Синдром Дюшенна I (Duchenne I) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 22.01.2026
Актуальность. Среди миодистрофий наиболее часто в детском возрасте диагностируются прогрессирующую мышечную дистрофию Дюшенна, Эмери-Дрейфуса, Эрба-Рота [2, 8]. Миодистрофия Дюшенна - наследственное заболевание, занимает второе место в мире по частоте встречаемости (1:5000 новорожденных мальчиков) [1, 4]. Мышечная дистрофия Дюшенна является наиболее разрушительной из всех мышечных дистрофий [5].
Генетический фактор - несовершенный ген - передается ребенку от матери, но у них самих нарушения не развиваются. Имеется 50-процентный шанс, что у каждого сына женщины-носительницы генетического фактора разовьется болезнь и 50% шанс, что каждая ее дочь станет носителем этого фактора [3, 6].
Несмотря на почти вековую историю изучения данной патологии, вопросы их патогенеза, достоверной диагностики и лечения остаются до сего времени неразрешенными. Существует большое количество классификаций, но отсутствие точных данных о первичном биохимическом дефекте не дает возможности построить ее по рациональному принципу [1, 7].
Поражения внутренних органов значительно утяжеляет течение основного заболевания. Пациенты теряют способность самостоятельно передвигаться между 7-ю и 13-ю годами, а погибают, нередко, в подростковом возрасте или на III декаде жизни; при этом вовлеченными в патологический процесс оказываются практически все органы и системы организма [3].
Таким образом, анализ данных литературы убеждает в необходимости проведения тщательного соматического обследования пациентов с прогрессирующей мышечной дистрофией (ПМД) с целью как можно раньше выявить нарушения и принять меры для своевременной коррекции и поддержания функции жизненно важных органов.
В связи с вышеизложенным, целью данного исследования явилось изучение клинико-диагностических особенностей мышечной дистрофии Дюшенна у детей.
Материалы и методы исследования. Проведен клинический анализ 37 детей с диагнозом прогрессирующая мышечная дистрофия Дюшенна. Возраст детей варьировал от 3 до 15 лет, средний возраст составлял 7,8±0,48 лет. Возраст к началу заболевания в среднем составлял 4,3±0,36 лет и варьировал в пределах от рождения до 8 лет.
Биохимическое исследование включало определение уровня ферментов крови трансаминаз (АЛТ и АСТ), креатинфосфокиназы (КФК), лактатдегидразы (ЛДГ).
Генеалогическим методом обследовано 240 родственников I степени родства (родители, сибсы) детей с ПМД. Составлена подробная родословная, куда входили сведения о заболеваниях в 3 поколениях семьи. Генеалогический материал собирался по обеим родительским линиям путём перекрёстного опроса обоих родителей, иногда бабушек и дедушек.
Результаты и их обсуждение. При анализе акушерского анамнеза было установлено, что беременность протекала в 32,4% на фоне анемии, в 10,8% - токсикоза. 10,8% матерей во время беременности перенесли острые респираторные вирусные инфекции (ОРВИ). Обострение хронических заболеваний во время беременности регистрировалось у 2,7% матерей больных детей. Возраст матери при рождении ребенка с ПМД в среднем составил 26,0±0,85 лет.
Число членов семей, у которых отмечалась ПМД составило 13,5%.
По результатам наших исследований в 4 (10,8%) родословных выявлены случаи ПМД у родных братьев и в 1 случае (2,7%) у дяди по материнской линии.
Из анамнеза жизни установлено, что в 13,5% регистрируется родственный брак. Так же было отмечено, что в большинстве случаев дети были рождены от 2 и 3 беременности (2,2±0,19) или 2-3 родов (2,15±0,17).
Двое детей были из двойни, хочется обратить внимание, что один из детей двойни был здоров.
Роды в 89,2% случаев протекали нормально, в 5,4% случаев стремительно и по 1 (2,7%) случаю наблюдались длительный безводный период и оперативные роды. Вес при рождении детей составлял в среднем 3130±107,3 гр. В асфиксии были рождены 8 детей, что составило 21,6%. Обвитие пуповиной регистрировалось у 5,4% детей.
Анализ развития у детей психомоторных навыков показал, что дети в большинстве случаев поздно начинали удерживать голову (48,6%), сидеть (после 9 месяцев и позже 51,3%). Психомоторное развитие в 35,1% случаев не соответствовало возрастной норме ещё до дебюта проявлений миодистрофии Дюшенна.
В 78,4% случаях в предварительном диагнозе выставлялся ПМД. Из 37 наблюдаемых больных только 1 пациент находился на диспансерном учете, остальные же обращались впервые.
По данным литературы симптомы обычно проявляются в возрасте 2-5 лет, когда мышцы нижних частей тела и позвоночника поражаются первыми. Согласно нашим данным, первые симптомы заболевания проявились в среднем 4,6±0,35 лет.
Основными клиническими симптомами мышечной дистрофии Дюшенна являются: нежелание ходить или замедленная ходьба; ненормальная ходьба, часто походка вперевалку или качающаяся походка; ходьба на пальцах ног; неспособность нормально прыгать или бегать; трудности подъема по лестнице, при входе или выходе из автомобиля; частые падения. В связи с ходьбой на пальцах ног у этих детей развивается переднее наклонное положение таза и, соответственно, деформация спины.
У 67,6% обследованных детей наблюдались проблемы с зубами, характеризующиеся расширением челюсти и расширение промежутка между зубами.
При осмотре наблюдалось увеличение объема мышц, особенно икр; ноги, как правило, поражаются симметрично. Наиболее часто псевдогипертрофии наблюдались у детей старше 6 лет и по мере дальнейшего прогрессирования болезни имели тенденцию к уменьшению.
У 21,6% наблюдаемых детей развились ранние мышечные контрактуры и ретракция пяточных (ахилловых) сухожилий.
Мышечные атрофии первоначально локализовались в мышцах тазового пояса, с максимальной выраженностью в проксимальных отделах нижних конечностей. С возрастом имели тенденцию к распространению в восходящем направлении на мышцы плечевого пояса, спины и проксимального отдела верхних конечностей.
При детальном неврологическом осмотре в 97,3% случаев наблюдается утиная походка, активные движения ограничены, с затруднением самостоятельной ходьбы в 72,9% случаев.
Обращает на себя внимание снижение и утрата коленных рефлексов при длительном сохранении ахилловых рефлексов. У детей в более старшем возрасте наблюдается снижение рефлексов с m.biceps et m.triceps.
Во всех наблюдениях отмечается тотальная мышечная гипотония и снижение мышечной силы, более выраженные в ногах. Мышечная сила в руках составляла в среднем 2,2±0,07, в ногах 1,25±0,03.
Судорожный синдром отмечен у 2 детей, что составило 5,4%, тремор у 1 ребенка - 2,7%.
Отличительной чертой миодистрофии Дюшенна является сочетание атрофии мышц с патологией костно-суставной, сердечно-сосудистой и нейроэндокринной систем.
В соматическом статусе у обследованных детей с миодистрофией Дюшенна выражена костная патология на ранних стадиях заболевания типичными нарушениями являются: поясничный лордоз (100%), кифосколиоз (94,6%), сколиоз (5,4%), деформации грудной клетки по типу "килевидной" или "ладьевидной" груди (32,4%), высокий свод стопы (100%).
По мере прогрессирования процесса развивается эквиноварусная деформация стоп и контрактуры крупных суставов.
Сердечно-сосудистые расстройства клинически проявлялись лабильностью пульса, артериального давления (АД), глухостью тонов сердца. На электрокардиограмме (ЭКГ) регистрировались изменения миокарда, блокада ножек пучка Гиса.
Наиболее частыми нарушениями со стороны сердца среди обследованных больных являлись: выраженная тахикардия, аритмии и развитие сердечной недостаточности.
На ЭКГ у больных с миодистрофией Дюшенна регистрировался глубокий зубец Q в отведениях 2, 3, aVF, V6. и высокий зубец в отведении V6.
Полученные данные свидетельствуют о поражении миокарда в области задне-нижней и латеральной стенок левого желудочка.
У больных в развернутой стадии заболевания наиболее часто выявляется гипертрофическая кардиомиопатия (51,4%) и дилятационная (27%), реже встречались - пролапс митрального клапана и миксома левого желудочка (21,6%).
У 40,5% больных наблюдались нейроэндокринные нарушения, из них у 53,3% регистрировался синдром Иценко-Кушинга и у 46,7% адипозогенитальная дистрофия.
У 35,1% обследованных детей наблюдались психомоторные навыки не соответствующие возрасту, а в более старшем возрасте умственная отсталость.
Важным показателем состояния нервно-мышечного аппарата служит М-ответ. У всех обследованных детей данный показатель ниже как по tibialis anterior D, так и tibialis anterior S по сравнению с контрольной группы почти в 3,5 раза (Р<0,001). Скорость распространения возбуждения по нерву (СРВ) не отличалась от показателей контрольной группы с обеих сторон.
Таким образом, у всех обследованных детей в проксимальных отделах мышц нижних конечностей по данным ЭНМГ отмечалось первично-мышечное поражение с тенденцией к прогрессии и недостаточной реинервацией, которые были более выражение с возрастом.
Показатели лабораторных исследований были определены в сравнении с показателями здоровых детей (n=20). Данный анализ показал, что уровень КФК в основной группе был повышен в 71 раз, АЛТ в 15,2 раза, АСТ 8,4 раза, ЛДГ в 4,4 раза (рис. 1).
Высокая активность плазменных ферментов, прежде всего КФК отражает темп деградации миофибрилл в мышечных волокнах. Параллельно с повышением КФК наблюдалось увеличение концентрации и других ферментов цитолиза - лактатдегидразы и трансаминаз.
Увеличение уровня ЛДГ в крови свидетельствует о накоплении лактата крови, что в свою очередь приводит к гипоксии и вызывает чувство мышечной усталости, нарушает процесс тканевого дыхания.
Увеличение уровня трансаминаз (АЛТ и АСТ) в крови говорит всегда о большем, чем в норме, разрушении клеток соответствующего органа, в нашем случае мышцах.
Заключение. Таким образом, диагностика прогрессирующих мышечных дистрофий является сложной задачей, для решения которой необходимы создание и использование алгоритмов диагностики.
Диагноз прогрессирующая мышечная дистрофия Дюшенна основывается на данных клинико-генеалогического анамнеза, клинических особенностей заболевания (начало в 2-5 лет, симметричная атрофия проксимальных групп мышц, их развитие в восходящем направлении, псевдогипертрофия икроножных мышц, грубые соматические и нейроэндокринные нарушения, быстрое злокачественное течение заболевания).
Рис. 1. Лабораторные показатели у обследованных детей в сравнительном аспекте
Имеют значение также данные биохимических исследований (повышение в сыворотке крови уровня КФК в 71 раз выше от нормы на фоне повышения АЛТ, АСТ и ЛДГ), электромиографии и патоморфологии, при которых определяют первично-мышечный тип поражения.
Список использованных источников:
2. Джурабекова А.Т., Хамркулова Ф.М., Юлдашева З.Т. Прогрессирующая мышечная дистрофия Дюшенна, новый подход к лечению// Умумий амалиёт доктори ахборотномаси. - 2006. - №1-2. - С.59-61.
3. Евтушенко С.К., Шаймурзин М.Р., Евтушенко Л.Ф. Медикаментозное и немедикаментозное лечение кардиомиопатии и пневмопатии при прогрессирующих нервно-мышечных заболеваниях у детей// Таврический медико-биологический вестник.-2009. - Т.12, №2. - С. 46.
4. Подагова Е.В., Мальмберг С.А., Дадали Е.Л. Псевдогипертрофические прогрессирующие мышечные дистрофии: алгоритмы диагностики // XIV Росс. нац. конгр. «Человек и лекарство». -М. - 2007.
5. Шаймурзин М.Р., Евтушенко С.К. Новые современные технологии в терапии нервно-мышечных заболеваний, направленных на замедление их прогрессирования// Вестник физиотерапии и курортологии.- 2010.- №6. - С.40-41.
6. Andersen S.P., Sveen M.L., Hansen R.S., Madsen K.L., Hansen J.B., Madsen M., Vissing J. Creatine kinase response to high-intensity aerobic exercise in adult-onset muscular dystrophy// Muscle Nerve. - 2013. - vol.10. - P. 1002.
8. Snow W.M., Anderson J.E., Jakobson L.S. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: A review// Neurosci Biobehav Rev. - 2013. - S0149-7634(13). - P. 76-86.
Подписано в печать: 01.09.2013
Лечится ли миодистрофия Дюшенна?

Почему пока у больных МДД есть только одна попытка патогенетической терапии в жизни
Мы живем в век развития генной терапии: ученые изобретают лекарства, которые действуют на уровне генома человека, замещают «поломанные» участки ДНК. Самые известные и самые дорогие в мире препараты такого рода - Спинраза, Золгенсма и Рисдиплам - применяются при спинальной мышечной атрофии (СМА) у детей. Золгенсма даже занесен в Книгу рекордов Гинесса как самое дорогое лекарство в истории.
Совсем недавно появились лекарства, замедляющие прогрессирование другого генетического заболевания - миодистрофии Дюшенна (МДД). Но с Дюшенном все гораздо сложнее. Что это за препараты и в чем сложность - рассказывает Татьяна Андреевна Гремякова, д.м.н. и президент благотворительного фонда «Гордей».
Лечится ли миодистрофия Дюшенна?
Миодистрофия Дюшенна встречается так же часто, как СМА (спинальная мышечная атрофия), но болеют практически только мальчики. Согласно статистике, один из 3500-5000 мальчиков в мире рождается с мышечной дистрофией Дюшенна.
Причина заболевания - «поломка» в Х-хромосоме, а точнее в гене, который отвечает за синтез белка дистрофина. Мутации этого гена и отсутствие дистрофина приводят к дегенерации мышц и прогрессирующей мышечной слабости. Такие дети живут обычно всего лишь 12-25 лет.
Кстати, подробно об этом заболевании: как оно возникает, почему педиатры путают его с гепатитом, и чем можно помочь пациентам, - мы рассказывали в статье «Миодистрофия Дюшенна: что это такое?».
Аналогия с препаратами против СМА, на этом заканчивается. Если Золгенсма, Спинраза, Рисдиплам - в идеальном варианте, при применении по схеме и как можно раньше, в младенческом возрасте, - позволяют любому ребенку с заболеванием развиваться с минимально возможными отклонениями, то Аталурен даже в идеальной ситуации сможет помочь лишь 13% детей с МДД (тем, чья болезнь вызвана нонсенс-мутацией), и принимать его нужно будет постоянно .
Миодистрофия Дюшенна: что это такое? Как часто и почему возникает это заболевание, почему педиатры нередко путают его с гепатитом, что делать, если вашему ребенку поставлен такой диагноз
Терапия СМА и Дюшенна: в чем разница?
При всей катастрофичности СМА молекулярная и клеточная основа этого заболевания проще и понятнее, чем у миодистрофии Дюшенна.
- Ген дистрофина в 1500 раз больше, чем «ген СМА»
Причина СМА - мутация в гене SMN1, отвечающем за работу моторных нейронов. Их относительно немного, они не делятся и все локализованы в пределах головного и спинного мозга. Ген, отвечающий за развитие СМА, небольшого размера - 1,5 кБ, легко помещается целиком в вирус, который доставляет его в поврежденную клетку, в 95% случаев это одна и та же мутация - делеция экзона 7.
Лекарство можно ввести в спинной мозг, оно не размывается по всему организму, не метаболизируется печенью и не выводится почками. Создается его локальная высокая концентрация. Одной инъекции хватает на несколько месяцев. А если это генотерапия, то полноценный замещающий генетический материал, однажды попав в больную нервную клетку, вылечивает ее, остается там надолго, если не на всю жизнь.
В случае миодистрофии Дюшенна все сложнее: ген дистрофина — один из самых больших. Он больше гена СМА почти в полторы тысячи раз, и в нем тысячи разных мутаций (делеции, дупликации, нонсенс и т.д.) в разных местах.
Ген не помещается целиком в вирус, поэтому используют только кусочки гена — мини- и микродистрофин, которые могут ограниченно восстановить функциональность мышечных клеток, перевести «Дюшенн в Беккер» — более легкую форму миодистрофии, при которой человек может прожить до 60 лет, сохранять дееспособность, работать.
- Нужно доставить лекарства в мышечные клетки, а они постоянно делятся
Мышечные клетки составляют 40% от всех клеток тела, они активно работают и постоянно заменяются. Доставить лекарство в мышечные клетки - трудная задача: оно должно с кровью попасть во все клетки, а раз с кровью, то с лекарством борется печень, и оно выводится через почки. Итог - низкая концентрация и ограниченное время действия.
- У 30% детей имеется предсуществующий иммунитет к вирусу, доставляющему в организм «лекарство» (кусочек гена)
Генотерапия мини- и микродистрофином - подводные камни на каждом этапе. У ребенка может быть имеющийся иммунитет к вирусу-переносчику гена, таких детей примерно 30%. Компании сейчас работают над тем, как убрать антитела к вирусу из крови. И пока пациентам доступна только одна попытка генотерапии в жизни, потому что после инфузии уже точно будет выраженный противовирусный иммунитет.
- Такая терапия пока подойдет только детям до 8 лет
Вирусов должно быть столько, чтобы хватило на все мышечные клетки во всем организме. Вирусы не будут делиться внутри клетки - сколько их в клетку проникло, столько и будет генов для синтеза укороченного дистрофина в клетке. Чем больше ребенок, тем больше доза требуется. А если он еще мал, то мышечные клетки будут делиться, в результате будет образовываться много клеток без гена дистрофина. На сегодняшний день определено достаточно узкое терапевтическое окно для даной генотерапии, поэтому и возраст, в котором ребенка рекомендуется лечить с помощью такой терапии, пока - 4-8 лет. Но это те результаты, которые есть на сегодняшний день. Начинаются новые исследования, где возрастной диапазон уже шире - от 2 до 14 лет.
Я верю, что скоро мальчики с Дюшенном будут жить не так, как сейчас Личная история семьи Татьяны Андреевны Гремяковой, президента благотворительного фонда «Гордей»
- Огромная вирусная нагрузка на организм: нужны высокие дозы стероидов
И еще, десять тысяч миллиардов вирусных частиц на килограмм веса тела ребенка - запредельное количество вирусов, мы не встречаемся в жизни с таким дозами, они токсичны, от такой дозы можно умереть. Поэтому нужны высокие дозы стероидных гормонов перед, во время и после инфузии, чтобы снизить иммунный и воспалительный ответы организма на вирусное вторжение.
Но результат стоит того, тем более с осложнениями врачи научились бороться. И, конечно, будут другие технологии, лучше и безопаснее, которые смогут преодолеть существующие проблемы.
Больные МДД как снежинки: все разные
Сделать лекарство для МДД сложно, один препарат не вылечит всех, как при СМА.
Больные МДД как снежинки: все разные, нет одинаковых, уже описано около десяти тысяч мутаций гена. Имеет значение и вся генетика ребенка. Поэтому болезнь проявляется по-разному, даже в одной семье у двух братьев с одной мутацией.
Мы живем во время орфанной революции, когда многие редкие генетические неизлечимые заболевания получают патогенетической лечение, и больные обретают возможность жить долгой полноценной жизнью. Новые лекарства могут превратить фатальную болезнь в хроническую, хоть и тяжелую, как, например, диабет.
Как получить лекарства?
Если у вашего ребенка выявили миодистрофию Дюшенна, и вы хотите получить терапию для него, нужно прежде всего иметь подтвержденный генетический диагноз с определением мутации, которая вызвала заболевание. Примерно в 1% случаев не удается выяснить тип мутации. В таком случае делают биопсию мышцы и определяют количество синтезируемого мышцами дистрофина.
В 2021 году в России создан фонд « Круг Добра », его цель - обеспечить лекарственными препаратами детей с орфанными заболеваниями. Можно обратиться туда и получить препараты бесплатно.
Другой путь - бесплатно получать лекарства по жизненным показаниям через региональные бюджеты здравоохранения. Аталурен фонд « Круг Добра ». Это не согласуется с законом, поэтому стоит добиваться.
Что такое Перечень жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП) Рассказываем, что такое перечень жизненно необходимых и важнейших лекарственных препаратов, и почему внесение в него изменений так важно.
Как участвовать в клинических исследованиях?
Сейчас активно проводятся клинические исследования нескольких лекарственных препаратов. О том, какие из них исследуются в России, можно узнать на сайте Минздрава РФ. Заполнив форму, введя название препарата (Аталурен), можно узнать, в каком учреждении проводится исследование. Родителям необходимо самостоятельно связаться с учреждением, чтобы попробовать попасть в число участников.
Информацию о новых препаратах и клинических исследований в области миодистрофии Дюшенна можно найти и в пациентских сообществах, например, ProДюшенн.
Паралич Дюшена-Эрба
Паралич Дюшена-Эрба — мышечно-тонические, чувствительные и трофические нарушения, развивающиеся при повреждении верхнего ствола плечевого сплетения. Наиболее распространен акушерский паралич Дюшена-Эрба, возникающий при травмировании сплетения в ходе родовспомогательных пособий. Заболевание характеризуется нарушением отведения, супинации и поднимания плеча, а также сгибания в локте при относительной сохранности движений в пальцах кисти. Подтверждением диагноза являются данные рентгенографии и УЗИ плечевого сустава, ЭМГ, ЭНГ, КТ-миелографии или МРТ. Лечится паралич Дюшена-Эрба консервативными методами (физиотерапия, фармакотерапия, массаж, рефлексотерапия, ЛФК), при их нецелесообразности или неэффективности — оперативным путем.
Общие сведения
Паралич Дюшена-Эрба получил свое название в честь 2 медиков, впервые подробно описавших его причину и клинику. Французский невролог Г. Дюшен в 1872 г. отметил у некоторых новорожденных поражение верхних отделов плечевого сплетения и связал это с проведенными во время их родов вспомогательными акушерскими манипуляциями. Немецкий врач В. Эрб в 1874 г. дал подробное описание и анатомическое обоснование верхнего паралича плечевого сплетения, доказал его прямую связь с проведением родовспомогательных пособий.
Сегодня паралич Дюшена-Эрба известен также как проксимальный верхний паралич. Наряду с ним выделяют дистальный нижний паралич Дежерин-Клюмпке, обусловленный травмой нижних отделов плечевого сплетения (С8-Th1) и тотальный паралич Керера (поражение уровня С5-Th1). Из этих заболеваний паралич Дюшена-Эрба встречается наиболее часто, в акушерской практике его частота составляет 1-2 случая на тысячу новорожденных. Несмотря на разработанные методы диагностики и лечения, данная патология продолжает оставаться актуальной проблемой целого ряда медицинских дисциплин — неврологии, ортопедии и травматологии, педиатрии, неонатологии, акушерства и гинекологии.
Причины возникновения
Паралич Дюшена-Эрба возникает при травматическом повреждении верхнего ствола плечевого сплетения, образованного спинномозговыми корешками С5 и С6. Травмирование ствола возможно при падении на руку, ударе сверху по плечу, сильном ушибе плечом о неподвижный предмет, резкой тракции за руку, резанном или огнестрельном ранении, проникающем в область плечевого сплетения. Но чаще всего верхний паралич развивается в результате травм плечевого сплетения новорожденного, происходящих во время родов. В таких случаях он носит название верхний акушерский паралич, или родовой паралич Эрба.
Чрезмерное растяжение верхнего ствола плечевого сплетения, приводящее к его разрыву, может возникнуть в результате проведения следующих родовспомогательных манипуляций: поворота за ножку, тракции за ручку, высвобождения плечека, тракции за таз. Необходимость применять подобные акушерские пособия возникает при тазовом предлежании плода, слабой родовой деятельности и затяжных родах, крупном плоде, узком тазе. При этом паралич Эрба может сочетаться с кривошеей, обусловленной повреждением грудинно-ключично-сосцевидной мышцы или травмой добавочного нерва.
В результате травмы происходит полный или частичный надрыв верхнего ствола сплетения, травматизация лестничных мышц и фасций, которая может сопровождаться кровотечением. Спустя некоторое время отмечается стихание острых посттравматических явлений (отека, воспаления), рассасывание гематом. Однако этот процесс сопровождается рубцеванием тканей, что приводит к сдавлению плечевого сплетения и кровоснабжающих его сосудов.
Симптомы паралича Дюшена-Эрба
В клинике паралича Эрба выделяют 3 переходящих одна в другую стадии. Острый период паралича наступает после травмы и длится 1 месяц. Обращает на себя внимание положение руки — она разогнута в локте, пронирована в плече и приведена к туловищу, кисть находится в положении ладонного сгибания. Невозможно сгибание в локтевом суставе, отведение от туловища, поднятие и супинация (разворот внутренней поверхностью кпереди) плеча при сохранности движений в пальцах кисти. При парезе отмечается снижение активных движений в руке; затруднение движений плечом, сгибания в локте и тыльного разгибания кисти. На стороне паралича отсутствует или снижен рефлекс с бицепса, у новорожденных — ладонно-ротовой рефлекс, ослаблен хватательный. Для детей с параличом Дюшена-Эрба типичным является свисание паретичной руки при удержании их в горизонтальном положении.
Отмечается гипотония мышц пораженной руки, ее кожа более холодная и бледная по сравнению с кожей здоровой конечности. Сенсорные нарушения включают снижение болевой чувствительности по латеральной поверхности всей руки. Поскольку у детей грудного возраста довольно сложно провести оценку сенсорной сферы, предположить расстройство чувствительности данной области можно по более вялой или наоборот более активной реакции ребенка в сравнении с исследованием на других участках кожи.
После острого посттравматического периода наступает восстановительный. Степень восстановления функции руки зависит от типа травмы, своевременности и адекватности проводимого лечения. При легком парезе возможно постепенное нарастание объема движений в руке до полного, восстановление чувствительности. Несмотря на это, при акушерском параличе примерно к трем годам становиться заметно некоторое укорочение пораженной руки и гипотрофия ее мышц.
Период остаточных явлений является следствием неполного восстановления иннервации верхней конечности. При среднетяжелой и тяжелой степени паралича Дюшена-Эрба формируется симптом «кукольной руки» - выраженная борозда, пролегающая между грудной клеткой и пораженным плечом. Пронированная и приведенная к туловищу установка плеча вызывает формирование внутриротаторной и пронаторной контрактуры мышц, нарушение локтевого сгибания — сгибательной контрактуры локтевого сустава. Наблюдается поворот лопатки с выстоянием ее позвоночного края. Кисть остается в состоянии ладонной флексии, затруднено разгибание пальцев. Атрофия мышц плечевого сустава может привести к подвывиху или вывиху плеча, развитию привычного вывиха плеча. Со временем из-за асимметричности плечевого пояса возникает искривление позвоночника в шейно-грудном отделе — сколиоз.
Диагностика паралича Дюшена-Эрба
Типичный вид повернутой внутрь и вытянутой ручки у детей грудного возраста, а также наличие в анамнезе указаний на полученную новорожденным родовую травму или осложненное течение родов, позволяют без затруднений поставить клинический диагноз. У взрослых паралич Дюшена-Эрба необходимо дифференцировать с параличом Дежерин-Клюмпке, в клинике которого преобладает поражение дистальных отделов руки.
Пациенты с парезом обязательно осматриваются неврологом и ортопедом. Акушерский паралич Дюшена-Эрба обычно диагностируется неонатологом, дополнительно проводятся консультации детского ортопеда и детского невролога. Рентгенография плечевого сустава позволяет выявить отклонения в формирующих его костных структурах. В первые месяцы жизни у ребенка с родовым параличом Эрба рентгенологически обнаруживается увеличение расстояния от суставной впадины лопатки до проксимального метафиза плеча. После формирования ядер окостенения сравнивается расстояние от впадины лопатки до ядра окостенения головки плечевой кости. У взрослых и у детей через несколько месяцев после травмы отмечаются признаки атрофии и остеопороза костей пораженной руки.
Для подтверждения диагноза и планирования лечебной тактики пациентам, у которых диагностируется паралич Дюшена-Эрба, показано проведение УЗИ плечевого сустава, компьютерной миелографии, электромиографии, электронейрографии, МРТ позвоночника. Оценка проведения импульса по стволу нервного сплетения, проводимая в рамках электронейрографии позволяет установить пре- или постганглионарный характер повреждения. В планировании оперативного лечения опираются преимущественно на данные КТ-миелографии. МРТ менее информативна, из-за наличия двигательных артефактов она может не выявить существующий отрыв корешка плечевого сплетения.
Лечение паралича Дюшена-Эрба
Консервативная терапия предусматривает иммобилизацию парализованной конечности специальной шиной, которую снимают лишь для осуществления лечебных и гигиенических манипуляций. Параллельно проводят массаж, лечебную физкультуру, рефлексотерапию, физиотерапию (УВЧ, соллюкс, электрофорез, электромиостимуляция, парафинотерапия, озокерит). Фармакотерапия может включать противовоспалительные и обезболивающие; антихолинэстеразные препараты (неостигмин. галантамин); медикаменты, улучшающие кровообращение тканей плечевого сплетения (папаверин, эуфиллин, пентоксифиллин); средства, повышающие метаболизм нервной ткани (гемодиализат из крови телят, витамины гр. В); рассасывающие препараты (алоэ, гиалуронидаза).
При отсутствии должного эффекта проводимой консервативной терапии пациент направляется к нейрохирургу для консультации относительно возможности хирургического лечения. Детям с акушерским парезом операция показана при тотальном характере повреждения; выявлении по данным КТ позвоночника отрыва от спинного мозга формирующих плечевое сплетение корешков; отсутствии у ребенка, имеющего акушерский паралич Дюшена-Эрба, к 3-месячному возрасту активных сгибательных движений в локте. Операция целесообразна на 1-ом году жизни, лучшие результаты дает хирургическое лечение в возрасте до 6-7 мес. Поэтому, если в течение 3-х мес консервативной терапии не было достигнуто значительных успехов в лечении, то следует решать вопрос о проведении операции. Хирургическое вмешательство заключается в пластике нервного ствола и проводится с применением микрохирургической техники.
В периоде остаточных явлений восстановление нервного ствола не представляется возможным. Для формирования более выгодного в функциональном плане положения пораженной конечности и некоторого увеличения объема ее функций могут проводиться реконструктивно-пластические ортопедические вмешательства.
Прогноз и профилактика
При частичном повреждении плечевого сплетения и раннем начале лечения возможно восстановление функции руки методами консервативной терапии. Отдельные данные говорят о положительной динамике в результате своевременно начатого консервативного лечения у 70% новорожденных, имевших паралич Дюшена-Эрба. Причем у 20 новорожденных из 100 имело место 100% восстановление. Это касается в основном легкой степени акушерского паралича. При полном разрыве ствола плечевого сплетения, надеяться на его самостоятельное срастание не приходится, следует прибегнуть к операции.
Профилактика родового паралича Эрба заключается в адекватном ведении беременности и родов, правильном выборе метода родоразрешения, позволяющем избежать необходимости применения акушерских родовспомогательных манипуляций. Профилактика у взрослых сводится преимущественно к предупреждению травматизма.
Прогрессирующая мышечная дистрофия Дюшенна
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Миодистрофия Дюшенна: что это такое?

Как часто и почему возникает это заболевание, почему педиатры нередко путают его с гепатитом, что делать, если вашему ребенку поставлен такой диагноз
Доктор медицинских наук, президент благотворительного фонда "Гордей" и бабушка Гордея, мальчика с диагнозом миодистрофия Дюшенна, Татьяна Андреевна Гремякова рассказывает о том, что это за болезнь, почему ее часто принимают за гепатит, и что необходимо делать, чтобы дети с этим диагнозом как можно дольше сохраняли активность и жили полной жизнью.
Мышечная дистрофия Дюшенна (МДД) — одно из наиболее распространенных среди редких (орфанных) генетических фатальных нейромышечных заболеваний. В большинстве случаев оно встречается только у мальчиков.
Заболевание связано с мутацией в Х-хромосоме: его частота составляет 1:3500 - 1:5000 новорожденных мальчиков и 1:50 000 000 у новорожденных девочек.
Заболевание развивается вследствие мутации гена, который отвечает за синтез белка дистрофина. У детей прогрессирует повреждение и дегенерация мышц. Со временем мышцы слабеют до такой степени, что дети/подростки не могут самостоятельно передвигаться — в возрасте 10-15 лет мальчики садятся в инвалидную коляску. Параллельно у них развиваются кардиореспираторные нарушения — проблемы с сердцем и дыханием. В возрасте 15-20 лет им уже требуется респираторная поддержка: вначале только ночью, а в дальнейшем - круглосуточно.
Клинические проявления со стороны скелетной мускулатуры и сердца могут различаться в каждом конкретном случае, но смерть, как правило, наступает из-за нарушения функции сердца или дыхания в возрасте 12-25 лет.
Сегодня, благодаря профилактике формирования контрактур, сколиоза и кардиомиопатии, применению стероидов, внедрению респираторной поддержки и другим превентивным мерам, удалось существенно продлить функциональную активность пациентов,
качество и продолжительность их жизни.
Ожидаемая продолжительность жизни мальчиков с МДД, рожденных в последние годы в развитых странах (при условии, что им доступна современная реабилитация, терапия и респираторная поддержка), — 30-40 лет.
Болезнь в большинстве случаев передается от матерей, но сами женщины от нее практически не страдают: они бессимптомные или малосимптомные носители дефектного гена. Ген может передаваться по женской линии многие поколения и никак не проявляться, поэтому рождение ребенка с дистрофией Дюшенна для семьи часто становится неожиданностью. В трети случаев болезнь - результат спонтанной мутации плода без семейной истории.
В начале болезнь распознать трудно: симптомы проявляются не сразу и нарастают постепенно. Чаще всего до четырех-пяти лет врачи и родители считают, что малыш здоров. У ребенка могут быть некоторые задержки двигательного и речевого развития, он может быть неуклюжим, часто падать и быстро уставать. Мальчик с МДД не бегает так быстро, как сверстники, не прыгает. Родителям зачастую видны некоторые особенные нюансы взросления, но никаких «красных флагов», которые позволили бы серьезно обеспокоиться, нет довольно долго.
Ребенок растет, со временем меняется его походка и осанка. Можно заметить, что он ходит на носочках. Ему трудно вставать с пола — поднимается, опираясь на руки (прием Говерса).
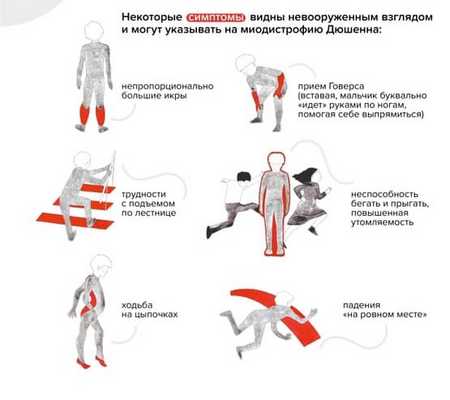
"Прием Говерса" и другие вероятные признаки наличия у ребенка миодистрофии Дюшенна / Фонд "Гордей"
Обращает на себя внимание увеличение икроножных мышц. Позднее трудности при ходьбе нарастают: ребенок с трудом поднимается по лестнице, у него совсем нет сил. В какой-то момент он вовсе перестает ходить и садится в инвалидную коляску.
Если есть сомнения относительно здоровья мальчика, то первое, что нужно сделать, — анализ крови на активность креатинфосфокиназы (КФК). Это фермент, содержащийся в скелетных мышцах, маркер их распада. Обычно при МДД он бывает выше нормы во много раз — например, несколько десятков тысяч единиц при норме в сотню.
МДД — редкое заболевание. Врачи могут ни разу за свою практику не столкнуться с ним и не знать, что делать. Часто ребенку ошибочно ставят диагноз гепатит, перинатальное поражение ЦНС и/или аутизм и назначают лечение, подчас ускоряющее прогрессирование Дюшенна (Рис. 1).
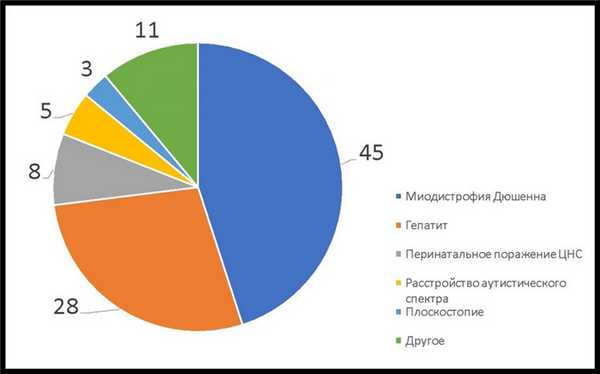
Рис. 1. Результаты опроса ~200 родителей (%): "Какой первый диагноз был поставлен мальчику с миодистрофией Дюшенна в первичном поликлиническом звене?"
Почему гепатит — самый частый из неверных диагнозов? При МДД в крови повышаются уровни трансаминаз, что ошибочно принимается за симптоматику гепатитов. Но эти ферменты в случае МДД мышечного, а не печеночного происхождения.
Если выявлено увеличение уровня КФК выше критического (более 2000 ед), необходимо делать генетический анализ и искать мутацию в гене дистрофина.
Информация для медицинских специалистов
Ген дистрофина — один из самых больших в человеческом организме. Он состоит примерно из 2,5 млн пар нуклеотидов, содержит 79 экзонов, размер гена 2,2 Мб. Уровень мутаций гена относительно высок: в одном из трех случаев МДД вызывается мутацией de novo (впервые возникшее изменение, в отличие от унаследованного - прим.ред). Таким образом, новые случаи возникают даже при наличии хороших инструментов пренатальной диагностики и семейного консультирования для известных случаев.
Высокая частота мутаций также лежит в основе большого разнообразия мутаций, выявленных у пациентов с МДД. Наиболее часто встречаются делеции (~68%), затем по частоте идут дупликации (~11%) одного или нескольких экзонов, реже обнаруживаются небольшие мутации (∼20% пациентов). Мутации могут происходить на любом участке гена, но делеции чаще сконцентрированы между экзонами 45-55, а дупликации — между экзонами 2-10. Тип и расположение мутации определяют ход течения заболевания и то, какое лечение требуется больному.
Сейчас в России применяется специфическая молекулярно-генетическая диагностика повреждения гена дистрофина, определение мутации в каждом конкретном случае.
Генетическое тестирование позволяет точно поставить диагноз. Ранняя и прецизионная диагностика больных МДД необходима для правильного сфокусированного назначения этиопатогенетической терапии. Фактически каждому такому ребенку требуется персонализированная терапия в зависимости от типа и локализации мутации гена.
Бесплатная генетическая диагностика проводится больным с диагнозом МДД, а также бессимптомным пациентам с высокими значениями КФК (более 2000 ЕД\л) с 2019 года. Ограничений по возрасту исследуемых больных нет.
Как попасть на генетическую диагностику?
Только в 30% случаев больной ребенок рождается в результате спонтанной мутации. В 70% случаев носителем мутации является мама ребенка, которая, как правило, не знает об этом.
При планировании беременности женщинам желательно сделать генетический тест на носительство МДД. В некоторых странах такой тест обязателен.
«Нет человека без патологических генов». Генетик Наталия Белова о редких болезнях и помощи семьям с больными детьми Можно ли избежать генетической поломки у ребенка и почему жизнь «редких» — пока еще борьба
Оценка функциональной активности ребенка позволяет понять его состояние и спрогнозировать дальнейшее течение заболевания. Для этого заболевания нет биохимических маркеров течения болезни. Изменение функциональной и физической активности у детей с МДД выражается в числовых значениях. Это позволяет понять степень выраженности болезни и динамику ее клинической симптоматики.
Если ребенок ходит, его состояние оценивают по методу The North Star (NSAA). Дополнительно оценивается расстояние, пройденное за 6 минут, и время подъема и спуска с четырех ступеней.
Для тех, кто потерял способность ходить, есть метод оценки работы рук — Performance of Upper Limb (PUL) test.
Физическая терапия и реабилитация — основа ухода за больными с МДД на протяжении всей жизни. Она нужна, чтобы поддерживать физическую активность и функциональность.
Современная реабилитация ориентирована:
- на защиту хрупких мышц, на сохранение и поддержание их оптимальной прочности;
- на сведение к минимуму, насколько это возможно, прогрессирования слабости;
- на предотвращение и сведение к минимуму прогрессирующих контрактур и деформаций;
- на поддержку оптимальной кардиореспираторной функции.
На более поздних этапах болезни это:
- использование адаптивного оборудования и вспомогательных технологий (например, коляски, вертикализатора, велотренажера, откашливателя, респираторного оборудования и др.);
- обезболивание;
- поддержка функций и функциональной независимости (поддержание функций рук, обслуживание себя, возможность занятости - работа с компьютером);
- учеба в школе, работа, участие в жизни семьи и общества, а также улучшение качества жизни.

Тутора для ребенка с миодистрофией Дюшенна / Фонд "Гордей"
Для сохранения способности ходить важно следить за постановкой ног ребенка на полную стопу, а также уделять внимание растяжке, особенно мышц голеностопа. Иначе через некоторое время мальчик сможет ходить только на цыпочках, и тогда ему понадобится коляска.
Кроме того, детям с МДД полезны занятия в бассейне с температурой воды 32-34 о С и велосипед (для маленьких детей - велосипед, а когда подросток теряет способность ходить и садится в коляску, тогда - велотренажер для ног и рук).

Занятия в бассейне / Фонд "Гордей"
Если не заниматься физической терапией, ребенок может потерять способность ходить рано и сесть в коляску в 7-8 лет.
В результате начнутся серьезные изменения, в первую очередь, позвоночника: сильный сколиоз, требующий операции на позвоночнике, пострадают сердце и дыхание.
Реабилитация и физическая терапия способны надолго продлить двигательную активность больного.
Позиционирование на стадии коляски фокусируется на том, чтобы сохранить правильное положение тела и ног, не допустить развития сколиоза и контрактур в суставах, сохранить максимальный объем движений рук.
В наиболее тяжелой стадии заболевания важна респираторная реабилитация.
Все о масках для неинвазивной вентиляции легких (НИВЛ) Виды масок, правила подбора, проблемы и осложнения при использовании маски, уход за маской
Медицинская помощь
Миодистрофия Дюшенна — прогрессирующее заболевание, которое со временем поражает многие системы и органы человека. А значит, вести пациента с МДД должна мультидисциплинарная команда врачей под руководством специалиста по нервно-мышечным заболеваниям (например, педиатр или детский невролог).
По мере взросления ребенка эта функция переходит к неврологу, работающему со взрослыми пациентами. Следует заранее определить необходимых специалистов, чья помощь понадобится уже взрослому пациенту.
В современной медицинской и паллиативной концепции ведения пациентов с МДД считается важным ориентироваться на его семью: общаться, координировать уход, информировать об ожидаемых изменениях, связанных с болезнью.
Какие врачи ведут пациента с МДД
| Специалист | Сфера ответственности |
|---|---|
| Невролог | Комплексное ведение болезни на протяжении всей жизни. |
| Реабилитолог | Упреждение развития контрактур и деформации, минимизация боли, продление функциональности и способности к передвижению. |
| Ортопед/Хирург | Поддержка двигательных функций как можно дольше, минимизация контрактур суставов, поддержка позвоночника в прямом положении. |
| Эндокринолог (поддержание костного здоровья) | Уменьшение прогрессирования остеопороза, восстановление при ранних признаках остеопороза. |
| Эндокринолог (рост и половое созревание) | Минимизация нарушений роста, коррекция пубертатного развития, развитие и предотвращение опасного для жизни криза надпочечников. |
| Пульмонолог | Уменьшение респираторных осложнений и сохранение функции дыхательных мышц. |
| Кардиолог | Максимальное продление работы сердца, упреждение формирования сердечной недостаточности и других отклонений. |
| Гастроэнтеролог и диетолог | Профилактика недостаточного или плохого питания, избыточного веса и ожирения. |
| Психолог | Психосоциальная поддержка на протяжении всей жизни, помощь в планировании будущего и формировании представлений о том, как пациенты будут активно участвовать в уходе за собой и ежедневной деятельности. |
| Паллиативная помощь | Симптоматическая терапия, предоставление технических средств реабилитации и медицинских изделий, социальная поддержка. |
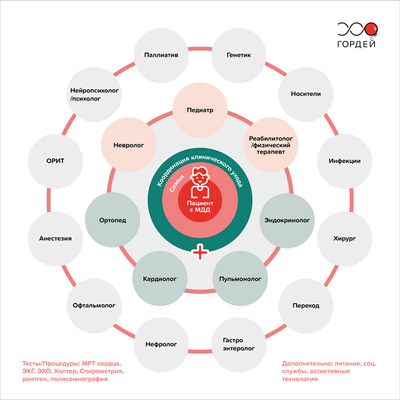
Рис. 2. Схема организации мультидисциплинарного ухода за пациентами с миодистрофией Дюшенна на протяжении всей жизни / Фонд "Гордей"
Транзит во взрослую медицину и жизнь
Человек с заболеванием, которое начинается в детском возрасте и ведется детскими врачами, взрослея, переходит во взрослую медицинскую и паллиативную службу. Переход обычно происходит в возрасте от 17 лет до 21 года, в зависимости от системы здравоохранения. Однако, чем старше становится больной с МДД, тем больше он нуждается в медицинской помощи и уходе — ведь болезнь прогрессирует. А значит, без поддержки семьи и медиков не обойтись.
К сожалению, не все больные МДД смогут перейти к самостоятельной и долгой взрослой жизни. Первая причина: около трети молодых взрослых с МДД испытывают психосоциальные трудности или страдают когнитивными нарушениями, которые ограничивают самостоятельность и независимость. Вторая причина: различное течение заболевания.
Обе причины зависят от множественности мутаций гена дистрофина: от чрезвычайно злокачественной, когда дети вовсе не научаются ходить, до доброкачественной, похожей по течению и сроку жизни на миодистрофию Беккера (Birnkrant DJ, Bushby KM, Bann CM, и др., Диагностика и ведение пациентов с мышечной дистрофией Дюшенна, часть 3: первичная помощь, неотложная помощь, психосоциальная помощь и преемственность в оказании помощи на протяжении жизни пациента., The Lancet Neurology, 2018; том 17: 445-55').
И все же для большинства молодых взрослых с МДД следует прогнозировать полноценное участие в планировании будущего и принятии решений.
Ребенку с инвалидностью скоро исполнится 18 лет. Как правильно подготовиться? Статус ребенка-инвалида и инвалида-взрослого сильно различаются. Чтобы не оказаться в трудной ситуации, надо подготовиться к совершеннолетию заранее.
Есть ли лекарство?
Сделать лекарство для МДД сложно: один препарат не вылечит всех, как при СМА. Больные МДД, как снежинки: нет одинаковых — уже описано около десяти тысяч мутаций гена. Поэтому болезнь по-разному проявляется — даже в одной семье у двух братьев.
Какие перспективы?
Кто поможет?
Фонд «Гордей» создала семья, в которой растет Гордей — семилетний мальчик с миодистрофией Дюшенна. Цель фонда - улучшить на системном уровне качество диагностики, ведения, ухода, реабилитации и лекарственного обеспечения больных МДД.

Гордей с мамой, Ольгой Гремяковой. Фото из личного архива
Одна из основных задач фонда - информирование и повышение осведомленности о миодистрофии Дюшенна врачебного и родительского сообщества. Чтобы практикующие и будущие педиатры, детские неврологи, медработники детских садов и школ знали о симптомах заболевания, а профильные специалисты имели возможности для обучения, стажировок, обмена опытом и в своей работе опирались на международные стандарты ухода и лучшие мировые практики.
Президент фонда «Гордей», доктор медицинских наук и бабушка Гордея, Татьяна Андреевна Гремякова ежемесячно проводит Школу для родителей вновь диагностированных детей с Дюшенном: отвечает на вопросы, помогает родителям понять, что именно они могут сделать для своего ребенка уже сегодня и что происходит в науке и медицине по данной нозологии.
Учредитель фонда «Гордей», психолог и мама Гордея, Ольга Гремякова два раза в месяц проводит «Передышку» — группу поддержки для мам и пап и вебинары, помогающие родителям развивать жизнестойкость, справляться со стрессом, возвращать себе контроль над своей жизнью. Для родителей вновь диагностированных мальчиков есть индивидуальные консультации с психологом.
С МДД также работают следующие организации:
- , г.Москва;
- Пациентская организация «Родительский проект по оказанию помощи пациентам с миодистрофией Дюшенна/Беккера»;
- ГАООРДИ, Ассоциация родителей детей-инвалидов, г.Санкт-Петербург.
Федеральные центры, работающие с МДД:
- ГБОУ ВПО РНИМУ им. Н.И. Пирогова МЗ РФ;
- ФГАУ Национальный медицинский исследовательский Центр Здоровья Детей;
- ФГБУ Центральная Клиническая Больница УДП РФ;
- ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России - Российская детская клиническая больница;
- ФГБУЗ Центральная клиническая больница РАН;
- ФГБОУ ВО Санкт-Петербургский государственный педиатрический медицинский университет;
- ФГБОУ ВО Сибирский государственный медицинский университет (г.Томск);
- ФГБНУ Томский национальный исследовательский центр РАН.
Читайте также:
- Синдром беспокойных ног: причины, симптомы и лечение
- Лечение протокового рака in situ молочной железы (DCIS) и ее эффективность
- Пространственная ориентация, постуральный контроль и их значение в развитии головокружения
- Диагностика порэнцефалической кисты по КТ, МРТ
- Местный остеопороз рук и его диагностика