Синдром Брауэра (Brauer) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 22.01.2026
наличие дополнительного тона, напоминающего по звучанию щелчок, в начальной фазе диастолы; признак спаечного перикардита.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Смотреть что такое "Брауэра симптом" в других словарях:
Брауэра симптом — (L. Brauer, 1865 1951, нем. врач; син.: перикардтон, перикардтон диастолический) наличие дополнительного тона, напоминающего по звучанию щелчок, в начальной фазе диастолы; признак спаечного перикардита … Большой медицинский словарь
перикардтон — (перикард + тон) см. Брауэра симптом … Большой медицинский словарь
перикардтон диастолический — см. Брауэра симптом … Большой медицинский словарь
Перикардто́н — (Перикард + Тон) см. Брауэра симптом … Медицинская энциклопедия
Перикардто́н диастоли́ческий — см. Брауэра симптом … Медицинская энциклопедия
Ладонно-подошвенные кератодермии: причины, симптомы, диагностика, лечение
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Ладонно-подошвенные кератодермии - большая группа заболеваний, очень разных по своей морфологии. Некоторые из них являются самостоятельным заболеванием, другие входят в состав многочисленных синдромов, третьи представляют собой одно из проявлений диффузных кератозов. Гистогенетически все многообразие клинических проявлений можно свести к нескольким гисгоморфологическим типам.
Все ладонно-подошвенные кератодермии имеют общие гистологические признаки: в разной степени выраженный акантоз. гиперкератоз, иногда очаговый паракератоз; изменения в базальном слое эпидермиса и базальной мембране отсутствуют. Воспалительной реакции в дерме, как правило, нет, лишь иногда встречаются небольшие периваскулярные инфильтраты в верхней ее части. К особенностям, позволяющим разделять ладонно-полошвенные кератодермии на различные типы, относятся изменения структуры зернистого и шиповатого слоев эпидермиса: гиперкератоз с увеличением количества слоев зернистого слоя (гранулез), эпидермолитический гиперкератоз. атрофия или отсутствие зернистого слоя. Гиперкератоз и гранулез отмечаются при подавляющем большинстве ладонно-подошвенных кератодермий, как при диффузных, так и при ограниченных формах.
К диффузным формам кератодермии относят следующие нозологические единицы.
Ладонно-подошвенная кератодермия Тоста-Унны
Наследуется по аутосомно-доминангному типу, характеризуется диффузным поражением ладоней и подошв. Описаны также изменения в области межфаланговых суставов кистей. Существует с рождения или развивается в течение 1-го года жизни, редко - в более позднем возрасте Имеется диффузный кератоз ладоней и подошв с полоской застойной зритемы но его краю. Часты болезненные трещины.
Патоморфология. Выраженный гиперкератоз, гранулез, гипертрофия потовых желез, иногда картина эпидермолитического гиперкератоза, но в таких случаях необходимо исключать ограниченную форму буллезной ихтиозиформной эритродермии. При электронно-микроскопическом исследовании выявлены атипичные кератогиалиновые гранулы двух типов - менее электронно-плотные гранулярного строения и более электронно-плотные, прикрепленные к первым.
Ладонно-подошвенная кератодермия Вернера
Наследуется по аутосомно-доминантному типу. Выявлена мутапия гена, кодирующего кератин 9, расположенного в локусе 17ql2-q21. Заболевание развивается в первые недели жизни. Клиническая картина сходна с ладонно-подошвенной кератодермией Тоста-Уины. Отмечаются гипергидроз и утолщение ногтевых пластинок. Описана самопроизвольная отслойка роговых масс, происходящая 1-2 раза в год.
Патоморфология. Сходна с таковой при врожденной буллезной ихтиозиформной эритродермии,. что подтверждено методом электронной микроскопии. Можно предполагать, что в основе гистогенеза заболевания лежит нарушения образовании тонофибрилл. Биохимический анализ выявил в эпидермисе появление низкомолекулярных кератинов, свидетельствующих о нарушении дифференцировки эпителиоцитов.
Мутилируюшая кератодермия
Наследуется по аутосомно-доминантному типу, характеризуется наличием кератоза с сотовидной поверхностью на ладонях и подошвах, кератотических очагов звездчатых очертаний на тыле кистей кистей и стоп, внутренней поверхности лучезапястных суставов, кольцевидных перетяжек пальцев (псевлоайнгум). Часто встречаются ониходистрофии, описана диффузная алопепия.
Сотовидный кератоз, но без перетяжек, наблюдается также при ладонно-подошвенной кератодермии, ассоциированной с глухотой, при которой, как и при мутилирующей ладонно-подошвенной кератодермии, на тыле кистей и стоп имеются кератотические очаги с переходом на внутреннюю поверхность лучезапястных суставов.
Патоморфология: гиперкератоз с гипергранулезом.
Диффузная ладонно-подошвенная кератодермия
С аутосомно-доминантным типом наследования (генный докус - 17q23-ater) может сочетаться с раком пищевода (синдром Howel-Evans), Кератоз развивается обычно в 5-15 лет, рак пищевода - после 30 лет. Одновременно могут наблюдаться множественные базалиомы.
Кератодермия острова Меледа
Син. болезнь острова Мелела наследуется по аутосомно-репессивному типу. Клинически характеризуется диффузным кератозом ладоней и подошв, выраженной воспалительной реакцией в виде эритематозного венчика вокруг кератотических очагов с выходом поражения на тыльную поверхность кистей и стоп, области коленных и локтевых суставов, нижней трети предплечий и голеней (в виде "перчаток и носков"). Часты контрактуры и сращения пальцев. Описано сочетание с псевдоайнгумом. Заболевание сопровождается гипергидрозом и изменениями ногтевых пластинок, возможны и лейкокератозы.
Патоморфология. При электронной микроскопии выявляют кератогиалиновые гранулы сложного строения, состоящие из менее плотной гранулярной сердцевины и более плотной периферической зоны, связанной с тонофиламентами. Подобные гранулы чаще располагаются в эпителиоцитах, расположенных в области устьев потовых желез.
Близка по клиническим проявлениям к болезни острова Меледа кератодермия, описанная A. Greither (1952). Однако эта форма наследуется по аутосомно-доминантному типу, отличается менее выраженным гиперкератозом, наличием на других участках кожного покрова изменений, сходных с наблюдаемыми при эритрокератодермиях, менее тяжелым течением, улучшением с возрастом.
Кератодермия Папийона-Лефевра
Син. синдром Папийона-Лефевра наследуется по аутосомно-рецессивному типу. Клиническая картина сходна с кератодермией острова Меледа. Поражение кожи сочетается с пародонтозом, воспалением десен и сосочков языка и подверженностью различным инфекционным заболеваниям. Иногда наблюдаются отставание в росте, гипотрихоз, кальцификация мозговых оболочек, сочетание с врожденными бронхоэктазами.
Патоморфология: массивный компактный гиперкератоз и гипергранулез; в эритематозно-сквамозных очагах в области крупных суставов и тыльной поверхности кистей и стоп гистологическая картина напоминает красный отрубевидный волосяной лишай (болезнь Девержи): гиперкератоз с чередованием участков орто- и паракератоза, неравномерный акантоз, незначительная периваскулярная воспалительная инфильтрация в сосочковом слое дермы.
Синдром Олмстеда
Представляет собой сочетание диффузной ладонно-подошвенной кератодермии с четкими краями, ониходистрофии, констрикции пальцев и периорифициального кератоза. В дополнение к перечисленным признакам описаны универсальная алопеция, лейкокератоз, аномалии зубов.
Ограниченные ладонно-подошвенные кератозы
Собирательный термин, использующийся для всех ограниченных (очаговых, линейных) форм кератодермии. Тип наследования аутосомно-доминантный. Клинические проявления заболевания могут появиться в юношеском возрасте или у взрослых. При крупноочаговых формах кератодермии на ладонях и подошвах обнаруживают монетовидные округлые кератотические очаги, наиболее выраженные на местах давления, и крупные, изолированные или сочетающиеся с линейными кератозами очаги в области сгибательных поверхностей пальцев. Может наблюдаться сочетание со спиралевидными курчавыми волосами. При электронно-микроскопическом исследовании в одном случае обнаружены отечность эпителиоцитов, повышение плотности тонофиламентов в супрабазальной области, вакуолизация шиповатых клеток, изменения в строении кератогиалиновых гранул и липидные капли в роговом слое.
Папулезные ладонно-подошвенные кератодермии отличаются рассеянным характером и меньшими размерами кератотических очагов. Развиваются в первые годы жизни (кератодермия Брауэра) или в 15-30-летнем возрасте (кератодермия Бушке-Фишера). Клинически характеризуются множественными плоскими, полушаровидными или веррукозными очажками ороговения, округлых или овальных очертаний, располагающимися обычно изолированно по всей поверхности ладоней и подошв, а не только в местах давления. После удаления роговых масс остается кратеро- или блюдцеобразное углубление. A. Greither (1978) считает перечисленные формы папулезных кератодермии идентичными.
Точечная врожденная акрокератодермия
Син. точечный кератоз ладоней и подошв характеризуется появлением на ладонях и тыле кистей мелких кератотических папул цвета нормальной кожи с гладкой блестящей поверхностью. Гистологически F.C. Brown (1971) выявил паракератотические столбики, сходные с наблюдаемыми при порокератозе Мибелли. D.G. Robestria и соавт. (1980) обнаружили при электронной микроскопии внутриядерные нарушения в виде множественных гипертрофированных ядрышек в клетках базального и шиповатого слоев, которые, по мнению авторов, способствуют развитию гиперкератоза. Описано сочетание этого заболевания с раком внутренних органов. M.J. Costello и R.C. Gibbs (1967) рассматривают папулезные и точечные кератодермии как синонимы.
Кератодермия с просвечивающими папулами представляет собой, возможно, разновидность точечной врожденной акрокератодермии. Наследуется также по аутосомно-доминантному типу, характеризуется желтовато-белыми, просвечивающими папулами с гладкой поверхностью, иногда с точечными углублениями в центре, сливающимися в бляшки. Сочетается с тонкими волосами на голове и атопией.
Точечный кератоз ладонных линий характеризуется наличием на ладонях и подошвах мелких гиперкератотических пробок, располагающихся в углублениях кожных линий, болезненных при надавливании.
Ладонно-подошвенная кератодермия с перекрученными волосами - аутосомно-доминантно наследуемое заболевание, характеризующееся наличием на ладонях и подошвах округлых очагов кератоза. Патологические изменения волос подтверждены методом сканирующей электронной микроскопии. В волосах гистохимически обнаружен дефицит цистеина.
Синдром Рнхнера-Ханхарта
Син.: кожно-глазной тирозиноз, тирозинемия II типа характеризуется болезненными ладонно-подошвенными кератотическими очагами, герпетиформной дистрофией роговицы и умственной отсталостью. Без лечения с возрастом развивается диффузная кератодермия, могут быть пузыри. Тип наследования аутосомно-рецессивный, поражен генный локус 16q22.1-q22. Гистологически, помимо общих для этой группы кератодермии признаков, выявляют эозинофильные включения в клетках шиповатого слоя. При электронно-микроскопическом исследовании находят увеличение количества тонофиламентов в шиповатых эпителиоцитах, тубулярные каналы а пучках тонофиламентов. В основе гистогенеза лежит дефицит фермента тирозина аминотрансферазы, что приводит к накоплению тирозина в крови и тканях. Предполагается, что молекулы L-тирознна мот способствовать образованию дополнительных поперечных связей. Это приводит к сгущению тонофибрилл в эпителиоцитах.
Ладонно-подошвешшя нумулярная кератодермия
Так называемые болезненные омозолелости наследуется по аутосомно-доминантному типу. Развивается в детском или молодом возрасте, характеризуется наличием ограниченных крупных гиперкератотических очагов. локализующихся в местах давления: на подошвах. у основания и на боковых поверхностях пальцев ног, на кончиках пальцев рук, болезненных при надавливании. Описаны пузыри по краям очагов, подногтевой или околоногтевой гиперкератоз, утолщение ногтевых пластинок и очаги гиперкератоза на голенях. Гистологически наблюдается эпидермолитический гиперкератоз.
Акрокератоэластоилоз Коста
Развивается в детском возрасте. Клинически проявляется мелкими, иногда сливающимися папулами птотноватой консистенции, сероватого цвета, просвечивающими, с блестящей поверхностью, располагающимися на ладонях и подошвах, по краям пальцев, в области пяточного сухожилия. Гистохимически в дерме в очагах поражения выявляются утолщение и фрагментация пластических волокон, электронно-микроскопически - изменения аморфной части их, нарушение расположения микрофибрилл. Изменения в зернистом слое отсутствуют.
Следует отметить, что большая группа ладонно-подошвенных кератодермяй до сих пор не классифицирован ни клинически, ни гистологически. В литературе имеются морфологические описания только отдельных случаев. В связи с этим диагностика, особенно дифференциальная, указанных заболеваний представляет большие трудности.
Различия в клинической характеристике высыпаний и типе наследования, особенности течения заболеваний внутри выделенных нами групп позволяют предположить неодинаковый их патогенез при сходной гистологической картине.
Кератодермия: разновидности и лечение
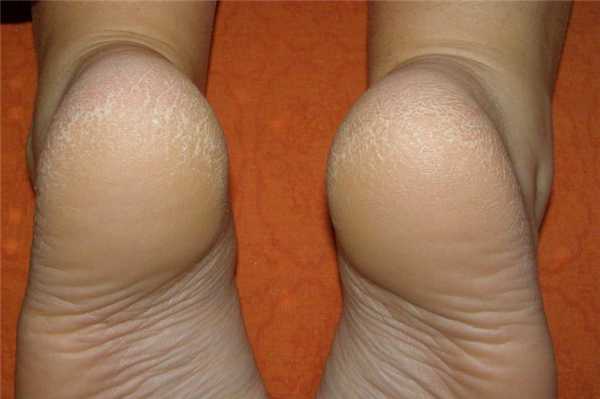
Кератодермия или ладонно-подошвенный кератоз — ряд заболеваний с нарушением формирования рогового слоя тыльной стороны кистей и стоп. Данная патология относится к хроническим.
Причины кератодермии
Причины возникновения ладонно-подошвенного кератоза:
- Мутации генов, отвечающих за образование кератина-механически прочного белка, из которого состоят волосы, ногти, кожа человека.
- Нарушение синтеза витамина А, который выполняет в организме биохимически важные функции. Одна из таких функций — формирование клеточных мембран.
- Дисфункция желез внутренней секреции. При нарушении выработки гормонов происходит сбой в процессах отмирания клеток эпидермиса.
- Инфекции и опухоли также могут повлечь за собой излишнее ороговение кожных покровов.
Виды кератодермий
Ладонно-подошвенные кератозы делятся по причине возникновения на наследственные и приобретенные. Наследственные кератодермии:
- Унны-Тоста наследуется аутосомно-доминантными генами и проявляется усиленным потоотделением и шелушением кожи.
- Кератодермия Меледа передается аутосомно-рецессивными генами, проявляется ближе к 16 годам толстыми роговыми наслоениями желтого цвета с сиреневой каймой по краям и трещинами тыльной стороны кистей и подошвы.
- Папийона-Лефевра относится к аутосомно-рецессивным заболеваниям. Болезнь протекает тяжело, затрагивает руки, ноги, кости, ногти и зубы. Кератоз начинает проявляться у детей в возрасте от 1 года до 4 лет.
- Мутилирующая кератодермия Фонвинкеля возникает при наличии доминантного гена, распространяется очень быстро на ладони, ступни, суставы (в том числе мелкие суставы пальцев) и волосы. Патология может привести к ампутации фаланг и облысению.
- Фигурная изменчивая эритрокератодермия передается аутосомно-доминантно, появляются зудящие очаги красновато-желтого цвета географической конфигурации. На места расчесов присоединяется вторичная инфекция.
- Фолликулярно-шиповидный кератоз передается аутосомно-доминантно и чаще проявляется у мальчиков возникновением роговых пробок и потерей волос.
- Кератодермия пятнистая диссеминированная Бушке—Фишера—Брауэра — рассеяно-точечный кератоз, унаследованный по доминантному типу. Болезнь обнаруживается в подростковом возрасте образованием кратероообразных ороговевших очагоы на ладонях, белых утолщений на деснах, нарушением форм и размеров языка.
- Болезнь Вернера — аутосомно-рецессивное заболевание, характеризующееся поражением ногтей.
- Поликератодермия Турена — эктодермальная дисплазия, локализованная на кистях и ступнях.
- Идиопатическая кератодермия манифестирует в пубертатный период и проявляется участками гиперкератоза на подошвах и ладонях.
- Инфекционная — результат перенесенных вирусных, бактериальных или грибковых заболеваний.
- Лучевая кератодермия является профессиональным заболеванием рентгенологов и рентгенотехников.
- Старческий кератоз возникает у пожилых людей. Сухие плотные бородавчатые очаги возникают на подошвах, ладонях, лице, предплечиях. Убирать ороговения опасно из-за риска кровотечений и присоединения инфекций.
Кератодермии делят по месту распространения на диффузные (сплошное поражение кожи) и локальные (небольшие участки).
Диагностика заболевания
Диагностика заболевания проводится дерматологом, дерматологом-онкологом, генетиком (если речь идет о наследственности). Врач назначает необходимые анализы, проведет внешний осмотр кожных покровов, соберет анамнез.
Лечение кератозов
Лечение кератозов проводится комплексно и включает в себя медикаметозные, физиотерапевтические и народные методы. К первой группе относят применение витаминов (витамины А, Е в капсулах), препараты кальция (кальция глюконат в ампулах или таблетках), ретиноиды (Неотигазон). Для увлажнения кожи назначаются крема и эмульсии с ароматическими ретиноидами (Ретиноевая мазь, Радевит актив). Снять воспаление и уменьшить ороговение помогают наружные средства с глюкокортикостероидами (Акридерм, Адвантан и т. д.). Физиотерапия включает лазеротерапию, криотерапию, термическую коагуляцию. К народным методам относят приготовление настоев и отваров для приема внутрь, а также примочки, ванночки и местное нанесение эфирные масел.
Кератоз — сложное в лечении заболевание, доставляющее дискомфорт и детям, и взрослым. Терапия кератодермии проводится длительно для снятия острых периодов и продления ремиссии заболевания. Для снижения частоты рецидивов необходимо увлажнять кожу, использовать солнцезащитные крема, своевременно лечить инфекции и принимать витаминные комплексы.
Брауэр Сергей

Сереже 18 лет, он человек творческий: любит слушать музыку, играет на гитаре, танцует.
Может вновь и вновь перечитывать любимые книги, например, про Незнайку или «Волшебник изумрудного города».
Вообще, если Сережу занятие увлекает, он будет постоянно к нему возвращаться, пока не закончит. Возможно, поэтому, конным спортом он занимается уже около шести лет, сначала с перерывами, а потом, благодаря последним спонсорам, стабильно. Удаются Сереже и занятия гольфом, жаль только, что они ограничены сезоном: лето и осень.
Зато, его радуют предстоящие Новогодний праздник и Рождество! Сережа путешествовал по Иерусалиму, поднимался на Святую гору! Мальчик мечтает стать священником, поэтому Рождество имеет для него особое значение.
Сережа знает, как он будет готовиться к праздникам: нужно чисто убрать дом и нарядить вместе со старшей сестрой елку. И обязательно написать письмо Деду Морозу! Интересно, что Сережа загадает под бой курантов? Пока это секрет! А мы можем пожелать, чтобы во всех своих делах ему сопутствовал успех, и тогда его мечты обязательно сбудутся!
Со слов мамы Сергея:
«Огромная благодарность спонсорам за возможность заниматься оздоровительной верховой ездой моему сыну Сергею Брауэру. Он получает большое удовольствие от общения с лошадьми и инструкторами. Серёжа занимается в любую погоду и перестал болеть простудными заболеваниями. У сына выправилась осанка, появилась работоспособность, улучшилась координация. Во время пандемии из-за изолированности от сверстников и ограничения передвижения иппотерапия для него единственная возможность поддержания здоровья и развития. Низкий Вам поклон!»
Все детские медицинские центры ГК «Вирилис» оказывают благотворительную помощь «Солнечным детям». У нас дети с синдромом Дауна получают лечение, профилактику, мы помогаем им заниматься иппотерапией
Синдром Стерджа-Вебера
Синдром Стерджа-Вебера — врожденный ангиоматоз, поражающий кожу, органы зрения и центральную нервную систему. Проявляется множественными врожденными ангиомами лицевой области, стойким эпилептическим синдромом, глаукомой, олигофренией, другими неврологическими и офтальмологическими симптомами. В ходе диагностики выполняется рентгенография черепа, КТ или МРТ церебральных структур, офтальмоскопия, измерение внутриглазного давления, гониоскопия, УЗИ глаза. Лечение включает противоэпилептическую терапию, консервативное и хирургическое лечение глаукомы, симптоматическую терапию. Прогноз во многих случаях неблагоприятный.
Общие сведения
Синдром Стерджа-Вебера — редко встречающееся врожденное ангиоматозное поражение церебральных оболочек, кожи и глаз. Распространенность находится на уровне 1 случай на 100 тыс. населения. Впервые пациента с таким синдромом описал в 1879 г. Стердж, затем в 1922 г. Вебер указал рентгенологические признаки, выявляемые при данном синдроме. В 1934 г. Краббе предположил, что у пациентов, наряду с ангиомами кожи, имеется ангиоматоз церебральных оболочек. В честь этих исследователей заболевание получило название синдром Стерджа-Вебера-Краббе. В связи с тем, что ангиомы лица локализуются в области иннервации кожи 1-ой и 2-ой ветвью тройничного нерва, в неврологии заболевание также известно под названием энцефалотригеминальный ангиоматоз. Наряду с нейрофиброматозом Реклингхаузена, синдромом Луи-Бар, туберозным склерозом, болезнью Гиппеля-Линдау и др., синдром Стерджа-Вебера входит в группу факоматозов — прогрессирующих нейрокожных заболеваний.
Причины синдрома Стерджа-Вебера
Синдром Стерджа-Вебера возникает в результате нарушений эмбрионального развития, приводящих к сбою дифференцировки экто- и мезодермальных листков. Большинство случаев составляет спорадическое появление синдрома, реже отмечается аутосомное (не сцепленное с полом) доминантное или рецессивное наследование с частичной пенетрантностью. Считается, что спорадические случаи обусловлены негативным воздействием на плод в эмбриональном периоде. Вредоносными факторами могут выступать экзо- и эндогенные интоксикации беременной, в том числе никотин, алкоголь, наркотики, различные медикаменты; внутриутробные инфекции; дисметаболические нарушения у будущей матери (например, некомпенсированный сахарный диабет или гипертиреоз).
Морфологическим субстратом заболевания является ангиоматоз — формирование и рост множественных сосудистых опухолей (ангиом), располагающихся на коже лица и в церебральных оболочках. Как правило, ангиоматоз оболочек затрагивает их конвекситальную часть, более часто наблюдается в затылочной и теменных зонах. Обычно ангиоматоз лица и ангиоматоз мозговых оболочек имеют гомолатеральное расположение, т. е. находятся на одной стороне. Однако нередко отмечается двусторонний характер поражения. В церебральных тканях, расположенных под ангиоматозно измененным участком оболочки развиваются дегенеративные процессы, происходит атрофия и избыточный рост глии, формируются кальцификаты.
Симптомы синдрома Стерджа-Вебера
Самым ярким признаком, характеризующим синдром Стерджа-Вебера, выступает ангиоматоз кожи лица. У всех пациентов сосудистое пятно является врожденным. Со временем оно может увеличиваться в размерах. Локализуется, как правило, в скуловой и подглазничной области. Бледнеет при надавливании. В начале имеет розовую окраску, затем приобретает ярко-красный или красно-вишневый оттенок. Внешний вид и распространенность ангиом различны, они могут представлять собой мелкие рассеянные очажки или сливаться в одно большое пятно, т. н. «пламенеющий невус». Ангиоматоз может охватывать полость носа, глотку, полость рта. В 70% случаев синдрома ангиоматоз является односторонним. В 40% изменения на лице сочетаются с ангиомами туловища и конечностей. Возможны и другие дерматологические симптомы: врожденные гемангиомы, локальные отеки мягких тканей, невусы, зоны гипо- и гиперпигментации. По некоторым данным в 5% случаев синдром Стерджа-Вебера протекает без характерного «пламенеющего невуса» на лице.
От 75% до 85% случаев энцефалотригеминального ангиоматоза протекают с судорожным синдромом, дебютирующим на первом году жизни. Характерны эпиприступы джексоновского типа, во время которых судороги охватывают конечности, контрлатеральные (противоположные) расположению ангиоматоза церебральных оболочек. Эпилепсия приводит к задержке психического развития, олигофрении, в отдельных случаях вплоть до идиотии. Может наблюдаться гидроцефалия, гемипарез и гемиатрофия контрлатеральных оболочечной ангиоме конечностей.
Со стороны органа зрения могут наблюдаться ангиомы сосудистой оболочки глаза, гетерохромия радужки, гемианопсия, колобомы. Примерно у трети больных диагностируется глаукома, которая вызывает помутнение роговицы, а в ряде случаев приводит к формированию гидрофтальма. В некоторых случаях синдром Стерджа-Вебера сочетается с дисплазией лицевого черепа, проявляющейся асимметрией лица; в других — с врожденным пороком сердца.
Патогномоничная триада проявлений синдрома («пламенеющий невус», нарушения зрения, неврологическая симптоматика) наблюдается лишь у пятой части больных. В остальных случаях сочетание симптомов значительно варьирует, в связи с чем на сегодняшний день выделено 10 клинических форм синдром Стерджа-Вебера. Нередки абортивные варианты синдрома, при которых клинические симптомы выражены лишь частично и в легкой степени.
Диагностика синдрома Стерджа-Вебера
Диагностировать синдром Стерджа-Вебера возможно по характерному сочетанию симптомов: наличию кожного лицевого ангиоматоза, эпиприступов и других неврологических проявлений, а также офтальмологической патологии (в первую очередь, глаукомы). Диагностический поиск проводится коллегиально неврологом, эпилептологом, офтальмологом и дерматологом.
При проведении рентгенографии черепа обнаруживаются зоны обызвествления коры, имеющие вид двойных контуров, как бы обводящих извилины в области церебрального поражения. КТ головного мозга визуализирует еще более обширные зоны кальцификации, чем показывает рентгенография. МРТ головного мозга выявляет участки дегенерации и атрофии церебрального вещества, истончения коры; позволяет исключить другие заболевания (внутримозговую опухоль, абсцесс головного мозга, церебральную кисту).
Электроэнцефалография дает возможность определить характер биоэлектрической активности мозга и диагностировать эпи-активность. Офтальмологическое обследование состоит из проверки остроты зрения, периметрии, измерения внутриглазного давления, офтальмоскопии и гониоскопии (при сохранности прозрачности роговицы), ультразвуковой биометрии глаза, АВ-сканирования.
Лечение и прогноз синдрома Стерджа-Вебера
В настоящее время синдром Стерджа-Вебера не имеет эффективного лечения. Терапия направлена на купирование основных проявлений. Проводится антиконвульсантная терапия вальпроатами, карбамазепином, леветирацетамом, топираматом.
Эписиндром зачастую оказывается резистентным к проводимому противоэпилептическому лечению, что требует перехода с монотерапии на прием комбинации из двух препаратов. В качестве одного из способов лечения применяется рентген-облучение черепа над пораженной ангиоматозом областью. По показаниям нейрохирургами может быть рассмотрен вопрос о проведении оперативного лечения эпилепсии.
Лечение глаукомы состоит в инстилляции глазных капель, снижающих секрецию внутриглазной жидкости: бримонидина, тимолола, дорзоламида, бринзоламид и пр. Однако подобная консервативная терапия зачастую оказывается малоэффективной. В таких случаях офтальмохирургами проводится хирургическое лечение глаукомы: трабекулотомия или трабекулэктомия.
К сожалению, при выраженной клинике синдром Стерджа-Вебера имеет неблагоприятный прогноз. Некупируемый эпилептический синдром приводит к выраженной олигофрении. Возможна потеря зрения, интракраниальные сосудистые нарушения, влекущие за собой вероятность развития инсульта.
Читайте также: