Синдром эритропатии ферментопенической - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 02.02.2026
МКБ-10: Q91 — Синдром Эдвардса и синдром Патау.
Синдром Патау представляет собой генетическую патологию, суть которой заключается в наличии дополнительной 13 хромосомы (трисомия). Это заболевание получило название в честь врача Клауса Патау, который впервые определил нарушение количества хромосом.
Стаж работы 13 лет.
Статья проверена заместителем генерального директора, врачом акушер-гинекологом Дмитриевым Дмитрием Викторовичем.
Данная статья не может быть использована для постановки диагноза, назначения лечения и не заменяет прием врача.
Причины и механизм развития
Синдром Патау стоит в одном ряду с другими хромосомными заболеваниями — синдромом Дауна ( трисомия 21 пары ) и синдромом Эдвардса ( трисомия 18 пары ). По сравнению с ними заболевания встречается несколько реже, но протекает тяжелее.
Согласно статистическим данным, синдромом Патау болеет 1 новорожденный из 7500.
Мальчики и девочки болеют с одинаковой частотой. У всех детей органы и ткани сильно недоразвиты, но это нельзя объяснить недоношенностью.
Диагностика хромосомных патологий
Причины синдрома Патау
Как и при других похожих заболеваниях, главная причина синдрома Патау — добавочная хромосома в 13 паре . Из-за излишка генетического материала нормальное развитие организма становится невозможным, возникают многочисленные аномалии. До сих пор ученые точно не могут сказать, почему она возникает. Этот вопрос является предметом научных дискуссий. Далеко не во всех случаях просматривается влияние факторов риска или других неблагоприятных обстоятельств.
По мнению генетиков, такой хромосомный дефект может спровоцировать:
- беременность женщины в возрасте старше 40 лет;
- прием некоторых медикаментов на ранних стадиях течения беременности;
- плохая экологическая обстановка, особенно радиоактивное загрязнение;
- воздействие промышленных ядов, радиации и других неблагоприятных факторов;
- близкородственные браки;
- отягощенный семейный анамнез — вероятность того, что может развиться синдром Патау, значительно повышается, если кто-либо из родственников пары страдал этой патологией.
Трисомия 13 хромосомы приводит к нарушению формирования внутренних органов и всех систем организма еще во время внутриутробного развития плода.
Признаки и симптомы синдрома Патау
Синдром Патау - это болезнь, которая проявляется сразу после рождения ребенка. Она характеризуется значительными пороками развития, которые затрагивают половую, сердечно-сосудистую и костно-мышечную систему. Наиболее часто они заключаются в неполном срастании верхнего неба («волчья пасть»), верхней губы («заячья губа»), недостаточным дозреванием органов половой системы и пороками сердца (отсутствие закрытия овального окна между предсердиями, триада или тетрада Фалло).
Вес большинства детей с синдромом Патау не превышает 2500 г.
Из-за недоразвитости головного мозга лоб у ребенка низкий, скошенный, черепная коробка уменьшенных размеров. Глазные щели узкие, расположены слишком близко друг к другу, вплоть до циклопии — состояния, при котором у больного виден один глаз, локализованный в области переносицы. Вместе с глубоко запавшей переносицей это придает ребенку довольно характерный внешний вид. Глаза недоразвиты, роговица помутневшая. Ушные раковины неправильной формы, располагаются низко. Шея короткая, количество пальцев на верхних и нижних конечностях больше пяти.
Кроме врожденных пороков сердца, при детальном обследовании детей выявляется ряд серьезных аномалий, среди которых:
- отклонения в развитии центральной нервной системы — головной мозг неразделен на полушария, мозжечок и мозолистое тело недоразвиты;
- атрофия зрительного нерва, и, как следствие, слепота;
- спинномозговые грыжи;
- фиброзные изменения во внутренних органах;
- увеличение и кистозные новообразования в почках.
Основные симптомы синдрома Патау хорошо заметны при ультразвуковом исследовании, которое проводится в рамках первого генетического скрининга на сроке 11-12 недель.
Симптомы синдрома Патау
Диагностика синдрома Патау
Часто болезнь приводит к смерти ребенка в течение нескольких недель после рождения. Поэтому для раннего выявления патологии выполняется диагностика, которая заключается в проведении генетического исследования беременной женщины.
В зависимости от времени проведения, диагностика синдрома Патау может проводиться на таких этапах планирования и рождения ребенка:
- до зачатия;
- после зачатия;
- после рождения.
На каждом этапе применяются различные диагностические методы, благодаря чему врач получает максимум полезных данных. На этапе планирования беременности пара должна посетить генетика , который изучит семейный анамнез, обнаружит факторы риска и обследует родителей.
Изучение семейного анамнеза — детальный опрос, в ходе которого выясняются все случаи патологии новорожденных в семье. Очень важно, чтобы будущие родители хорошо подготовились к консультации и собрали как можно больше полезной информации о своих предках. Особое внимание следует обратить на случаи мертворождения, самопроизвольные выкидыши. Наличие случаев синдрома Патау у родственников значительно повышает вероятность заболевания у будущих детей.
Чем больше неблагоприятных факторов выявит генетик, тем больше вероятность рождения у пары ребенка с патологией . Таким родителям следует крайне внимательно отнестись к предстоящему генетическому скринингу, который рекомендуют пройти беременным.
Генетическое исследование родителей — инструментальный метод диагностики, который проводится в высокотехнологичных лабораториях. Он предоставляет информацию о хромосомном наборе обоих родителей, о наличии дефектных генов, их локализации и количестве. В период вынашивания малыша всем парам из группы риска настоятельно рекомендуют пройти генетический скрининг, который включает ультразвуковую диагностику и биохимический анализ крови. В ходе УЗИ врач может увидеть аномалии, свидетельствующие о хромосомной патологии у плода.
К основным из них относятся:
- внешние дефекты;
- задержка внутриутробного развития;
- ускоренное сердцебиение;
- многоводие (наблюдается в половине случаев);
- увеличенный мочевой пузырь у плода;
- аномалии головного мозга;
- пуповинная грыжа;
- пороки других органов и систем.
На основании данных анализов, при помощи компьютерной программы генетик рассчитает риск рождения больного ребенка. Сопоставив полученные результаты, делают выводы о состоянии здоровья будущего ребенка. Если вероятность хромосомных аномалий высока, паре рекомендуют более детальное обследование — амниоцентез, кордоцентез или биопсию хориона.
- Амниоцентез.
Малоинвазивная процедура, в ходе которой врач прокалывает амниотический мешок и отбирает небольшое количество околоплодных вод. В них содержатся клетки кожи ребенка, что позволяет подтвердить или исключить заболевание, обнаружив главные причины синдрома Патау — дополнительные хромосомы. - Кордоцентез.
Предусматривает забор крови из пуповины эмбриона. Прокол делают тонкой иглой, под контролем ультразвуковой аппаратуры. В результате врач получает кровь развивающегося плода. Выделив из нее лейкоциты (белые кровяные клетки) можно изучить набор хромосом будущего ребенка и сделать выводы о его здоровье. На сегодняшний день это самый точный метод диагностики синдрома Патау. Частота осложнений при его применении достигает 2%. - Биопсия хориона.
Инвазивное вмешательство, которое позволяет получить небольшой кусочек околоплодных структур. В нем содержатся клетки плода вместе с генетическим материалом. После его изучения генетик дает заключение о количестве и качестве хромосом малыша.
Диагностика синдрома Патау
Цитогенетическая диагностика синдрома Патау позволяет выявить такие разновидности заболевания:
- простая трисомия — наличие дополнительной 13 хромосомы;
- транслокация Робертсона — аномалия, при которой происходит слияние двоих хромосом с образованием одной. Носители мутации в большинстве случаев — это здоровые люди, но вероятность того, ребенку от такого родителя будет поставлен диагноз «синдром Патау» очень высока. В некоторых случаях они испытывают проблемы с наступлением естественной беременности;
- транслокации других видов — изменения в генетическом материале, которые возникают при делении и созревании половых клеток;
- мозаичная форма , при которой дополнительную хромосому имеют только часть клеток. Развивается после зачатия, чаще всего при воздействии неблагоприятных факторов внешней среды;
- изохромосома — состояние, при котором часть одной хромосомы из пары утрачена, а аналогичная часть второй — продублирована.
После рождения ребенка поставить диагноз «синдром Патау» можно на основании внешних признаков заболевания. Предварительное заключение делают практически сразу после рождения, окончательное — после генетического исследования образцов крови новорожденного.
Если симптомы синдрома Патау, внешние отклонения видны при осмотре, то для диагностики аномалий развития внутренних органов применяют инструментальные и лабораторные тесты:
- общеклинические анализы;
- биохимический анализ крови;
- ультразвуковое исследование;
- компьютерная и магнитно-резонансная томография;
- рентгенография.
Некоторые методы обследования применяют несмотря на высокую вероятность осложнений, ведь она значительно меньше, чем потенциальная польза полученных данных.
Лечение синдрома Патау
В связи с тем, что хромосомный дефект затрагивает все клетки организма плода или родившегося ребенка, лечение синдрома Патау на сегодняшний день является невозможным. Приоритетным является симптоматическое лечение синдрома Патау, направленное на максимальное устранение дефектов и пороков развития различных органов и систем.
Семье, в которой родился больной ребенок, нужно быть готовой к таким действиям:
- тщательному контролю здоровья малыша и своевременному обнаружению осложнений синдрома Патау;
- частным диагностическим тестам, направленным на поиск внутренних пороков развития и оперативным вмешательствам, цель которых — устранить выявленные аномалии;
- постоянному уходу за ребенком, проведению общеукрепляющей терапии, поддержанию работы внутренних органов и систем.
Ввиду того что заболевание не поддается терапии, при его выявлении на ранних сроках беременности рекомендуется ее прерывание по медицинским показаниям. К сожалению, прогноз при данном заболевании неблагоприятный. Примерно 95% детей умирают до года. В развитых странах, благодаря тщательному уходу около 15% больных доживают до пяти лет, 2-3% — до 10 лет. Выжившие дети сильно отстают в умственном и физическом развитии. Основная причина смерти - многочисленные пороки и связанные с ними осложнения синдрома Патау.
Профилактика синдрома Патау
Несмотря на то что синдром Патау — это серьезное заболевание с очень высокими показателями смертности , специфических профилактических мер по отношению к нему не разработано. Основная профилактика синдрома Патау — это тщательное планирование беременности, диагностика заболевания на ранних сроках внутриутробного развития и принятия решения о целесообразности сохранения беременности.
Далеко не второстепенную роль играет неспецифическая профилактика синдрома Патау , которая заключается в правильном питании, исключении вредных привычек, своевременном лечении инфекционных заболеваний. Эти простые мероприятия помогут предотвратить не только хромосомные патологии, но и многие другие заболевания.
При проведении ЭКО эту хромосомную аномалию можно выявить еще до наступления беременности. На этапе культивирования эмбрионов большинство клиник предлагают родителям сделать ПГД - преимплантационную генетическую диагностику.
Это анализ, направленный на выявление мутаций - как генных, так и хромосомных. Если болезнь обнаружена в одном из эмбрионов, он не подсаживается в матку. Для этой цели выбирается другой, здоровый эмбрион. Таким образом, родители, использующие ЭКО для зачатия ребенка, находятся в более выгодном положении, так как способны предотвратить множество врожденных заболеваний у своего потомства.
- Алтынник H.A. Значение ультразвуковой оценки толщины воротникового пространства плода в ранние сроки беременности для прена-тальной диагностики хромосомных аномалий: Дисс. . канд. мед. наук. М., 2002. - 105 с.
- Алтынник H.A. Ультразвуковые пренатальные маркеры врожденных и наследственных заболеваний в ранние сроки беременности: Дисс. . докт. мед. наук. М., 2012. -211 с.
- Берешева А.К. Роль молекулярно-цитогенетической диагностики в генетическом консультировании супружеских пар с нарушением репродуктивной функции. // Автореф канд диссер, Харьков, 1995.-17 с.
- Ворсанова С.Г., Юров Ю.Б., Александров И.А., Демидова И.А., Миткевич С.П., Доронин Л.Г. Молекулярно-цитогенетическая диагностика наследственных болезней, связанных с различными аномалиями хромосом X. // Педиатрия, 1989.-1.-76-80.
- Давиденкова Е.Ф., Берлинская Д.К., Тысячник С.Ф. Клинические синдромы при аномалиях половых хромосом. // JI. Медицина, 1973.-198с.
У Вас есть вопросы? Получите квалифицированный ответ от ведущих специалистов клиники.
Эритремия
Эритремия — это опухолевое клональное заболевание кроветворной системы, при котором отмечается пролиферация эритроидного, гранулоцитарного и мегакариоцитарного ростков кроветворения с преимущественной активацией эритропоэза. При этом в крови отмечается повышение уровня эритроцитов и гемоглобина, тромбоцитоз и лейкоцитоз. Практически все больные эритремией являются носителями мутации JAK2V617F.
Как развивается эритремия
Причины возникновения эритремии до сих пор не известны. Считается, что это многоэтапное заболевание, при котором под действием внешних факторов происходит повреждение генома нормальной клетки, что приводит к ее злокачественной трансформации и образованию опухолевого клона клеток, который замещает нормальный гемопоэз.
Практически у всех больных обнаруживается мутация в гене JAK2. Обычно страдает 14 экзон, у 90-96% больных отмечается мутация V617F. У 2% больных имеются мутации в 12 экзоне. Очень редко обнаруживаются повреждения других генов, в частности MPL, CALR. Все эти генетические патологии являются специфичными для эритремии, поэтому их определение необходимо для подтверждения диагноза.
Итак, молекулярно-генетические нарушения вызывают активацию JAK-STAT сигнального пути, что приводит к повышению пролиферации ростков гемопоэза и увеличению количества форменных элементов крови.
Моноциты и мегакариоциты (предшественники тромбоцитов) вырабатывают множество цитокинов — биологически активных молекул, которые стимулируют фиброзные изменения, образование новых кровеносных сосудов, что в конечном итоге приводит к остеосклерозу и фиброзу костного мозга. Кроме того, массивная продукция цитокинов способствует развитию опухолевой интоксикации, что усугубляет общее состояние больных.
Также присутствует нарушение связи стволовых клеток с микроокружением. Это провоцирует образование очагов кроветворения вне костного мозга. В первую очередь страдают печень и селезенка.
Симптомы эритремии
Клинически эритремия проявляется двумя синдромами:
- Плетора (полнокровие). Этот синдром характеризуется увеличением числа циркулирующих эритроцитов. Симптоматически проявляется головными болями, головокружением, приступами учащенного сердцебиения, зудом кожи и зрительными нарушениями. Кожа и слизистые имеют синюшный оттенок. Также возможны сосудистые осложнения: тромбозы, эритромелалгия (покраснения пальцев конечностей, боли и чувство жжения в них).
- Миелопролиферативный синдром — развивается в результате гиперплазии ростков кроветрения. Симптоматически проявляется слабостью, повышением температуры, потливостью, зудом кожи, болями в костях. При распаде гранулоцитов отмечается нарушение уратового обмена, что приводит к развитию подагры, камней в почках и уратовому диатезу.
Стадии эритремии
В процессе своего развития эритремия проходит несколько стадий. Первая, она же начальная, может продолжаться более 5 лет. В этот период в основном присутствуют умеренные проявления плеторального синдрома. В анализе крови отмечается умеренный эритроцитоз, в костном мозге отмечается усиление пролиферации всех гемопоэтических ростков за исключением лимфоцитарного. Селезенка не увеличена, осложнения развиваются редко.
Вторая стадия эритремии — это полицитемия. Она делится на 2 подстадии А и В. Стадия А длится 5-15 лет. Для нее характерно повышение количества форменных элементов крови. В результате образуется ярко выраженный плеторический синдром, который осложняется тромбозами, кровотечениями, увеличением размеров печени и селезенки. В анализе крови нарастает цитоз, в костном мозге помимо пролиферации эритропоэтического, гранулоцитарного и тромбопоэтического ростков отмечаются рубцовые изменения.
При эритремии в стадии В продолжает нарастать цитоз и клиническая симптоматика. Образуются очаги опухолевого роста в селезенке, в костном мозге прогрессируют рубцовые изменения.
3 стадия эритремии — анемическая. Здесь уже развивается фиброз костного мозга, который приводит к его истощению и снижению уровня клеток крови. Возможна трансформация заболевания в острый лейкоз.
Диагностика эритремии
В рамках диагностики эритремии пациент проходит комплексное обследование, которое включает следующие мероприятия:
- Сбор анамнеза и физикальный осмотр, во время которого проводится оценка окраски кожи лица и конечностей. Обязательно выполняется определение размеров печени и селезенки.
- Развернутый анализ крови, включающий подсчет количества форменных элементов крови и значений эритроцитарных индексов.
- Трепанбиопсия костного мозга с последующим гистологическим и гистохимическим исследованием.
- Определение уровня эритропоэтина.
- Молекулярно-генетическое тестирование на предмет наличия специфических мутаций V617F.
- УЗИ печени и селезенки для точного определения их размеров.
Диагноз эритремия выставляется согласно критериям ВОЗ 2018 года, где выделяют три больших критерия и один малый.
Большие критерии эритремии:
- Уровень гемоглобина больше 160 г/л у женщин и больше 165 г/л у мужчин.
- Гиперплазия трех ростков миелопоэза в трепанате костного мозга.
- Наличие мутации в гене JAK2.
Малый критерий — снижение уровня эритропоэтина.
Для постановки диагноза эритремия необходимо либо наличие всех трех больших критериев, либо 1-2 больших и одного малого.
Лечение эритремии
Основные цели лечения эритремии следующие:
- Предотвращение образования тромбов и лечение уже развившихся тромбозов и тромбоэмболий.
- Снижение уровня опухолевой интоксикации и контроль связанных с этим симптомов (повышение температуры, кожный зуд, потеря веса).
- Снижение риска трансформации в острый миелобластный лейкоз или миелофиброз.
- Предупреждение развития осложнений при необходимости хирургических вмешательств или беременности.
Для оценки вероятности развития тромбозов при эритремии, проводят стратификацию риска по следующим критериям:
- Возраст старше 60 лет.
- Наличие тромбозов в прошлом.
- Сердечно-сосудистые факторы риска: артериальная гипертензия, лишний вес, диабет, гиподинамия.
Если у пациента нет перечисленных факторов, то его относят к группе низкого риска, при наличии сердечно-сосудистых факторов — к промежуточной, и при возрасте более 60 лет и/или наличии тромбозов в анамнезе выставляется высокий риск тромбоэмболических осложнений. Уровень тромбоцитов на риск развития тромбозов не влияет, но он играет определенную роль при развитии кровотечений. Для предотвращения развития тромбозов необходимо устранение сердечно-сосудистых факторов риска, а также назначение антиагрегантов.
Кроме того, для лечения эритремии применяются следующие методы:
- Удаление избытка эритроцитов в крови. Может осуществляться либо с помощью гемоэкскурсии (обычное кровопускание), либо эритроцитафереза (удаление непосредственно эритроцитов).
- Циторедуктивная терапия. Применяются цитостатики и интерфероны.
- Лечение осложнений уже развившихся тромбозов.
Гемоэкскурсии (кровопускание) при эритремии
Кровопускание применяется для уменьшения объема циркулирующей крови. Объем гемоэкскурсии составляет 250-500 мл за одну процедуру, после чего недостающий объем жидкости восполняют физраствором. Либо вторым вариантом является предварительная инфузия антиагрегантов вместе с физраствором в объеме, превышающем удаляемый объем крови. Сеансы проводят через день до достижения гематокрита до 40-45%. Пожилым пациентам сеансы выполняют 2 раза в неделю, либо снижают объем гемоэкскурсии.
Эритроцитоферез
Эритроцитаферез относится к методам экстракорпоральной детоксикации, который пришел на смену кровопусканию. В его основе лежит удаление эритроцитов с последующим возвратом плазмы и восполнением объема растворами кристаллоидов или коллоидов. За один сеанс можно удалить до 400 мл эритроцитов.
Препараты ацетилсалициловой кислоты
Препараты ацетилсалициловой кислоты, или аспирина, назначаются для предотвращения тромбических осложнений. Аспирин должен назначаться каждому больному эритремией при отсутствии противопоказаний. Если они есть, назначают клопидогель или тиклопидин.
Циторедуктивная терапия
Гидроксимочевина
Теоретически гидроскимочевина может применяться в рамках терапии первой линии у пациентов любого возраста. Но из-за того, что она обладает генотоксичностью и может спровоцировать лейкозогенный эффект, пациентам младше 50 лет и беременным женщинам такое лечение в рамках первой линии не рекомендуется. Также требуется отмена лечения при непереносимости и неэффективности терапии. Критерием непереносимости гидроксимочевины является наличие хотя бы одного из этих симптомов:
- Превышение гематокрита более 45% через 3 месяца после лечения гидроксимочевиной в дозировке 2000 мг/день.
- Отсутствие контроля миелопролиферации. Об этом может свидетельствовать уровень тромбоцитов более 400×10 9 и лейкоцитов выше 10×10 9 через 3 месяца лечения.
- Сохранение увеличенной селезенки (более 10 см ниже края реберной дуги) или невозможность устранения симптомов спленомегалии.
- Миелопения при применении минимальных дозировок препаратов — уровень тромбоцитов ниже 100×10*9 или гемоглобина ниже 100 мг/л.
- Язвы на голенях.
- Поражение кожи и слизистых оболочек.
- Нарушение работы ЖКТ.
- Пневмониты.
- Лихорадка.
Интерферон альфа
Интерферон альфа обладает высокой эффективностью в отношении эритремии и позволяет у части пациентов добиться молекулярного ответа. Также он хорошо купирует плеторальный синдром, снижая выраженность зуда. Однако его широкое применение ограничивает плохая переносимость. В основном его рекомендуют пациентам младше 50 лет.
Бусульфан
С помощью бусульфана возможен активный контроль эритремии, однако при длительном его применении повышается риск развития вторичных лейкозов. Поэтому его применяют у пациентов старше 70 лет при непереносимости гидроксимочевины и интерферонов.
Прогноз при эритремии
В целом прогноз при эритремии относительно благоприятный. Общая 10-летняя выживаемость составляет около 75%. Но заболевание может трансформироваться в острый миелобластный лейкоз (риск 5%) либо в миелофиброз (риск чуть менее 10%).
Симптомы эритремии (головные боли, боли в костях, парестезии) могут ухудшать качество жизни пациентов. Основной причиной гибели пациентов являются тромбозы, кровотечения и тяжелые инфекции, риск которых увеличивается при трансформации патологии в острый миелобластный лейкоз или в миелофиброз.
Лечение эритремии в клинике «Евроонко» проводится с применением современных лечебных протоколов. В сложных случаях решение принимается коллегиально консилиумом специалистов. Важное значение мы уделяем наблюдению пациента в период ремиссии. Это также позволяет достигать увеличения эффективности проводимого лечения.
Синдром (болезнь) Рейтера
Синдром (болезнь) Рейтера - ревматическое заболевание, характеризующееся сочетанным поражением урогенитального тракта (уретритом и простатитом), суставов (моно- или полиартритом) и слизистой глаз (конъюнктивитом), развивающимися последовательно или одновременно. В основе синдрома Рейтера лежит аутоиммунный процесс, вызванный кишечной или мочеполовой инфекцией. Диагностическими критериями являются связь с перенесенной инфекцией, лабораторное выявление возбудителя и характерных изменений крови, клинический симптомокомплекс. Лечение включает антибиотикотерапию инфекции и противовоспалительную терапию артрита. Синдром Рейтера имеет тенденцию к рецидивам и хронизации процесса.
Общие сведения
В 80% случаев болезнь Рейтера атакует молодых мужчин от 20 до 40 лет, реже - женщин и исключительно редко - детей. Ведущим этиологическим агентом синдрома Рейтера служит хламидия - микроорганизм, способный к длительному паразитированию в клетках хозяина в виде цитоплазматических включений. Кроме того, синдром Рейтера может развиваться после перенесенного колита, вызванного шигеллой, иерсинией, сальмонеллой, а также провоцироваться уреаплазменной инфекцией. Предполагается, что перечисленные возбудители благодаря своей антигенной структуре вызывают определенные иммунологические реакции у генетически склонных лиц.
В течении синдрома Рейтера выделяют две стадии: инфекционную, характеризующуюся нахождением возбудителя в мочеполовом или кишечном тракте, и иммунопатологическую, сопровождающуюся иммунокомплексной реакцией с поражением конъюнктивы и синовиальной мембраны суставов.
Классификация синдрома (болезни) Рейтера
С учетом этиофактора различаются спорадическая и эпидемическая (постэнтероколитическая) формы заболевания. Спорадическая форма, или болезнь Рейтера, развивается после перенесенной мочеполовой инфекции; эпидемическая - синдром Рейтера - после энтероколитов различной этиологической природы (дизентерийных, иерсиниозных, сальмонеллезных, недифференцированных).
Течение болезни или синдрома Рейтера может быть острым (до 6 месяцев), затяжным (до года) или хроническим (длительнее 1 года).
Клиника синдрома (болезни) Рейтера
Для болезни (синдрома) Рейтера специфическими являются поражение урогенитального тракта, глаз, суставных тканей, слизистых и кожи. При болезни Рейтера первым манифестирует уретрит, сопровождающийся дизурическими расстройствами, скудным слизистым отделяемым, ощущениями дискомфорта и гиперемией в области наружной уретры. При бессимптомной клинике наличие воспаления определяется на основании увеличения числа лейкоцитов в мазке. Вслед за уретритом при синдроме Рейтера развивается глазная симптоматика, чаще имеющая форму конъюнктивита, реже - ирита, увеита, иридоциклита, ретинита, кератита, ретробульбарного неврита. Явления конъюнктивита могут быть мало продолжительными и слабо выраженными, незаметными для пациента.
Определяющим признаком синдрома Рейтера является реактивный артрит, который дебютирует спустя 1-1,5 месяца после урогенитальной инфекции. Для синдрома Рейтера типично асимметричное вовлечение суставов ног - межфаланговых, плюснефаланговых, голеностопных, коленных. Артралгии более выражены утром и по ночам, кожа в области суставов гиперемирована, в полости суставов образуется выпот.
Синдром Рейтера отличается последовательным лестничным (от проксимальных к дистальным) вовлечением суставов в течение нескольких дней. При урогенном артрите развиваются отеки, сосискообразные дефигурации пальцев; кожа над ними приобретает окраску синюшно-багрового цвета. При болезни Рейтера может развиваться тендинит, пяточный бурсит, пяточные шпоры, поражение крестцово-подвздошных суставов - сакроилеит.
Слизистые оболочки и кожные покровы при синдроме Рейтера поражаются у 30-50% пациентов. Характерны язвенные изменения слизистой рта (глоссит, стоматит) и полового члена (баланит, баланопостит). На коже появляются красные папулы, эритематозные пятна, очаги кератодермии - участки гиперемии кожи с гиперкератозом, шелушением и трещинами преимущественно на ладонях и стопах. При синдроме Рейтера возможно развитие лимфаденопатии, миокардита, миокардиодистрофии, очаговой пневмонии, плеврита, полиневритов, нефрита и амилоидоза почек.
При осложненной форме синдрома Рейтера развиваются дисфункции суставов, расстройства зрения, эректильные нарушения, бесплодие. В поздней фазе болезни Рейтера могут поражаться почки, аорта, сердце.
Диагностика синдрома (болезни) Рейтера
В ходе диагностики пациент с подозрением на синдром Рейтера может быть направлен на консультацию ревматолога, венеролога, уролога, офтальмолога, гинеколога. Общеклинические анализы при синдроме Рейтера выявляют гипохромную анемию, рост СОЭ и лейкоцитоз крови. В пробах мочи (трехстаканной, по Аддису-Каковскому и Нечипоренко) определяется лейкоцитурия. Микроскопия простатического секрета показывает увеличение лейкоцитов (>10) в поле зрения и снижение числа лецитиновых телец. Изменения биохимии крови при синдроме Рейтера характеризуются повышением α2- и β-глобулинов, фибрина, сиаловых кислот, серомукоида; наличием С-реактивного протеина, отрицательной пробой на РФ.
Цитологические исследования соскобов уретры, шейки матки, конъюнктивы, синовиального экссудата, спермы, секрета простаты с окрашиванием по Романовскому-Гимзе обнаруживает хламидии в виде внутриклеточных цитоплазматических включений. В диагностике синдрома Рейтера широко используется метод обнаружения ДНК возбудителя в биоматериале (ПЦР). В крови хламидийные и др. антитела выявляются с помощью серологических реакций - ИФА, РСК, РНГА. Специфическим признаком синдрома Рейтера является носительство антигена HLA 27.
В анализе синовиальной жидкости, взятой путем пункции сустава, определяются воспалительные изменения - рыхлость муцинового сгустка, лейкоцитоз (10-50×109/л), нейтрофилез свыше 70%, наличие цитофагоцитирующих макрофагов, хламидийных антител и антигенов, повышенная активность комплемента, РФ не выявляется. При рентгенографическом исследовании суставов выявляются признаки несимметричного параартикулярного остеопороза, уменьшения размеров суставных щелей, эрозивной деструкции костей стоп, наличия пяточных шпор и шпор пястных костей, тел позвонков, у трети пациентов - односторонний сакроилеит.
При диагностике синдрома Рейтера принимаются во внимание анамнестические сведения (связь заболевания с урогенитальной или кишечной инфекцией); наличие симптомов конъюнктивита, реактивного артрита, кожных проявлений; лабораторное подтверждение возбудителя в эпителиальных соскобах.
Лечение синдрома (болезни) Рейтера
Тактика лечения синдрома Рейтера предусматривает проведение антибиотикотерапии (для обоих половых партнеров), иммунокоррекции, противовоспалительного курса и симптоматической терапии. Антибиотикотерапия включает 2-3 последовательных курса (по 2-3 недели) препаратами из различных фармакологических групп: тетрациклинами (доксициклин), фторхинолонами (ломефлоксацин, офлоксацин, ципрофлоксацин) и макролидами (кларитромицин, азитромицин, эритромицин и др.). При хламидийной инфекции предпочтение отдается доксициклину. Одновременно с антибиотикотерапией назначается противогрибковые препараты, поливитамины, гепатопротекторы, протеолитические ферменты (панкреатин, трипсин, химотрипсин).
Иммунокоррегирующая терапия при синдроме Рейтера включает использование иммуномодуляторов (препаратов тимуса), адаптогенов, индукторов интерферона ( оксодигидроакридинилацетата натрия, акридонуксусная кислота в комбинации с N-метилглюкамином), а также УФОК, надвенной и внутривенной квантовой терапии. При тяжелых артралгических атаках и высокой активности воспаления проводится дезинтоксикационная и антигистаминная терапия. В целях дезинтоксикации при болезни Рейтера показана экстракорпоральная гемокоррекция - проведение плазмафереза, каскадной фильтрации плазмы и криоафереза.
Для подавления внутрисуставного воспаления при синдроме Рейтера используются НПВС (рофекоксиб, целекоксиб, нимесулид, мелоксикам), глюкокортикостероиды (бетаметазон, преднизолон), базисные препараты (сульфасалазин, метотрексат). При наличии внутрисуставного экссудата проводится лечебная пункция сустава с введением пролонгированных глюкокортикоидов (бетаметазона, метилпреднизолона). Местно накладываются компрессы с раствором диметилсульфоксида, обезболивающими и противовоспалительными мазями.
Стихание явлений острого артрита при синдроме Рейтера позволяет подключить физиотерапевтические сеансы фонофореза с протеолитическими ферментами, глюкокортикоидами, хондропротекторами; УВЧ, диатермию, магнитотерапию, лазеротерапию, массаж, грязелечение, сероводородные и радоновые ванны. В комплексе с терапией собственно синдрома Рейтера проводится лечение других экстрагенитальных очагов воспаления.
Прогноз и профилактика синдрома (болезни) Рейтера
Динамика течения синдрома Рейтера преимущественно благоприятная. У большей части пациентов через полгода заболевание переходит в стойкую ремиссию, что, однако, не исключает обострения болезни Рейтера много лет спустя. У четверти пациентов артрит переходит в хроническую фазу, приводя к дисфункции суставов, атрофии мышц, развитию плоскостопия. Исходом синдрома Рейтера может служить амилоидоз и другие висцеропатии.
Профилактика синдрома (болезни) Рейтера включает предупреждение кишечных и урогенитальных инфекций, проведение своевременной этиотропной терапии уретритов и энтероколитов.
Хронический энтерит или синдром малабсорбции?
В терапевтической практике у больных с тонкокишечной формой диареи весьма часто диагностируется "хронический энтерит". Этот привычный для российских врачей диагноз не имеет четких критериев и не может быть реально подтвержден (например, гистологически). В классификации ВОЗ "хронический энтерит" не упоминается, а к хроническим воспалениям кишечника относят только болезнь Крона и язвенный колит. По-видимому, целесообразнее использовать диагноз "синдром малабсорбции" с уточнением его причин в каждом конкретном случае. Статья известного петербургского гастроэнтеролога, д-ра мед. наук, профессора кафедры пропедевтики внутренних болезней СПбГМУ им. акад. И.П. Павлова Евгения Симоновича Рысса посвящена этиологии, клинике и диагностике синдрома малабсорбции (СМ).
Термин "малабсорбция" (от лат. "mal" - болезнь, "аb" - от, из и "sorbeo" - поглощаю) в буквальном переводе означает "плохое всасывание". Однако правильнее трактовать это понятие как симптомокомплекс, обусловленный нарушением переваривания (малдигестия) и собственного всасывания (малабсорбция) в тонкой кишке одного или нескольких питательных веществ. СМ проявляется хронической диареей, приводит к расстройствам питания и тяжелым метаболическим сдвигам. При этом происходит нарушение транспорта пищевых веществ, электролитов и витаминов через энтероциты в лимфатические и кровеносные сосуды ворсинок тонкой кишки.
Этиология и патогенез
Общепринятой международной классификации синдрома малабсорбции нет. Существует множество причин, приводящих к СМ (таблица). Принято различать врожденную, первичную и вторичную малабсорбцию. К врожденному СМ относятся различные ферментопатии, к первичному СМ - патология абсорбирующего эпителия тонкой кишки (целиакия, тропическая спру и др.), к вторичному СМ - большая группа заболеваний, при которых (вторично) поражаются отдельные слои или все стенки тонкой кишки, а также другие органы. Конечно, такое разделение удобно, но во многом условно. Основными звеньями патогенеза СМ являются:
- нарушение полостного пищеварения,
- недостаточность мембранного (пристеночного) пищеварения,
- нарушение собственного всасывания и транспорта нутриентов через кишечную стенку.
Среди заболеваний тонкой кишки, приводящих к развитию СМ, центральное место занимают интестинальные ферментопатии (энзимопатии) - патологические состояния, обусловленные отсутствием или снижением активности одного или нескольких кишечных ферментов. Дисахиридазы тонкой кишки (лактаза, мальтаза, инвертаза, трегалаза) обеспечивают процессы гидролиза углеводов-дисахаридов. Недостаточность этих ферментов клинически проявляется синдромом непереносимости и нарушением всасывания продуктов питания, содержащих соответствующие углеводы. Наиболее часто встречается врожденный или приобретенный дефицит лактазы, расщепляющей молочный сахар, что сопровождается непереносимостью молока и молочных продуктов. Дефицит трегалазы, расщепляющей трегалозу, которая содержится в грибах, встречается достаточно редко и, как правило, бывает врожденным. Крайне важную роль в развитии СМ играет кишечный дисбактериоз. Изменение микроэкологии тонкой кишки затрагивает основные процессы ассимиляции пищевых веществ. Выделяют три основных механизма влияния микробной флоры на состояние слизистой оболочки тонкой кишки (схема).
Таблица. Причины синдрома малабсорбции по Е.А. Белоусовой, А.Р. Златкиной, 1998)
I. Гастрогенные (и агастральные): хронические гастриты с секреторной недостаточностью, резекция желудка, демпинг-синдром
II. Гепатогенные: хронические гепатиты, циррозы печени, холестаз
III. Панкреатогенные: хронический панкреатит, муковисцероз, резекция поджелудочной железы.
IV. Энтерогенные:
I. Неинфекционные ферментопатии (недостаточность дисахаридаз, лактазы, сахаразы, трегелазы и др.), целиакия (глютеновая болезнь); тропическая спру; экссудативная энтеропатия; язвенный колит, болезнь Крона; кишечный дисбактериоз.
II. Инфекционные: бактериальные, вирусные, паразитарные, глистные инвазии
V. Сосудистые: хроническая интенстициальная ишемия (гликемический энтерит, ишемический колит).
VI. Системные заболевания с висцеральными проявлениями: амилоидоз, склеродермия, болезнь Уиппла, лимфома, васкулиты
VII. Эндокринные: диабетическая энтеропатия
VIII. Лекарственные, радиационные, токсические (алкоголь, уремия).
Схема. Пути влияния микробного обсеменения тонкой кишки на процессы переваривания и всасывания
Клиника
Клиническая картина СМ весьма многообразна и зависит от основной патологии. В развитии клинических симптомов имеет значение степень компенсации нарушенных функций различых органов, выраженность дисбактериоза. Проявления СМ весьма вариабельны - от полного отсутствия видимых признаков до глубоких расстройств питания, обусловленных нарушениями всасывания основных питательных веществ - белков, жиров и углеводов. Для СМ характерны слабость, анорексия, утомляемость, метеоризм, урчание в животе - на фоне прогрессирующего похудания. Все эти симптомы малоспецифичны и могут встречаться при других патологических состояниях. Ведущий клинический симптом - диарея - является следствием нарушения процессов переваривания и всасывания основных нутриентов.
Для диареи, сопровождающей заболевания тонкой кишки, характерно наличие жидкого, пенистого, водянистого, объемного стула без патологических примесей, от 3 до 5 раз в сутки, чаще во второй половине дня. Так как при тонкокишечной диарее в патологический процесс не вовлечена толстая кишка, такие поносы протекают без боли. Однако могут отмечаться ноющие и тупые боли вокруг пупка, связанные с нарушением моторики кишечника. Обычно выделяют 4 типа диареи: осмотический, секреторный, моторный, экссудативный. При СМ диарея носит преимущественно осмотический характер. В просвете тонкой кишки происходит накопление осмотически активных непереваренных нутриентов. По градиенту концентрации происходит выход воды в просвет кишки, увеличивается масса жидкого химуса и, как следствие, возникает диарея. Под влиянием бактериальных энтеротоксинов иногда присоединяется и секреторный компонент, усугубляющий диарею.
При СМ диарея, как правило, сопровождается стеатореeй, которая обусловлена либо дефицитом панкреатической липазы, либо снижением ее активности, связанной с преждевременнои деконъюгацией желчных кислот.
Из-за резкого нарушения всасывания триглицеридов в процессе СМ может возникать дефицит жирорастворимых витаминов. Прежде всего это выражается в недостаточности витамина В (стоматит, глоссит), а затем и прочих витаминов этой группы (A, D, Е, К). Необходимо принимать во внимание, что витамин Е является одним из наиболее мощных антиоксидантов, витамин D регулирует всасывание кальция в кишечнике, витамин К относится к факторам свертывания крови, поэтому даже скрытые их дефициты требуют обязательной коррекции. Часто при тяжелых формах СМ возникает анемия смешанного характера, связанная с нарушениями железа в тонкой, а витамина В12 - в подвздошной кишке. Нередко при СМ развиваются и нарушения белкового обмена, обусловленные расстройствами полостного переваривания и всасывания белков или увеличением экскреции их в просвет кишки, из-за повышения проницаемости кишечной стенки. В итоге развивается та или иная степень гипо- и диспротеинемии.
Лабораторная и инструментальная диагностика
Помимо анализа анамнестических данных, для диагностики СМ обязательным является полное обследование желудочно-кишечного тракта - желудка, поджелудочной железы, гепатобилиарной системы, тонкой и толстой кишки.
Рентгенологические методы включают исследование желудка, двенадцатиперстной кишки с барием, проведение ирригоскопии. В ходе рентгенологического исследования тонкой кишки очень важно определить время пассажа контраста по кишечнику, а также оценить рельеф слизистой его оболочки. В последние годы стала постепенно внедряться интестиноскопия (еюноскопия) с обязательной биопсией для гистологического и гистохимического анализа. Этот метод особенно информативен при целиакии, болезни Крона и Уиппла.
Для оценки переваривания и всасывания жиров, белков и углеводов должна широко использоваться обычная копрограмма.
Обнаружение даже единичных капель нейтрального жира является достоверным признаком стеатореи, поскольку он может появиться в кале лишь при 90% дефиците панкреатической липазы. Следствием нарушения переваривания белков служит креаторея (большое количество непереваренных мышечных волокон), а непереваренные зерна крахмала (амилорея) являются следствием снижения активности панкреатической амилазы. Все эти три симптома объединяются в полный энтеральный синдром.
Для экспресс-диагностики тонкокишечного дисбактериоза используется качественный метод - водородный тест. У больных с избыточным ростом бактерий в тонкой кишке отмечается не только повышенное, но и более раннее выделение водорода с выдыхаемым воздухом после углеводной нагрузки (50 г глюкозы). Часто используемый микробиологический метод исследования фекалий неинформативен, так как отражает "бактериальный пейзаж" только толстой кишки. Для диагностики дисахаридазной недостаточности и оценки всасывания дисахаридов прибегают к нагрузочным пробам с сахарозой, лактозой и мальтозой (50 г углевода внутрь) с последующим определением уровня глюкозы в крови каждые 15, 30 и 60 минут после нагрузки. Отсутствие повышения уровня глюкозы в течение первого часа указывает на нарушение расщепления и всасывания тех или иных углеводов.
Классическим методом оценки всасывательной функции тонкой кишки является тест с Д-ксилозой, которая всасывается неповрежденной слизистой оболочкой тонкой кишки, поступает в кровь и выводится почками. При нормальной абсорбции после приема внутрь 25 г ксилозы, выведение углевода с мочой должно составлять 5 г в течение 5 часов. При СМ вся принятая Д-ксилоза выделяется только с калом и в моче не определяется.
Со специальной целью используются радиоизотопные методики (оценка всасывания жиров или альбумина, меченных J 131 ).
Особенности формулировки диагноза. Прогноз
СМ либо является врожденной патологией, либо присоединяется ко многим заболеваниям пищеварительной системы. Поэтому при формулировке диагноза необходимо указывать вначале основную патологию, а затем уже и сам симптомокомплекс. Например:
1. Хронический панкреатит в фазе обострения. Синдром малабсорбции (полный энтеральный синдром; анемия смешанного происхождения).
2. Хронический гепатит с синдромом холестаза в фазе обострения. Синдром малабсорбции (стеаторея).
По клиническому течению выделяют латентную форму СМ (выявляющуюся только с помощью функциональных нагрузочных тестов) и клинически выраженную форму с легкими, средне-тяжелыми и тяжелыми проявлениями.
Как правило, СМ течет волнообразно годами и десятилетиями, что определяет относительно благоприятный прогноз для жизни пациента. Однако при этом существенно страдает качество жизни, резко снижается работоспособность, физическая активность. Это особенно четко проявляется у лиц, занятых физическим трудом. При хроническом тяжелом течении СМ, вследствие дистрофических изменений внутренних органов, развивается жировая инфильтрация печени (стеатоз), прогрессирующая мышечная атрофия, полигландулярная недостаточность (гипотиреоз, вторичная недостаточность надпочечников, дисфункция яичников), кахексия. Кроме того, при тяжелых формах СМ может развиваться энцефалопатия с психическими нарушениями - вплоть до психозов.
Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
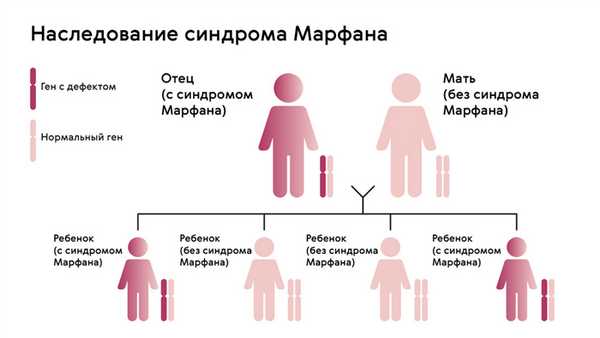
Рисунок. 1. Схема наследования синдрома Марфана. Источник: МедПортал
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5-10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25-50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
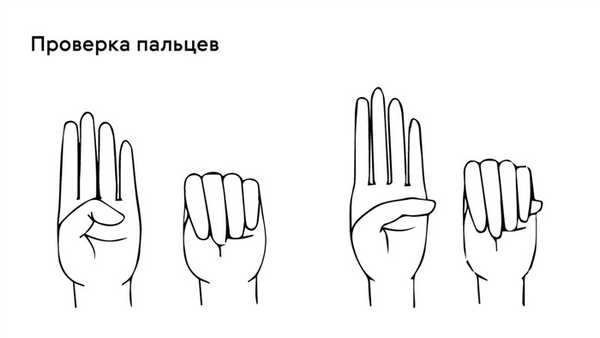
Рисунок 4. Проверка на арахнодактилию. Источник
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65-100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7-18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Читайте также:
- Анатомия: Слезный аппарат. Слезная железа, glandula lacrimalis. Слезный мешок, saccus lacrimalis.
- Техника операции при невриноме яремного отверстия основания черепа
- Нарушения речи при миастении. Клиника и диагностика миастенической реакции
- Консистенция слепой кишки. Объем и форма слепой кишки
- Этиология остановки сердца. Механизм остановки сердца.