Синдром Дитлена (Dietlen) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 21.01.2026
Синдром Прадера-Вилли- врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 25000-10000 новорожденных. Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание. Заболевание характеризуется выраженной мышечной гипотонией при рождении, сохраняющейся в течение первого года жизни ребенка. Сосательный и глотательный рефлексы снижены, что затрудняет кормление ребенка. Из-за гипотонии у таких детей задерживается развитие двигательных функций: они с трудом учатся держать голову, сидеть и т. д. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает.
Позднее, к второму-четвертому году жизни появляются постоянное чувство голода и отсутствие насыщения, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей. Из-за тяжелого ожирения грозным осложнением является обструктивное апноэ (остановка дыхания) во сне.
Рост больных нередко снижен. Часто отмечается долихоцефалия (удлиненная форма головы), миндалевидный разрез глаз, низко расположенные ушные раковины, широкая переносица, маленький рот с тонкой верхней губой. Стопы и кисти больных диспропорционально маленькие (акромикрия). У 75% детей наблюдается слабая пигментация кожи, волос и радужки.
У мальчиков при рождении отмечается недоразвитие полового члена, мошонки,крипторхизм, а у девочек недоразвитие половых губ, иногда и матки. В дальнейшем заболевание проявляется задержкой или отсутствием полового созревания, бесплодием.
Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед.). Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарем, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, косоглазие.
Продолжительность жизни больных может достигать 60 лет и более. Нередко у таких детей развивается сахарный диабет.
Лечение
Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным.
Младенцы со сниженным мышечным тонусом должны получать массаж и другие виды специальной терапии. Комплекс лечебных мероприятий включает также диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). Рекомендуется терапия гормоном роста.
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Медико-генетическое консультирование
Родителям ребенка с синдромом Прадера-Вилли рекомендуется пройти генетическое обследование, прежде чем планировать дальнейшую беременность, поскольку существует риск того, что следующий ребенок у тех же родителей родится также с синдромом Прадера-Вилли, что зависит от механизма, вызвавшего генетический сбой.
Болезнь де Кервена ( Стенозирующий лигаментит , Стенозирующий тендовагинит , Хронический тендосиновит )
Болезнь де Кервена - это сужение канала, в котором проходят сухожилия большого пальца. Сопровождается воспалением сухожильных влагалищ. Возникает вследствие постоянной повышенной нагрузки на кисть, нередко - в связи с выполнением профессиональных обязанностей. Обычно развивается постепенно. Течение хроническое. Для заболевания характерна боль в основании I пальца и небольшой местный отек. Из-за боли у пациентов снижается или утрачивается способность выполнять ряд движений с участием как I пальца, так и всей кисти. Диагноз выставляется на основании предъявляемых жалоб и осмотра больного, дополнительные исследования не требуются. Консервативная терапия обеспечивает эффект примерно в 50% случаев. Радикальным методом лечения является операция.
МКБ-10
Общие сведения
Болезнь де Кервена (хронический тендосиновит, стенозирующий тендовагинит, стенозирующий лигаментит) - сужение (стеноз) канала, в котором расположены сухожилия I пальца кисти. Причина заболевания - постоянная травматизация канала при движении в нем сухожилий. Заболевание развивается постепенно и протекает хронически. Женщины страдают чаще мужчин, пожилые люди - чаще молодых. Обычно выявляется связь заболевания с характером работы или повышенной нагрузкой на кисть при выполнении бытовых обязанностей.
Причины
В современной травматологии и ортопедии доминирует мнение, что болезнь де Кервена носит преимущественно профессиональный характер. Заболевание, как правило, наблюдается у пианистов, домработниц, доярок, прачек, швей, слесарей, скорняков, каменотесов, полевых рабочих, маляров, намотчиц, утюжильщиц и т. д. Вместе с тем, эта патология может выявляться и у неработающих женщин. В последнем случае развитие болезни связано с выполнением домашних обязанностей и ношением маленьких детей на руках.
Патогенез
I палец - самый активный. Он участвует практически во всех мелких движениях кисти и играет существенную роль при выполнении целого ряда более крупных операций, например, фиксации предметов или инструментов. При постоянном выполнении движений, связанных с длительным напряжением большого пальца и отклонением кисти в сторону мизинца, и без того немалая нагрузка на канал и сухожилия еще больше увеличивается. Создаются благоприятные условия для развития стеноза и сопутствующего воспаления. По мере прогрессирования болезни из-за сужения канала сухожилия начинают все сильнее тереться о его стенки, в сухожильных влагалищах возникает воспаление (тендовагинит), и они отекают, приводя к еще большему повреждению канала при движениях и стимулируя дальнейшее развитие стеноза.
Симптомы болезни де Кервена
Заболевание развивается постепенно. Обычно больные впервые приходят на прием через несколько дней или недель после возникновения симптомов. Примерно в 7% случаев наблюдается острое начало, связанное с предшествующей травмой кисти. При сборе анамнеза заболевания выясняется, что вначале пациентов беспокоила боль только при значительном разгибании и отведении большого пальца, а также при резком отведении кисти в сторону мизинца. В последующем болевой синдром прогрессирует и возникает даже при незначительных движениях.
Пациенты жалуются на боль в нижней части предплечья и проекции лучезапястного сустава на стороне большого пальца. Боли могут возникать исключительно во время движений или быть давящими, ноющими, постоянными, не исчезающими даже в покое. При случайных неловких движениях возможно также возникновение резкой боли во сне. Более чем в половине случаев боли отдают вниз, по наружной поверхности I пальца или вверх, по предплечью, локтевому суставу и плечу.
Диагностика
Осмотр обязательно проводится в сравнении обеих кистей - это позволяет точно выявить порой не слишком сильно выраженные, но абсолютно характерные для болезни де Кервена изменения со стороны больной кисти. В области лучезапястного сустава со стороны I пальца определяется незначительный или умеренный местный отек. Анатомическая табакерка сглажена или не выявляется из-за припухлости. Кожа над пораженной областью не изменена, местного повышения температуры нет. Редкие случаи шелушения, покраснения и местной гипертермии обусловлены не самим заболеванием, а самостоятельным лечением, которое иногда проводят пациенты, прежде чем обратиться к врачу.
При пальпации выявляется болезненность в области поражения, достигающая максимума в проекции шиловидного отростка лучевой кости. Надавливание на область сухожилий I пальца безболезненно. Чуть ниже шиловидного отростка прощупывается плотное и гладкое образование округлой формы - тыльная связка, утолщенная в области канала. После исследования пораженной области больного просят положить руки ладонями вниз и отклонить кисти поочередно в сторону мизинца и большого пальца. Кисти пациента практически одинаково отклоняются в сторону I пальца. При отклонении в сторону мизинца выявляется ограничение движений на 20-30 градусов по сравнению со здоровой кистью, а движение сопровождается выраженной болезненностью.
Кроме того, на больной руке определяется ограничение отведения большого пальца. Для выявления симптома пациента просят поставить кисти на ребро ладонями друг к другу. При движениях заметно значительное ограничение отведения (разница между больной и здоровой стороной составляет от 40 до 80 градусов). Разница при разгибании I пальцев не так разительна, однако тоже видна невооруженным глазом.
Еще одним исследованием, позволяющим подтвердить диагноз, является тест Финкельштейна. Пациент прижимает большой палец к ладони и плотно сжимает его остальными пальцами, а затем отводит кисть в сторону мизинца. Движение сопровождается резкой болью в области поражения. Также при данном заболевании выявляется нарушение способности удерживать предметы с помощью I пальца. Пациента просят одновременно взять какие-то предметы (например, ручки или спичечные коробки) I и II пальцами обеих рук. При потягивании за предмет выявляется боль и слабость при удерживании с больной стороны. Диагноз болезни де Кервена выставляется на основании клинических данных. Дополнительные исследования не требуются.
Лечение болезни де Кервена
Лечение осуществляется ортопедом или травматологом. Консервативная терапия проводится амбулаторно. Пациенту накладывают гипсовую или пластиковую шину сроком на 1-1,5 месяца, обеспечивая покой пораженной конечности, а в последующем рекомендуют носить специальный бандаж для I пальца. Кроме того, больному назначают нестероидные противовоспалительные средства (ибупрофен, напроксен и т. д.). При выраженном болевом синдроме выполняют местные блокады.
При неэффективности консервативной терапии показано хирургическое лечение. Операция проводится в стационарных условиях в плановом порядке. Обычно используют местную анестезию. До начала обезболивания врач отмечает самую болезненную точку, а после введения новокаина выполняет косой или поперечный разрез над областью шиловидного отростка, проходящий через эту точку. Затем он тупым крючком осторожно отводит в сторону подкожную клетчатку вместе с венами и поверхностной ветвью лучевого нерва и обнажает тыльную связку. Связка рассекается и частично иссекается.
Прогноз и профилактика
Прогноз благоприятный. При консервативном лечении удовлетворительный эффект отмечается в 50% случаев. После операций обычно наблюдается хорошее восстановление. Следует учитывать, что заболевание обусловлено хроническим патологическим процессом в области кольцевидной связки. Если пациент после операции по-прежнему перегружает руку, заболевание может рецидивировать. Поэтому больным обычно рекомендуют изменить характер профессиональной деятельности и уменьшить нагрузку на руку при выполнении бытовых обязанностей.
Что такое синдром Лойса - Дитца у ребенка?

Синдром Лойса-Дитца — это новая болезнь, которую описали менее 20 лет назад. Эксперты утверждают, что даже не все врачи знакомы с таким заболеванием и нередко неверно диагностируют его как синдром Марфана. Что же это за новый синдром и как он проявляется у детей?
Что такое синдром Лойса-Дитца?
Это генетическое заболевание, которое разрушает соединительные ткани. Те самые, что поддерживают и придают гибкость мышцам, кровеносным сосудам и костям.
Изменения в соединительных тканях влияют на формирование костей, а также на развитие артерий. Обычно симптомы проявляются в подростковом возрасте, но постановка точного диагноза может быть затруднена до взрослого возраста.
Каковы генетические причины болезни?

Существует пять различных версий синдрома Лойса-Дитца в зависимости от того, какой из генов мутировал. Хотя каждая версия патологии вызывается мутацией разных генов, но все пять из этих генов вовлечены в один и тот же клеточный сигнальный путь — путь трансформирующего бета фактора роста.
Этот путь контролирует, как клетки функционируют во время развития ребёнка. Он также помогает в развитии внеклеточного матрикса — сети белков и других молекул, соединяющих клетки.
Мутировавшие гены производят «сломанные» белки, которые не работают должным образом. Симптомы синдрома Лойса-Дитца являются результатом этих нарушений.
Все мутации при Лойса-Дитца являются доминантными, а это означает, что ребёнку достаточно унаследовать только одну мутировавшую копию от одного родителя, чтобы иметь синдром.
Однако по новым данным исследования самих первооткрывателей этого синдрома, врачей Dietz H и Loeys B, в 75% всех случаев данные мутации возникают спонтанно, поэтому семейного анамнеза болезни может и не быть.
Каковы симптомы синдрома Лойса-Дитца?
Четыре основные характеристики предполагают, что у ребёнка это заболевание. Эти особенности обычно не возникают все вместе при других расстройствах соединительной ткани как основные характеристики. Признаки включают:
- Аневризмы (расширение артерий), которые можно наблюдать с помощью методов визуализации. Они чаще всего наблюдаются в корне аорты (основание артерии, ведущее от сердца), но их можно увидеть в других артериях по всему телу.
- Артериальная извилистость (скрученные или спиральные артерии), чаще всего встречающиеся в сосудах шеи.
- Гипертелоризм — необычно широко расположенные глаза на лице.
- Разделенный или широкий небный язычок, или увула (маленький кусочек плоти, который свисает в задней части рта).
Важно: эти симптомы не всегда наблюдаются у всех пациентов, однако присутствуют в большинстве случаев.
Кроме этого набора у детей с диагнозом синдрома Лойса-Дитца могут быть отличительные признаки во внешности: плоские щеки (маларская гипоплазия), краниосиностоз (раннее зарастание родничков), расщелина неба, синеватые склеры глаз, маленький или отсутствующий подбородок, очень длинные пальцы и/или их срастание, деформации, сколиоз, полупрозрачная, очень мягкая кожа с быстро возникающими синяками и широкими шрамами
Синдром Лойса-Дитца также вызывает врожденные пороки сердца, грыжи, близорукость, частые пищевые аллергии и болезни желудка и кишечника, астму, мигрень и так далее.
Как ставят диагноз «синдром Лойса-Дитца» у ребёнка?

Если есть подозрение на синдром Лойса-Дитца, в первую очередь рекомендуется консультация генетика, который знаком с расстройствами соединительной ткани. Во время первоначального визита будет проведен сбор семейного анамнеза и истории болезни, комплексное физическое обследование для оценки скелетных, черепно-лицевых и связанных с кожей признаков.
Если подозрение на болезнь продолжается, должна быть выполнена эхокардиография (ультразвуковая визуализация сердца), чтобы оценить, есть ли увеличение аорты и/или другие структурные дефекты сердца, которые согласуются с диагнозом. Консультация с кардиологом потребуется, чтобы помочь интерпретировать результаты обследования.
Врач также может предложить дальнейшую визуализацию артерий по всему телу. Ее проводят с помощью КTA ( КТ-ангиография, или обследование сосудов при компьютерной томографии) или МРА (магнитно-резонансная томография в ангиорежиме) всего артериального дерева (голова, шея, грудь, таз и живот). Эти исследования помогут обнаружить аневризмы в других артериях.
Генетическое тестирование на мутации в генах TGFBR1, TGFBR2, SMAD3, TGFB2 и TGFB3 проводятся, если есть высокое подозрение наличия синдрома. Если у ребёнка обнаружена мутация гена, обычно рекомендуется проверить родителей на ту же мутацию, чтобы дать точную информацию о риске рецидива.
Ожидаемая продолжительность жизни людей с синдромом Лойса-Дитца по данным экспертов (Genetics in Medicine) оценивается примерно в 37 лет, но некоторые люди с этим расстройством могут жить намного дольше — иногда до семидесяти лет.
Доступное лечение синдрома Лойса-Дитца: что возможно?
Терапия при синдроме Лойса-Дитца зависит от симптомов ребёнка: если что-то беспокоит или грозит развитием патологии, специалисты концентрируют усилия в этих направлениях. Российские ученые из Санкт-Петербурга уточняют, что основной командой специалистов для ребёнка с такой болезнью должны стать педиатр, кардиолог, невролог и ортопед, а обследование сосудов сердца надо проходить как минимум ежегодно, даже если ничего не беспокоит.
К общим целям лечения относятся:
- Снижение нагрузки на артерии.
- Управление различными скелетными и мышечными проблемами, которые развиваются, и болью, которую они могут вызвать.
- Управление любыми проблемами иммунной системы — с помощью образа жизни, диеты, лекарств, вакцинации.
В случаях, когда у ребёнка развивается аневризма, врач будет в зависимости от размера и локализации советовать либо оперативное лечение, либо наблюдение.
Если у ребёнка синдром Лойса-Дитца, ему следует избегать напряженных, повторяющихся действий, таких как приседания и отжимания. Однако сама физическая нагрузка полезна, с учетом основной рекомендации: на пике усилий ребёнок должен дышать достаточно эффективно, чтобы спокойно поддерживать разговор. Полезна также целенаправленная закалка организма. Как ее проводить, читайте в статье «Закаливание детей: виды, принципы, рекомендации».
Болезнь де Кервена
Лечение болезни де Кервена современными эффективными методами, с использованием инновационной аппаратуры, под руководством врачей со специальными навыками и по лояльным расценкам.

Одним из тяжелых недугов, сопровождающихся ощутимым болевым синдромом, отёчностью и постепенным сокращением количества движений в области кисти, считается болезнь де Кервена. Этой патологии подвержены лица, занимающиеся монотонной работой в течение продолжительного времени. При своевременном визите к специалисту удается избежать серьезных последствий, связанных с полной утратой двигательной активности в зоне большого пальца.
В медцентре «Здоровье Плюс» работают физиотерапевты и хирурги с внушительным стажем, что гарантирует быстрый выбор подходящих лечебных мероприятий, а также получение действенного и долгосрочного результата. Мы выполняем тщательную диагностику. При необходимости производим оперативное вмешательство. Однако наиболее приоритетным методом воздействия, реализуемым в нашей клинике, остается ударно-волновая терапия. У нас данная процедура осуществляется на инновационной технике, под руководством врачей со специальной подготовкой и по демократичной стоимости.
Далее Вы подробно узнаете обо всех тонкостях устранения стенозирующего тендовагинита. Надеемся, предложенная информация позволит сделать правильный вывод о необходимости своевременного обращения к специалисту и надлежащего выполнения необходимых лечебных процедур.
Что собой представляет болезнь де Кервена?
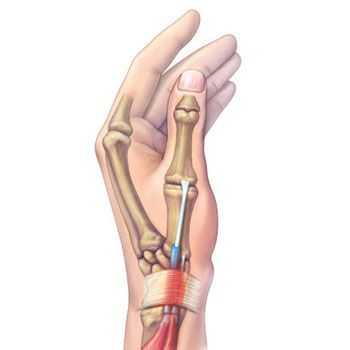
Данная патология характеризуется появлением воспалительного процесса и сужением футляра, охватывающего соединительнотканную часть двух мышц. Речь идет о разгибателе большого пальца кисти и длинной мышцы, отвечающей за его отведение. Заболевание было названо в честь известного врача из Швейцарии Фрица де Кервена, который занимался его изучением в конце прошлого столетия.
Болезнь де Кервена возникает на фоне длительного напряжения мышц руки и большого пальца, а также в результате сильного травмирования кисти. Кроме того, существенную роль играет анатомия футляра, окружающего сухожилия у каждого отдельного пациента (наличие дополнительных фиброзных образований, присутствие узких разветвлений и др.). Нередко болезнь де Кервена и симптомы данного заболевания проявляются у людей, осуществляющих определенную профессиональную деятельность (копирайтеры, программисты, пианисты и т. д.).
Если говорить о наиболее распространенных признаках недуга, то к их числу относятся болевые проявления и отечность местного характера. Как правило, для этого патологического процесса характерно медленное течение. Острые симптомы возможны в случае травмирования кисти руки. На начальной стадии больной констатирует эпизодические боли, появляющиеся во время отведения большого пальца или при внезапном развороте запястья в сторону пятого пальца.
Усиление болевых проявлений при болезни де Кервена наблюдается не сразу. Однако со временем даже незначительная двигательная активность кисти начнет доставлять пациент ощутимый дискомфорт. В отдельных ситуациях болевой синдром распространяется в область плеча или ногтевой пластины. Также могут появляться отёки в зоне сухожильного канала. В запущенных случаях боль приобретает интенсивный характер, что препятствует нормальному шевелению пальца.
Какие категории пациентов входят в группу риска?

Нередко вышеуказанную патологию называют стенозирующим тендовагинитом или «запястьем матери». При этом в число наиболее подверженных заболеванию лиц входят пациенты, выполняющие однотипные движения кистью в течение длительного периода времени (толкание, хватание, использование клавиатуры или мыши для компьютера).
Более того, вызвать болезнь де Кервена и нарушить функции большого пальца могут занятия шитьем, вязанием или игрой на фортепиано. Также спровоцировать недуг может работа, связанная с частым применением строительных инструментов. Достаточно часто болезнь де Кервена и нарушение движений в кисти руки проявляется в результате травмирования области запястья или на фоне воспалительного заболевания суставов. Что касается названия «запястье матери», то его происхождение часто связывают с продолжительным ношением женщинами малышей на руках или с выполнением ими домашних обязанностей.
Диагностика болезни де Кевена
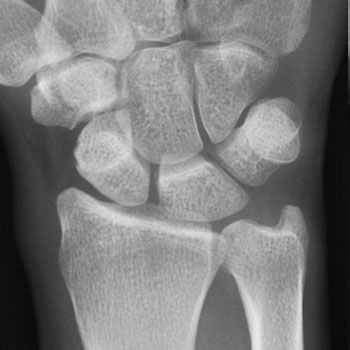
Обязательным этапом диагностики является осмотр кистей, что позволяет выявить нарушения, характерные для зоны поражения. Следует отметить, что вокруг больной зоны не наблюдается повышение температуры и другие изменения со стороны кожных покровов. Однако известны определенные методы, позволяющие четко констатировать у пациента стенозирующий лигаментит:
- Тест Финкельштейна. Применяется с целью обнаружения данного недуга при наличии у пациента болевого синдрома в области лучезапястного сустава. В ходе диагностики человека просят прижать первый палец к ладони, затем соединить его с другими пальцами, и в завершении - согнуть кисть в направлении пятого пальца. При этом во время двигательной активности пациента отмечает резкий дискомфорт в пораженной области. Кроме того, вышеуказанный недуг может привести к утрате возможности удерживать предметы посредством большого пальца. Проверить этот момент можно, если дать пациенту сразу несколько вещей (например, коробку со спичками и ручки): при наличии заболевания он не сможет держать предметы первым и вторым пальцами;
- Для подтверждения диагноза болезнь де Кервена может потребоваться рентген. На полученных снимках можно заметить наличие кальциноза в области первого костно-фиброзного канала;
- МРТ. При помощи этого метода удается исключить наличие какого-либо другого недуга.
Если у пациента констатирована болезнь де Кервена, лечение начинают с консервативных способов:
- Снижение нагрузок;
- Рекомендации, связанные с применением мази, обладающей противовоспалительным действием;
- Использование специальной шины для фиксации первого пальца.
В некоторых случаях могут потребоваться стероидные препараты, которые необходимо вводить в область сухожилия.
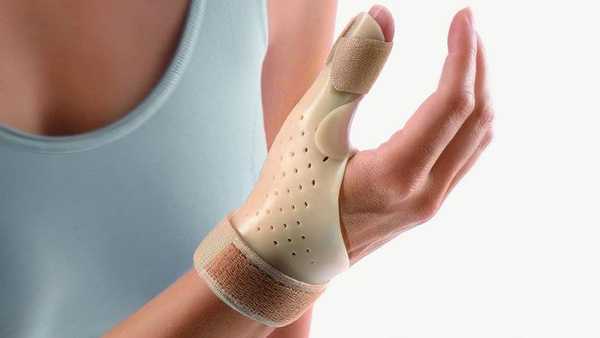
Если диагностирована болезнь де Кервена, а лечение мазями не дает ожидаемого результата в течение 2-х недель, переходят к другим способом устранения недуга. Причем специалисты склоняются к применению гормональных средств с противовоспалительным эффектом. В наиболее острых ситуациях выполняют оперативное вмешательство.
При выборе тактики во внимание принимаются следующие моменты: наличие хронических патологий и характерных симптомов.
Если говорить о практической стороне и опыте сотрудников, то получить необходимый положительный эффект можно только при своевременном консервативном лечении. Ключевая цель данного способа заключается в купировании воспалительного процесса, устранении болевых проявлений, укреплении мышц и связок, а также в предотвращении дальнейшего развития болезни.
Также приоритетным способом является использование инъекции. При сильном болевом синдроме и значительном воспалении назначают кортикостероидные средства. При этом последствия при применении подобных препаратов часто бывают негативными. В целом свыше 90% пациентов отмечают положительную динамику при применении лекарственных средств.
Снижение нагрузки с кисти при болезни де Кервена
За счет соблюдения эргономического режима удается минимизировать нагрузки на кисть. Чтобы достигнуть подобного результата, следует отказаться от повторяющихся движений. Также требуется сократить период осуществления монотонной деятельности. Желательно уменьшить время пребывания за ПК и выполнения спортивных тренировок. Кроме того, следует исключить длительное использование смартфона. Верхняя конечность должна чаще пребывать в спокойном состоянии.
Физиотерапия при болезни де Кервена
Физиотерапевтические методы направлены на купирование болевых проявлений и устранения воспаления. Однако при наличии тахикардии, повышенной температуры, сбоях в работе кровеносной системы и онкологических процессах такие процедуры проводить запрещается.
Особой эффективностью для пациентов, у которых была диагностирована болезнь де Кервена и симптомы данной патологии, обладают: воздействие ударными волнами, лазерная терапия, лечение холодом, применение статического магнитного поля.
Часто активное развитие патологии связывают с наличием ревматоидного артрита. В подобных ситуациях, если у пациента подтверждена болезнь де Кервена, лечение в Москве будет включать следующие мероприятия:
- Воздействие статическим магнитным полем;
- Введение в глубокие слои кожи активных веществ под действием ультразвука высокой частоты;
- УФО;
- Нанесение парафиново-озокеритовой аппликации;
- УВТ.
Лечение болезни де Кервена методом ударно-волновой терапии
При воздействии на пораженный участок ударными волнами удается быстро устранить спаечные процессы, при этом здоровые ткани остаются нетронутыми. Также обеспечивается активизация работы иммунной системы, купируются симптомы воспаления, формируются новые кровеносные сосуды, улучшаются обменные и восстановительные процессы, нормализуется кровоток.
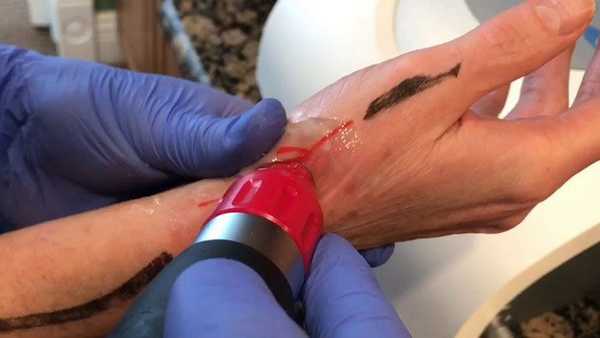
Как правило, полный курс УВТ включает несколько процедур, с перерывом 6-7 дней. Пациенты начинают ощущать облегчение уже после первых двух сеансов. Снижается интенсивность болевых проявлений и уходит отёчность.
После выполнения завершающей процедуры восстанавливаются движения в области кисти. Однако ключевой задачи такой терапии является полная нормализация работы сухожильного аппарата, и устранение последствий сужения канала.
Для эффективной реализации ударно-волнового метода не требуется госпитализация больного. Процедуру выполняют в амбулаторных условиях. После удобного расположения пациента на кушетке, специалист наносит на область поражения гелеобразное вещество, усиливающее действие ударных волн. Также в ходе УВТ применяется специальный датчик.
Длительность одной процедуры не превышает 20 минут. При этом некоторые пациенты могут ощущать небольшие покалывания с последующим покраснением кожных покровов, которое через несколько часов после завершения сеанса.
Ортезирование и тейпирование
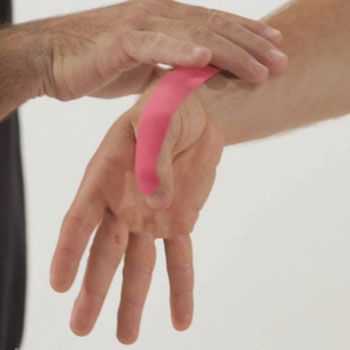
Одним из обязательных этапов устранения недуга считается ортезирование, что предполагает использование специальных приспособлений, позволяющих зафиксировать кисть и пальцы в правильном положении. Длительность иммобилизации может достигать 35-40 дней, в зависимости от скорости купирования болевых ощущений.
Данная методика является незаменимой для пациентов, у которых имеются ограничения для применения кортикостероидных гормонов.
Хирургическое вмешательство
В случае, если консервативные способы не дали ощутимого эффекта, следует прибегнуть к помощи хирурга. Оперативное вмешательство выполняется в стационаре с использованием анестезирующих средств местного действия. После завершения манипуляции на руку накладывают повязку косыночного типа. Снятие швов осуществляют через 9-10 суток, а восстановление работоспособности верхней конечности наблюдается через 14 дней после вмешательства. В период реабилитации некоторые пациенты отмечают появление мурашек и утрату чувствительности в области первых трех пальцев кисти. Эти проявления исчезают через 14-20 суток после хирургической манипуляции.
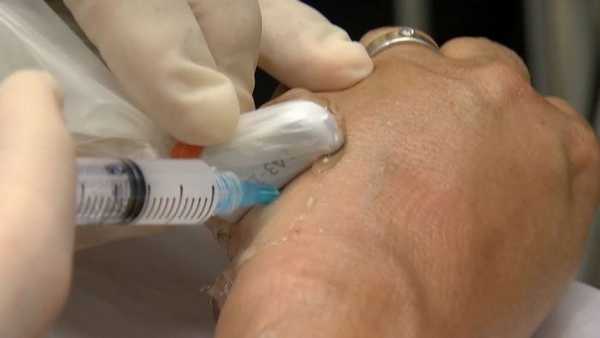
В нашей клинике осуществляется оперативное вмешательство под руководством опытных специалистов. Однако наиболее приоритетным направлением деятельности является реализация процедуры УВТ. При этом используется аппаратура европейского качества, а на первую консультацию предоставляется 20%-ная скидка.
Прогноз выздоровления
Как правило, при своевременном и грамотном лечении специалисты гарантируют положительную динамику. В случае реализации консервативных процедур рассчитывать на благоприятный прогноз удается в 50% ситуаций. После оперативного вмешательства удается добиться максимального положительного эффекта. Однако, если после хирургической манипуляции пациент продолжает перегружать кисть, может наблюдаться рецидив заболевания. Это говорит о необходимости изменения характера трудовой деятельности больного после завершения лечебных мероприятий.
Эксперт статьи:
Татаринов Олег Петрович

Врач высшей категории, врач невролог, физиотерапевт, специалист УВТ, ведущий специалист сети «Здоровье Плюс»
Синдром Дежерина
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Под синдромом Дежерина подразумевают заболевание, которое встречается достаточно редко. Оно основано на генетической предрасположенности. Синдром Дежерина также называют гипертрофической невропатией. Можно сразу сказать, что заболевание неизлечимо, потому что все заболевания, причиной которых являются различные мутации и изменения в генах вылечить невозможно.
Первое описание заболевания принадлежит французскому неврологу Дежерину, который изначально предполагал, что заболевание корнями своими уходит глубоко в генетику. Он заметил, что заболевание передается из поколения в поколение, наблюдается в пределах одной семьи. Он также провел экспериментальные исследования, которые позволили заключить, что за передачу заболевания отвечают доминантные гены. Таким образом, в генетической консультации можно заранее просчитать, родится ли ребенок здоровым, или у него будет развиваться синдром Дежерина.
К сожалению, предотвратить его развитие нельзя никак. Если заболевание передалось ребенку, оно неизбежно будет развиваться.
Код по МКБ-10
Эпидемиология
Разновидностей синдромов Дежерина в настоящее время наблюдается множество. Однако все они имеют сходные черты - проявляются в возрасте от рождения до 7 лет. При этом на первом году жизни проявляется примерно 20 % случаев заболевания. На втором году жизни заболевание дает о себе знать в 16% случаев.
По частоте встречаемости преобладает синдром Дежерина-Сотта. Он регистрируется примерно в 43% случаев. Примерно в 96% случаев заканчивается полной инвалидностью, человек прикован к инвалидному креслу.
Второе место принадлежит синдрому Дежерина-Клюмпке, он встречается примерно в 31% случаев. Третье место отводится синдрому Дежерина - Руссо, частота встречаемости которого составляет примерно 21% случаев. При этом для синдрома Дежерина - Русси свойственно формирование устойчивых симптомов в течение года у тех пациентов, которые перенесли инсульт, или другое нарушение мозгового кровообращения в острой форме.
Причины синдрома Дежерина
Главная причина синдрома Дежерина - генная мутация, которая передается по аутосомно-генетическому типу. Вместе с тем, многочисленные генетические факторы могут оказывать воздействие на формирование патологии. Они влияют на человека, его головной мозг. К числу основных причин заболевания относят:
- травмы, повреждения, другие негативные воздействия. Особенно это касается черепно-мозговых нервов. Также заболевание может быть следствием сотрясений мозга;
- переломы костей, расположенных у основания черепа;
- воспаление мозговых оболочек, которое проявляется в острой форме. Воспаление может быть различной природы. Может быть обусловлено инфекционными агентами, воспалением, аллергической реакцией. Также развитие синдрома может быть следствием травмы;
- воспаление мозговых оболочек различной природы, перешедшее в хроническую форму;
- повышенное внутричерепное давление.
Факторы риска
Существуют определенные факторы риска, которые способны провоцировать заболевание. Люди, подверженные воздействию этих факторов, сильнее подвержены заболеванию, чем другие. К факторам риска относят также некоторые заболевания, которые сопровождают патологию.
К группе риска принадлежат пациенты, страдающие опухолями мозга. Фактором риска можно считать опухоль, которая оказывает давление на продолговатый мозг. Также в эту группу относятся различные туберкулемы, повреждения сосудов, а также саркоидозы. Повреждение мозга происходит в результате давления на головной мозг. Повреждение сосудов головного мозга может иметь различный характер. Прежде всего, это касается геморрагических поражений, эмболий, тромбозов, аневризмов, мальформаций.
Также одним из факторов, способствующих развитию синдрома Дежерина, являются такие сопутствующие заболевания, как полиэнцефалит, рассеянный склероз, полиомиелит. Опасность могут представлять и те заболевания, которые сопровождаются нарушением нормального функционирования головного мозга, нарушением его кровоснабжения. В первую очередь следует опасаться нарушений тока крови в артериальном русле. К группе риска относят также пациентов, подверженных нарушению кровоснабжения двенадцатого нерва, его ядра, медиальной петли, пирамиды.
Развитию заболевания также способствуют сирингобульбии, бульбарные параличи. Эти факторы представляют собой большой риск, поскольку им свойственно постоянное прогрессирование.
Также в качестве фактора риска можно рассматривать опухоли мозжечка различного характера.
К группам риска отнесены врожденные пациенты с врожденными аномалиями головного мозга. Если человек с такой аномалией подвергается воздействию инфекционных, токсических, дегенеративных агентов, риск развития заболевания существенно увеличивается. Такие факторы, как едкие химические вещества, радиоактивные вещества могут провоцировать развитие патологии. Они могут вызвать генную мутацию. Поэтому женщины, подверженные воздействию токсических, химических веществ, а также проживающие в зоне повышенной радиации, могут попасть в группу риска. Предрасположенность к заболеванию в таком случае резко возрастает.
Патогенез заболевания обусловлен генной мутацией. Она способствует нарушению строения оболочек стволовых нервов. При развитии заболевания отмечается чрезмерное разрастание соединительных оболочек, которые входят в состав нервной ткани. В результате соединительная ткань гипертрофируется, между нервными соединениями откладывается мукозное вещество. Это приводит к существенному утолщению стволов нервов, спинномозговых корешков и мозжечковых путей. Меняется их форма. Дегенеративные процессы охватывают нервную ткань и спинномозговые нервы.
Симптомы синдрома Дежерина
Синдром Дежерина может проявлять себя абсолютно по-разному. Необходимо понимать, что существует много разновидностей этого заболевания, и каждая из них проявляет себя абсолютно не похожими друг на друга признаками. Поэтому имеет смысл говорить о признаках, свойственных каждому отдельному виду этого синдрома.
Вместе с тем, существует ряд ранних признаков, которые в общих чертах могут подсказать о вероятности развития у ребенка патологии. На ранних стадиях различные виды могут иметь многочисленные сходные черты.
Первые признаки
В большинстве случаев заболевание уже в полной мере проявлено в дошкольном возрасте. Однако первые его признаки можно заподозрить практически с рождения ребенка. Если ребенок развивается медленнее, чем его сверстники, это может быть первым тревожным признаком. Нужно обратить особое внимание на ребенка, который в положенном возрасте еще не сидит, поздно делает первый шаг, начинает самостоятельно передвигаться.
Внешний вид ребенка также может о многом сказать. Обычно у ребенка опущены мышцы лица. Руки и ноги постепенно начинают деформироваться. Они становятся менее чувствительными, практически ни на что не реагируют. Это состояние может все время усугубляться, вплоть до тех пор, пока мышцы не атрофируются.
Как только ребенок начинает развиваться неправильно, нужно обращаться к врачу. Необходима консультация невролога.
При проведении осмотра врач обнаруживает дополнительные признаки, которые указывают на синдром. Наблюдается фибриллярное подергивание мускулатуры. Многие сухожильные рефлексы не проявляются. Зрачки могут быть сужены и в большинстве случаев не проявляют реакции на свет. Врач подтверждает признаки ослабления мимической мускулатуры.
Стадии
Различают легкую (начальную) стадию, среднюю, тяжелую. На начальной стадии появляются первые признаки заболевания. Эта стадия приходится обычно на младенческий возраст.
Средняя стадия - ярко выраженная задержка речевого и двигательного развития, различные двигательные расстройства, нарушение чувствительности, выпадение некоторых рефлексов, нарушение зрительных реакций.
Тяжелая стадия - нейросенсорная тугоухость, скелетные деформации, нарушение тонуса мышц, нистагмы. Прогрессирование болезни. Заканчивается инвалидностью.
Формы
Существует огромное множество разновидностей синдрома Дежерина, в зависимости от типа и тяжести поражения. Наиболее часто встречаются альтернирующий синдром, синдром Дежерина Сотта, синдром Дежерин Клюмпке, синдром Дежерина Руссе.
Альтернирующий синдром Дежерина
Если у ребенка альтернирующий синдром, у него в первую очередь парализуется язык. Причем поражается не весь язык, а только часть его. На противоположной стороне развивается гемипарез. Чувствительность к вибрации достигает глубинных слоев. Тактильные ощущения ребенок практически не различает. Причиной является тромбоз или окклюзия базилярной артерии. Именно это нарушает иннервацию и кровоснабжение продолговатого мозга.
Синдром Дежерин Клюмпке
При синдроме Дежерина Клюмпке парализуются нижние ветви плечевого соединения. Параличу подвергается не вся конечность, а только ее часть. Постепенно развивается парез и паралич кистей. Резко снижается чувствительность соответствующих участков. Изменяется состояние сосудов. Зрачковые реакции неправильны.
Паралич постепенно распространяется и на глубинные слои мышечного каркаса. Наблюдается сильное онемение. Сначала немеют кисти, потом предплечья, локти. В тяжелых случаях может поражаться даже грудной нерв. Также развиваются многочисленные птозы и миозы.
Синдром Дежерина Русси вмеде
Для этого синдрома свойственно поражение перфорирующих артерий. Также повреждаются участки вокруг артерии, и те зона головного мозга, которые иннервируются пораженной артерией. Также этот синдром называют синдромом хронической боли, или синдромом таламической (постинсультной) боли.
Это название объясняется тем, что синдром сопровождается интенсивной болью, постоянной, пронизывающей. Боли часто бывают невыносимые. Также заболевание сопровождается чувством ломоты, выкручивания всего тела. Также наблюдается гиперпатия, в результате которой одни мышцы приходят в чрезмерный тонус. Однако чувствительность при этом резко снижается. Также для болезни свойственны приступы панического, неестественного плача, крика, или смеха.
При этом повреждениям подлежит преимущественно одна сторона. Это может быть одна нога, или одна рука. В пораженных участках в первую очередь наблюдается сильная боль, ощущение жжения. Боль изматывает пациента. Может усиливаться под действием различных факторов. Боль усиливать могут как положительные, так и отрицательные эмоции. Под действием тепла, холода, различных движений также боль может усиливаться.
Часто заболевание бывает сложно дифференцировать, отделить от других заболеваний. Оно имеет множество признаков, сходных с другими невралгическими поражениями. Иногда окончательно установить можно только после того, как полностью сформировался болевой синдром.
Синдром Дежерина Сотта
Синдром Дежерина Сотта - разновидность заболевания. Заболевание генетическое. В ходе этого заболевания нарушается толщина стволовых нервов. Заболевание можно диагностировать на самых ранних этапах беременности при помощи генетического консультирования. При рождении ребенок ничем не отличается от здорового ребенка. Потом, по мере роста и развития, становится заметно, что ребенок очень медленно развивается. Скудные движения, речь несформирована. Мышцы очень расслаблены, ребенок не способен держать голову, шею, туловище. Нарушены зрительные реакции. Ребенок очень отстает в развитии от сверстников. Прогрессирует снижение чувствительности, мышцы постепенно атрофируются. Полноценного развития не происходит. Постепенно атрофия переходит на костную систему. Заканчивается инвалидностью.
Синдром Нери Дежерина
При синдроме Нери Дежерина постоянно раздражаются задние корешки спинного мозга. Причиной этому становится остеохондроз, различные опухоли, которые поражают головной мозг, и давят на него. Грыжи, защемления, травмы также способствуют давлению на корешки. Кроме того, это может происходить из-за сильных костных разрастаний. Основным проявлением является сильная боль в том месте, где происходит давление на мозг и его корешки.
В большинстве случаев этот синдром является не основным, а сопутствующим, при различных других патологиях и заболеваниях. Например, традиционно сопровождает остеохондроз. Отличительной чертой является резкая боль в зоне поясницы, и тянущая боль в области шеи, головы, которая не дает возможности человеку поднять полностью голову из лежачего положения. Постепенно эта зона затвердевает, чувствительность постепенно утрачивается. Также наблюдается мышечный спазм. Постепенно конечности подвергаются патологическим изменениям.
Синдром Ландузи Дежерина
Синонимом является миопатия. Название заболевания указывает на ослабление мышц, которое все время прогрессирует. Параллельно наблюдается развитие различных патологий в мышцах, дистрофические процессы. Можно сказать, что это не отдельное заболевание, а целая группа болезней. Поражается плечевая, лопаточная и лицевая сторона. Заболевание является генетической патологией, передается из поколения в поколение.
Развивается в несколько этапов. На первом этапе развивается мимическая слабость, в результате которой мышцы лица не только ослабляют, но и теряют форму, искажаются. В результате лицо приобретает неправильные, искаженные черты. Чаще всего распознать заболевание можно по округленному рту и опущенной нижней и верхней губе.
Постепенно заболевание настолько прогрессирует, что человек уже не может закрыть рот. Он оставляет рот открытым сначала во время сна, потом даже в дневное время. Постепенно мышечная слабость затрагивает мышцы плечевого пояса.
В редких случаях может ослабляться мускулатура глоточных мышц, языка. Но этот признак не имеет диагностического значения и выражен не настолько ярко, как остальные признаки.
На самом тяжелом этапе у человека развивается слабость скелетных мышц. В первую очередь ослабевают руки, потом ноги. Прогноз неутешительный - инвалидность.
Читайте также: