги. Также, как и при гастрите, кальция карбонат нейтрализует желудочную кислоту.
Патогенез
В основе синдрома Бернетта лежат гиперкальциемия, метаболический алкалоз и почечная дисфункция. Заболевание развивается поэтапно. Активное употребление молока, прием витаминно-минеральных комплексов и антацидных средств приводит к избыточному всасыванию кальция из кишечника и развитию гиперкальциемии легкой степени. Повышение уровня кальция в плазме способствует усилению экскреции натрия через почки. Нарастает дегидратация, подавляется секреция паратиреоидного гормона - регулятора концентрации Ca+ в кровотоке. Увеличивается почечная реабсорбция бикарбоната, формируется алкалоз - накопление щелочных соединений и увеличение pH крови. Снижение скорости клубочковой фильтрации и алкалоз стимулируют процессы повторного всасывания кальция почками, гиперкальциемия становится умеренной или тяжелой.
Ч. Бернетт и коллеги выделили и описали две формы синдрома: обратимую, проявления которой редуцируются сразу после снижения дозировки кальция, и необратимую, симптомы которой сохраняются в течение многих лет, несмотря на отмену кальцийсодержащих препаратов и молока. В настоящее время используется схожая классификация, в основе которой лежит характер течения болезни:
- Острая форма. Развивается спустя несколько дней после начала молочно-растительной диеты, приема антацидов и препаратов кальция. Характерна умеренная либо тяжелая гиперкальциемия, алкалоз, умеренная азотемия, легкое повышение фосфатов крови. Симптомы обратимы.
- Хроническая форма. Прогрессирует постепенно, в течение нескольких месяцев, лет. Гиперкальциемия тяжелая и стойкая, нарушения работы почек необратимые, в коже, суставах, мышцах образуются кальцификаты. Существует риск летального исхода вследствие почечной недостаточности.
На начальных этапах синдром проявляется общей слабостью, заторможенностью, отсутствием аппетита, тошнотой и рвотой. Пациенты испытывают чувство отвращения при виде и запахе молока или молочных блюд. Одним из симптомов является полиурия - обильные частые мочеиспускания. При отсутствии медицинской помощи развивается обезвоживание, усиливается жажда, появляется сухость во рту, кожный зуд, головные боли. Отложение в тканях солей кальция провоцирует боли в мышцах, суставах и сухожилиях, образование плотных узелков под кожей. У некоторых больных уплотняются хрящевые ткани, подкожные сосуды, появляются беловатые вкрапления на конъюнктиве и склере. Кожа покрывается шелушащейся псориазоподобной сыпью. Крупные суставы болезненны, увеличены из-за кальцификатов и отеков. Тяжелое течение синдрома сопровождается изменениями в работе ЦНС, возникновением атаксии, помрачения сознания.
Осложнения
Продолжительная гиперкальциемия провоцирует развитие нефрокальциноза. Активный приток кальция повышает нагрузку на почки. Кальцинаты накапливаются в почечной паренхиме. Эпителий, выстилающий почечные канальцы, атрофируется. Клетки погибают, солевые отложения образуются внутри канальцев. Разрастаясь, они превращаются в цилиндры, которые полностью закупоривают просвет. Отложения солей стимулируют деление клеток соединительной ткани, которая постепенно замещает функциональную паренхиму. Почечные клубочки сморщиваются, развивается нефросклероз. На фоне этих осложнений возникают воспалительные и инфекционные процессы, например, пиелонефрит, мочекаменная болезнь.
Первичное обследование проводится врачом-эндокринологом. При опросе пациенты сообщают о непереносимости запаха и вида молока, тошноте, рвоте, общей слабости, суставных и мышечных болях, зуде и сухости кожи. В анамнезе часто определяется гастрит, язва желудка, остеопороз, продолжительное употребление гидрокарбоната натрия (20 г/сут.), молока (2-3 л/сут.). Для подтверждения диагноза синдрома Бернетта и его дифференциации с гипервитаминозом D, хроническим нефритом, гиперпаратиреозом и нефрокальцинозом выполняются специфические диагностические процедуры:
- Общий осмотр. При пальпации определяется уплотнение ушных раковин, стенок подкожных сосудов. На коже - шелушение, псориазоподобная сыпь. При ощупывании крупных суставов пациенты отмечают боль, внешне наблюдается отечность, пальпируются костные узелки. Типичный признак - болезненность ахилловых сухожилий.
- Анализ крови. По результатам биохимического исследования крови диагностируется повышенный уровень кальция, чрезмерная концентрация остаточного азота (не обязательно), алкалоз (избыток щелочных оснований, показатель pH более 7). По данным общего анализа - увеличенная СОЭ (40-60 мм/час).
- Анализ мочи. Характерны повышенные показатели белка и лейкоцитов, единичные эритроциты и цилиндры. Гиперкальциурия обычно отсутствует, реакция мочи щелочная. При тяжелом течении синдрома с осложнениями возможна лейкоцитурия.
- Краниография. На рентгенограммах черепа имеются признаки уплотнения черепных костей. Ячеистость исчезает, понижается прозрачность пазух.
- Офтальмологический осмотр. Длительное течение заболевания провоцирует кальциноз с поражением глаз. Выявляется отложение кальцинатов в конъюнктиве, реже - в роговице.
Лечение синдрома Бернетта
Первичные лечебные мероприятия проводятся на базе стационара, после улучшения самочувствия пациента переводят на амбулаторное наблюдение. Терапия нацелена на устранение причин алкалоза и гиперкальциемии, восстановление водно-электролитного равновесия, коррекцию и продолжение лечения основного заболевания, осложнений. В схему мероприятий включены:
- Отмена препаратов кальция. Больным с гастритом, язвенной болезнью желудка и 12-перстной кишки вместо всасывающихся антацидов назначают H2-блокаторы, невсасывающиеся соли. Отменяются кальцийсодержащие БАДы, снижается дозировка тиазидных диуретиков, эстрогенов, витаминов D и A.
- Изменение диеты. Для пациентов, имеющих высокую кислотность желудочного сока, корректируются принципы питания. Сокращается объем молока, молочных продуктов в суточном рационе. Остальные правила сохраняются.
- Регидратация. Выполняется внутривенное введение 5% раствора глюкозы, раствора натрия хлорида, калия хлорида (при гипокалиемии). Процедуры восстанавливают баланс электролитов, уменьшают симптомы обезвоживания, способствуют восстановлению функции почек, рассасыванию узелков и уплотнений под кожей и в суставах.
Прогноз и профилактика
В большинстве случаев проявления синдрома Бернетта обратимы, сокращение поступающего через ЖКТ кальция является наиболее эффективным методом лечения. Для профилактики заболевания пациентам, принимающим кальцийсодержащие препараты, необходимо строго соблюдать дозировку, назначенную лечащим врачом, периодически контролировать концентрацию кальция крови. Всасывающиеся антациды рекомендуется заменить на невсасывающиеся (препараты солей алюминия и магния), сократить количество молока, а также изготовленных из него продуктов.
3. The milk-alkali syndrome/ E. Clinton Texter Jr., H. C. Laureta// The American Journal of Digestive Diseases - 1966 - Vol. 11, Issue 5.
Синдром Жильбера - симптомы и лечение
Что такое синдром Жильбера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, гастроэнтеролога со стажем в 15 лет.
Над статьей доктора Васильева Романа Владимировича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Жильбера — это генетический пигментный гепатоз с аутосомно-доминантным типом наследования, протекающий с повышением уровня неконъюгированного (свободного) билирубина, чаще проявляющееся в период полового созревания и характеризующийся доброкачественным течением [1] .
Краткое содержание статьи — в видео:
Синонимы названия болезни: простая семейная холемия, конституциональная или идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха.
По распространённости данное заболевание встречается не менее, чем у 5 % населения, в соотношении мужчин и женщин — 4:1. Впервые заболевание описал французский терапевт Августин Жильбер в 1901 году.
Чаще синдром Жильбера проявляется в период полового созревания и характеризуется доброкачественным течением. Основным проявлением этого синдрома является желтуха.
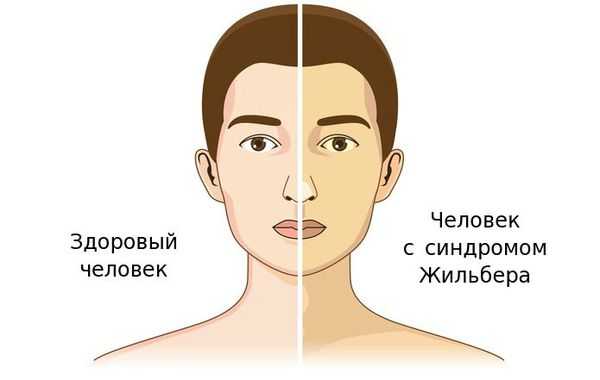
К провоцирующим факторам проявления синдрома можно отнести:
- голодание или переедание;
- жирную пищу;
- некоторые лекарственные средства;
- алкоголь;
- инфекции (грипп, ОРЗ, вирусный гепатит);
- физические и психические перегрузки;
- травмы и оперативные вмешательства.
Причина заболевания — генетический дефект фермента УДФГТ1*1, который возникает в результате его мутации. В связи с этим дефектом функциональная активность данного фермента снижается, а внутриклеточный транспорт билирубина в клетках печени к месту соединения свободного (несвязанного) билирубина с глюкуроновой кислотой нарушается. Это и приводит к увеличению свободного билирубина.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Жильбера
Некоторые специалисты трактуют синдром Жильбера не как болезнь, а как физиологическую особенность организма.
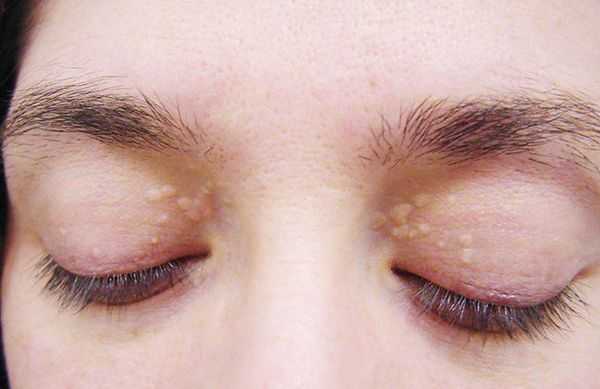
До периода полового созревания данный синдром может протекать бессимптомно. Позже (после 11 лет) возникает характерная триада признаков:
- желтуха различной степени выраженности;
- ксантелазмы век (жёлтые папулы);
- периодичность появления симптомов [1] .
Желтуха чаще всего проявляется иктеричностью (желтушностью) склер, матовой желтушностью кожных покровов (особенно лица), иногда частичным поражением стоп, ладоней, подмышечных впадин и носогубного треугольника.
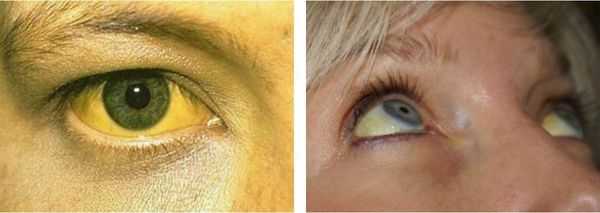
Заболевание нередко сочетается с генерализованной дисплазией (неправильным развитием) соединительной ткани.
Усиление желтухи может наблюдаться после перенесения инфекций, эмоциональной и физической нагрузки, приёма ряда лекарственных препаратов (в частности, антибиотиков), голодания и рвоты.
Клиническими проявлениями заболевания общего характера могут быть:
- слабость;
- недомогание;
- подавленность;
- плохой сон;
- снижение концентрации внимания.
В отношении ЖКТ синдром Жильбера проявляется снижением аппетита, изменением привкуса во рту (горечь, металлический привкус), реже возникает отрыжка, тяжесть в области правого подреберья, иногда наблюдается боль ноющего характера и плохая переносимость лекарственных препаратов.
При ухудшении течения синдрома Жильбера и существенном повышении токсичной (свободной) фракции билирубина может появляться скрытый гемолиз, усиливая при этом гипербилирубинемию и добавляя в клиническую картину системный зуд.
Патогенез синдрома Жильбера
В норме свободный билирубин появляется в крови преимущественно (в 80-85 % случаев) при разрушении эритроцитов, в частности комплекса ГЕМ, входящего в структуру гемоглобина. Это происходит в клетках макрофагической системы, особенно активно в селезёнке и купферовских клетках печени. Остальная часть билирубина образуется из разрушения других гемсодержащих белков (к примеру, цитохрома P-450).
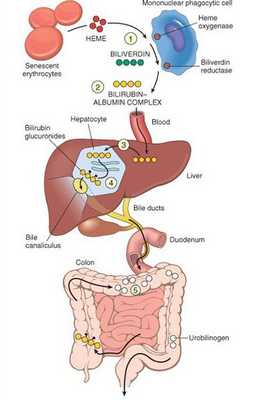
У взрослого человека в сутки образуется приблизительно от 200 мг до 350 мг свободного билирубина. Такой билирубин слаборастворим в воде, но при этом хорошо растворяется в жирах, поэтому он может взаимодействовать с фосфолипидами ("жирами") клеточных мембран, особенно головного мозга, чем можно объяснить его высокую токсичность, в частности токсичное влияние на нервную систему.
Первично после разрушения комплекса ГЕМ в плазме билирубин появляется в неконъюгированной (свободной или несвязанной) форме и транспортируется с кровью при помощи белков альбуминов. Свободный билирубин не может проникнуть через почечный барьер за счёт сцепления с белком альбумином, поэтому сохраняется в крови.
В печени несвязанный билирубин переходит на поверхность гепатоцитов. С целью снижения токсичности и выведения в клетках печени свободного билирубина при помощи фермента УДФГТ1*1 он связывается с глюкуроновой кислотой и превращается в конъюгированный (прямой или связанный) билирубин. Конъюгированный билирубин хорошо растворим в воде, он является менее токсичным для организма и в дальнейшем легко выводится через кишечник с желчью.
При синдроме Жильбера связывание свободного билирубина с глюкуроновой кислотой снижается до 30% от нормы, тогда как концентрация прямого билирубина в желчи увеличивается.
В основе синдрома Жильбера лежит генетический дефект — наличие на промонторном участке A(TA)6TAA гена, кодирующего фермент УДФГТ1*1, дополнительного динуклеотида ТА. Это становится причиной образования дефектного участка А(ТА)7ТАА. Удлинение промонторной последовательности нарушает связывание фактора транскрипции IID, в связи с чем уменьшается количество и качество синтезируемого фермента УДФГТ1, который участвует в процессе связывания свободного билирубина с глюкуроновой кислотой, преобразуя токсичный свободный билирубин в нетоксичный связанный.
Вторым механизмом развития синдрома Жильбера является нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой, которая доставляет свободный билирубин к микросомам клеток печени.
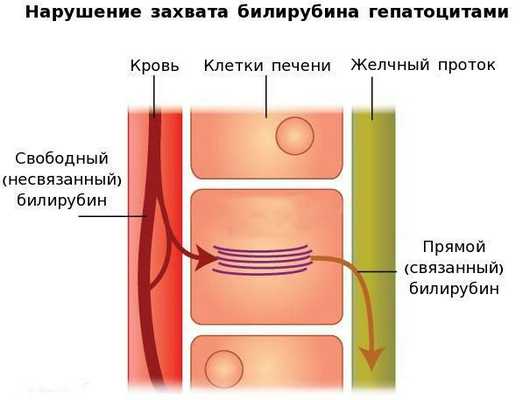
В конечном итоге вышеперечисленные патологические процессы приводят к увеличению содержания свободного (несвязанного) билирубина в плазме, что обуславливает клинические проявления заболевания [6] .
Классификация и стадии развития синдрома Жильбера
Общепринятой классификации синдрома Жильбера не существует, однако условно можно разделить генотипы синдрома по полиморфизму.
Синдром Туретта - симптомы и лечение
Что такое синдром Туретта? Причины возникновения, диагностику и методы лечения разберем в статье доктора Диордиева Максима Борисовича, психиатра со стажем в 8 лет.
Над статьей доктора Диордиева Максима Борисовича работали литературный редактор Вера Васина , научный редактор Владимир Вожжов и шеф-редактор Маргарита Тихонова
Синдром Туретта (Tourette's syndrome) — это заболевание нервной системы, при котором возникают множественные двигательные и вокальные тики. Для постановки диагноза они должны присутствовать дольше года.

Впервые заболевание, похожее на синдром Туретта, было описано в 1486 году в книге «Молот ведьм». Там упоминался священник с моторными и вокальными тиками, считавшийся одержимым. В конце XIX века симптомы заболевания на примере нескольких пациентов описал вместе с коллегами французский невролог Жорж Жиль де ла Туретт, в честь которого и назван синдром [1] .
Обычно синдром Туретта проявляется уже в детстве, но часто заболевание выявляют поздно или не диагностируют вовсе, так как родители не обращают на тики должного внимания. Из-за этого маленькие пациенты не получают своевременной помощи и могут страдать не только от самих тиков, но и от психологических проблем, связанных с заболеванием. В Европе от возникновения первых симптомов синдрома Туретта до постановки диагноза проходит в среднем более 5 лет [2] .
Распространённость синдрома Туретта
Синдром Туретта очень распространён — он встречается примерно у 10 из 1000 детей. В России его диагностируют у 8 человек на 10 000 населения, им могут страдать до 5 % школьников [3] . Мужчины болеют чаще, чем женщины: соотношение между ними составляет примерно 3 к 1.
Причины синдрома Туретта
Синдром Туретта — это генетическое расстройство, которое передаётся от родителей. Однако точный механизм наследования и ген, ответственный за болезнь, не известны. Риск передачи заболевания ребёнку составляет около 50 %. В прошлом, в начале XX века, тики считались следствием психотравм, но современная медицина это отвергает, так как такое предположение не удалось доказать [4] . Психосоциальные факторы и аутоиммунные заболевания не являются причиной синдрома Туретта, но могут влиять на тяжесть течения болезни.
Существует теория, что недостаток магния в организме и связанные с ним нарушения обмена веществ могут влиять на развитие синдрома Туретта. Косвенным доказательством этого служит то, что препараты с некоторыми соединениями магния могут улучшать состояние больных. Однако большие исследования на эту тему не проводились [5] .
Симптомы синдрома Туретта
Синдром Туретта проявляется тиками — быстрыми, внезапными, повторяющимися навязчивыми движениями или произнесением звуков. Чаще всего они возникают у мальчиков в возрасте от 4 до 11 лет. Тяжесть тиков доходит до пика примерно в 10-12 лет и ослабляется в подростковом возрасте. Большинство тиков исчезают спонтанно, но примерно у 1 % детей они сохраняются во взрослой жизни [7] .
Выделяют две основные группы тиков:
- Моторные тики — это непроизвольные движения частей тела. Самый распространённый из них — усиленное моргание. Также могут возникать подпрыгивания, постукивания по себе, развороты и повороты тела, гримасы, плевки, нецензурная жестикуляция и повторения чужих движений (копропраксия и эхопраксия).
- Вокальные, или звуковые, тики — это навязчивое произношение звуков, реже слов. Может проявляться кашлем, покашливанием, кряхтением. Иногда таких детей ошибочно лечат от бронхитов, трахеитов и бронхиальной астмы. Синдром Туретта часто ассоциируется с копролалией — внезапным высказыванием нецензурных фраз или слов, которое зачастую сопровождается копропраксией. Однако копролалия возникает только у 10 % пациентов [6] . Помимо копролалии, они могут повторять чужие слова, собственное слово или фразу (эхолалия и палилалия).
При синдроме Туретта моторные тики обязательно сочетаются с вокальными. Если присутствуют моторные тики, но нет вокальных, то стоит заподозрить другие заболевания: органическое поражение головного мозга, эпилепсию, синдром дефицита внимания (СДВГ), обсессивно-компульсивное расстройство (ОКР).
У многих детей и подростков с синдромом Туретта также отмечается СДВГ, ОКР, повышенная агрессивность, тревожность и склонность к депрессиям.
При синдроме Туретта интеллектуальные способности не нарушаются. Дети с этим заболеванием могут сильно расстраиваться от подшучиваний своих сверстников. При эмоциональном напряжении, вызванном пристальным вниманием или насмешками окружающих, тики могут усиливаться.
Форма тиков при синдроме Туретта может меняться в течение суток или недели, например от лёгких единичных моторных тиков утром или в начале недели до сложных и множественных по вечерам или под конец учебной недели. Видимо, их выраженность зависит в том числе от психоэмоциональных нагрузок.
Иногда дети пытаются сдерживать тики, но такой контроль возможен лишь в некоторой степени. Когда ребёнок старается подавить тики, симптомы могут усилиться. Попытка сдержать тик вызывает выраженный дискомфорт, из-за чего возрастает тревога — тикозные движения, наоборот, немного успокаивают. Из-за стресса, тревожных состояний и усталости тики могут учащаться и усиливаться.
Патогенез синдрома Туретта
Патогенез синдрома Туретта до конца не изучен. Известно лишь, что расстройство вызвано генетическими причинами. Скорее всего, при определённых генетических факторах нарушается работа нейромедиаторных систем в подкорковых образованиях и лобной коре.
Помимо генетических факторов, в патогенезе может участвовать и органическое повреждение головного мозга, например при патологии беременности и родов, черепно-мозговых травмах или нейроинфекциях.
Основная роль в патогенезе заболевания, вероятно, принадлежит дисфункции лобных долей. Считается, что большую роль в развитии синдрома Туретта играет правая лобно-височная область, сенсомоторные отделы орбитофронтальной коры, моторная область, базальные ганглии и поясная извилина. Также важное значение имеют нарушения в кортико-стрио-таламо-кортикальном контуре — нейронных цепях, связывающих кору, базальные ганглии и таламус.

Нарушения в работе этих структур также характерны для детей с ОКР и СДВГ — эти заболевания часто сопутствуют синдрому Туретта. Даже известны генетически связанные с синдромом Туретта формы ОКР (преимущественно ОКР с навязчивыми действиями — F42.1).
Предположительно, при синдроме Туретта нарушается работа дофаминергической системы. Изменения, вероятно, затрагивают серотонин-, норадреналин-, глутамат-, холин-, ГАМКергическую и опиоидную системы. Косвенно на связь синдрома Туретта с дофамином указывает то, что тики уменьшаются при лечении препаратами, которые воздействуют на передачу нервного импульса, вызванную дофамином ( например, путём блокады постсинаптических D2-рецепторов) [2] .
Классификация и стадии развития синдрома Туретта
Синдром Туретта — это разновидность хронических тиковых (или тикозных) нарушений. В Международной классификации болезней (МКБ-10) заболевание кодируется как F95.2 Комбинированные голосовые и множественные двигательные тики.
В следующей Международной классификации болезней (МКБ-11) тики и синдром Туретта из психических расстройств перенесены в неврологические [1] .
Согласно классификации Американской психиатрической ассоциации (DSM-IV), тики подразделяются на следующие группы:
- по виду — двигательные или голосовые;
- по продолжительности — преходящие или хронические.
Преходящее тиковое расстройство — это множественные двигательные, голосовые или тики обоих видов, которые длятся от 1 до 12 месяцев. Хронические тиковые расстройства присутствуют больше года. Они могут быть одиночными или множественными, двигательными или голосовыми, но не оба вида сразу. Синдром Туретта относится к хроническому тиковому расстройству. Для постановки диагноза необходимо, чтобы множественные двигательные тики и хотя бы один голосовой тик наблюдались более года.
Стадии синдрома Туретта не выделяют. Но обычно расстройство начинается с преходящих двигательных тиков, как правило подёргивания лица, которые длятся до года. Часто это гримасничание, затем покашливание и шипение. Постепенно тики распространяются на руки, ноги и мышцы шеи. Затем, обычно через год, присоединяются вокальные тики, которые осложняют картину болезни.
Осложнения синдрома Туретта
Синдром Туретта может сопровождаться депрессией, тревожным расстройством, ОКР и СДВГ, что осложняет прогноз. Депрессия возникает из-за того, что детей с этим синдромом часто обижают и унижают. В результате у них формируется чувство одиночества и может развиться аутоагрессия, вплоть до попытки суицида. Поэтому важно не оставлять ребёнка один на один с этим расстройством: ему особенно необходима поддержка родителей и друзей.
При симптоме копролалии дети выкрикивают нецензурную брань, из-за чего могут подвергаться агрессии со стороны окружающих. Поэтому для таких пациентов очень важно организовать правильную социальную среду [7] .
Диагностика синдрома Туретта
Синдром Туретта диагностирует врач-психиатр или невролог, основываясь на наблюдении и сборе сведений об истории болезни, условиях жизни и перенесённых заболеваниях. Сейчас разрабатываются генетические карты, которые позволят с самого рождения определять совокупность генов, характерных для синдрома Туретта, но пока этот метод недоступен.
Чтобы установить диагноз «синдром Туретта», состояние должно соответствовать следующим критериям:
- присутствуют множественные двигательные тики и как минимум один голосовой тик;
- тики возникают много раз в день, почти ежедневно;
- расстройство длится более года, но необязательно непрерывно, ремиссии продолжаются меньше двух месяцев;
- симптомы появились в возрасте до 18 лет [8] .
Очень важно отличать тики при синдроме Туретта от вторичных тиков, которые появились на фоне инфекций или черепно-мозговых травм при беременности, родах или в раннем детстве. Их различают только на основе анамнеза и физикального обследования. Другие методы, например магнитно-резонансная томография и анализы крови, для диагностики синдрома Туретта не используются.
Лечение синдрома Туретта
Перед врачом всегда стоит выбор — назначать ли препараты при синдроме Туретта. При этом важно ориентироваться на состояние пациента, так как более чем в половине случаев симптомы исчезают без приёма медикаментов.
Психологическая помощь
Если тики не мешают человеку общаться, учиться и работать, то, скорее всего, принимать препараты не нужно. В такой ситуации будет полезно психологическое консультирование ребёнка и родителей. Важно рассказать родителям, что от ребёнка ни в коем случае нельзя требовать, чтобы он перестал кашлять и гримасничать, — это только усилит эмоциональное напряжение и, соответственно, тики.
В зарубежных странах, особенно в США, хорошо зарекомендовал себя метод под названием «Habit reversal training», т. е. тренировка отмены привычки [10] . Пациента учат отслеживать ощущение, предшествующее тикам, и пытаться заменить их на более приемлемые действия. Метод не избавляет от тиков, но заметно уменьшает их проявления.
Также для коррекции поведения используется когнитивно-поведенческая психотерапия [12] .
Медикаментозное лечение
Для лечения синдрома Туретта могут применяться антипсихотики (нейролептики) в невысоких дозировках, которые воздействуют на дофаминэргическую активность. Наиболее эффективны Арипипразол, Рисперидон и Галоперидол. Из них лучше всего переносится Арипипразол. Однако в российских инструкциях по лечению синдрома Туретта это лекарство не упоминается, хотя в США оно одобрено Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) и активно используется в терапии. В России назначают Галоперидол и Рисперидон, а также ряд ноотропов (чаще всего гопантеновую и аминофенилмасляную кислоту) без достаточной доказательной базы.
Есть данные об эффективности Клонидина, но этот препарат опасен при передозировке. Среди возможных побочных эффектов — коллаптоидные состояния, т. е. резкое падение артериального давления, которое может привести к развитию обморока.
Существуют не подтверждённые данные об эффективности инъекции ботулотоксина в мышцы лица.
При возникновении сопутствующих заболеваний, таких как тревожно-депрессивное расстройство и ОКР, применяются селективные ингибиторы обратного захвата серотонина: Сертралин, Пароксетин, Эсциталопрам. При выраженной агрессии, направленной на себя или окружающих, назначаются нормотимики (препараты лития и другие) [12] .
Транскраниальная магнитная стимуляция
В настоящее время изучается влияние транскраниальной магнитной стимуляции на синдром Туретта как у детей, так и у взрослых, но пока недостаточно данных об эффективности этого метода [11] .
Нейрохирургическое лечение
При тяжёлом течении заболевания и выраженной устойчивости к медикаментам может применяться нейрохирургический подход — глубокая стимуляция подкорковых структур головного мозга (бледного шара и таламуса). Её используют для взрослых пациентов.
Прогноз. Профилактика
Прогноз благоприятнее и заболевание чаще заканчивается ремиссией или выздоровлением, если тики появились в возрасте до 7 лет.
Синдром Туретта часто вызывает у людей страх и ассоциации, что эта особенность мешает нормально общаться с другими. Но если грамотно подойти к терапии и исключить провоцирующие факторы, расстройство может протекать вполне благоприятно. Примером служит певица Билли Айлиш. Из-за тяжести заболевания она не ходила в школу и обучалась дома. Это не помешало певице в 2019 году записать сингл, завоевавший первые места в мировых хит-парадах, а в 2021 году войти в список 100 наиболее влиятельных людей года по версии журнала Time. Также синдромом Туретта, предположительно, страдал Вольфганг Моцарт [13] .
Синдром Туретта — это генетическое заболевание, поэтому предупредить его развитие нельзя. Однако если своевременно обратиться к врачу, можно снизить тяжесть болезни и предотвратить развитие депрессии, аутоагрессии и обсессивно-компульсивного расстройства.
При сильных тиках, не поддающихся лечению и мешающих общаться, учиться или работать, пациент может пройти медико-социальную экспертизу: специалисты оценят тяжесть состояния и, при необходимости, определят группу инвалидности.
Читайте также:
- Техника, этапы операции дренирования плевральной полости (введения торакостомической трубки)
- Рецепторная теория действия лекарств. Комплекс агонист-рецептор
- Исследование активных и пассивных движений
- Общие сведения о хроническом гепатите
- Инфекционное поражение сердца на фоне ревматизме. Нагноение в полости рта при ревматизме