Синдром Форсиуса-Эрикссона (Forsius-Eriksson) - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 28.01.2026
Что такое синдром Леннокса — Гасто? Причины возникновения, диагностику и методы лечения разберем в статье доктора Поздняковой А. А., детского невролога со стажем в 5 лет.
Над статьей доктора Поздняковой А. А. работали литературный редактор Вера Васина , научный редактор Сергей Макаров и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Синдром Леннокса — Гасто (Lennox — Gastaut Syndrome) — это тяжёлая форма эпилепсии, которая начинается в детстве.
Заболевание проявляется выраженным снижением интеллекта и частыми приступами трёх видов:
- тоническими — внезапное напряжение всех мышц, основной признак заболевания;
- атоническими — кратковременное расслабление мышц с потерей сознания;
- атипичными абсансами — сокращение мышц, потеря сознания, автоматические действия; атипичные абсансы длятся дольше типичных, более постепенно начинаются и заканчиваются, их сложнее контролировать и лечить [14] .
Приступы при синдроме Леннокса — Гасто вызваны внезапными вспышками аномальной электрической активности в головном мозге и с трудом поддаются лечению противосудорожными препаратами.
Распространённость синдрома Леннокса — Гасто
Синдром Леннокса — Гасто — это редкое заболевание. Его выявляют у 2 из 100 000 детей, чаще у мальчиков. Среди всех форм эпилепсии на этот синдром приходится 2-5 % случаев [4] .
Заболевание проявляется в возрасте от года до 9 лет, чаще в 2-4 года [1] .
Причины синдрома Леннокса — Гасто
Выделяют криптогенный и симптоматический варианты синдрома.
Примерно у 40 % детей причина болезни неясна, в таких случаях синдром называют идиопатическим, или криптогенным [4] .
Вторичный, или симптоматический, вариант синдрома Леннокса — Гасто — это проявление диффузного поражения головного мозга (диффузные поражения, в отличие от очаговых, затрагивают весь мозг, а не отдельный участок). Причинами такого поражения могут быть:
- тяжёлые генетические заболевания;
- энцефалит и менингит;
- черепно-мозговые и родовые травмы;
- пороки развития коры головного мозга;
- метаболические нарушения (митохондриальные заболевания);
- опухоли головного мозга; .
При симптоматическом варианте прогноз хуже, чем при идиопатическом. У 30 % детей развитию заболевания предшествуют инфантильные спазмы (синдром Веста) [4] . Спазмы не являются причиной синдрома Леннокса — Гасто, но у таких детей болезнь протекает тяжелее.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Леннокса — Гасто
Для синдрома Леннокса — Гасто характерны различные типы приступов: тонические и атонические припадки, атипичные абсансы.
При тонических приступах напрягаются мышцы всего тела, рук и ног, чаще с разгибанием, чем со сгибанием. Эти приступы непродолжительны: не более полминуты, обычно около 10 секунд. Часто они возникают по ночам и могут остаться незамеченными для родителей ребёнка.
Атонические приступы состоят из миоклонического и атонического компонентов. При миоклоническом приступе пациент непроизвольно вздрагивает, выбрасывает руки вперёд и подгибает ноги. Затем следует атонический приступ, при котором слабеют мышцы, человек теряет сознание и падает, что нередко приводит к травмам. Как правило, атонический приступ длится всего несколько секунд.
Атипичные абсансы проявляются кратковременной потерей сознания, не больше чем на несколько минут. Во время приступа подёргиваются мышцы лица, пациент высовывает язык, кивает и часто моргает. Также человек может совершать автоматические действия, например включить свет, о чём в дальнейшем забывает.
У взрослых пациентов могут возникать генерализованные тонико-клонические приступы. Они проявляются потерей сознания, судорогами по всему телу, прикусом языка, непроизвольным мочеиспусканием. У детей таких приступов, как правило, не бывает.
Бессудорожный эпилептический статус часто встречается при синдроме Леннокса — Гасто. Это состояние может длиться от пары часов до нескольких недель. Проявляется в двух основных формах: спутанным сознанием и тоническими приступами. У пациента снижается психическая и двигательная активность, вплоть до ступора, лицо становится похожим на маску, слабеют мышцы, возникает слюнотечение, временами теряется ясность сознания. Такое состояние трудно распознать при тяжёлых когнитивных нарушениях — эпизоды спутанного сознания можно принять за нарушения интеллекта. Вероятно, бессудорожный статус усугубляет уже имеющиеся интеллектуальные нарушения [12] .
Умственная отсталость тоже относится к классическим проявлениям синдрома Леннокса — Гасто. У некоторых пациентов она наблюдается ещё до начала болезни. В течение пяти лет после начала заболевания умственная отсталость развивается у 90 % пациентов, но в литературе описаны редкие случаи нормального интеллекта при синдроме Леннокса — Гасто.
Также у многих пациентов наблюдается агрессивность, импульсивность и аутичные черты (задержка речи, повторяющиеся стереотипные движения, трудности в общении, ограниченный круг интересов) [3] .
Патогенез синдрома Леннокса — Гасто
Патогенез синдрома Леннокса — Гасто до конца не изучен. Это связано с разнообразием симптомов и причин заболевания, а также с ограниченными данными о генетических причинах болезни.
В основе синдрома Леннокса — Гасто лежит повышенная возбудимость коры головного мозга. Предполагается, что у детей с этим заболеванием нарушено формирование синапсов между нервными клетками, из-за чего в дальнейшем повышается их возбудимость. К такому нарушению может приводить повреждение как корковых, так и подкорковых структур (кортикоретикулярных связей и таламуса).
Нарушения в работе этих структур могут быть вызваны различными заболеваниями: гипоксически-ишемической энцефалопатией, менингоэнцефалитом, нейрокожными синдромами, опухолями и пороками развития головного мозга. Синдром Леннокса — Гасто может развиться при любом типе повреждения головного мозга.
Обычно в начале заболевания возникают тонические приступы, затем присоединяются атипичные абсансы, миоклонические и тонико-клонические приступы. Тонический компонент связан с поражением мезэнцефальной ретикулярной формации, а клонический — с поражением переднего мозга. Отсюда следует, что присоединение атипичных абсансов и клонических приступов вызвано возрастающим участием коры головного мозга.

При некоторых состояниях центральной нервной системы, например при кровоизлиянии или дефиците кислорода, повышается возбудимость в коре головного мозга. Неконтролируемая электрическая активность, возникающая при повышенной возбудимости, проявляется симптомами синдрома Леннокса — Гасто [5] .
Классификация и стадии развития синдрома Леннокса — Гасто
Согласно классификации Международной противоэпилептической лиги (ILAE), синдром Леннокса — Гасто относится к генерализованной криптогенной или симптоматической формам эпилепсии, т. е. болезнь вызвана аномальной электрической активностью в обоих полушариях, но причина может быть как выявлена, так и неизвестна.
Согласно Проекту классификации ILAE 2001 года, синдром Леннокса — Гасто входит в группу детских эпилептических энцефалопатий. К тяжёлым детским энцефалопатиями также относят синдромы Веста, Драве и Дуза [2] .
В Международной классификации болезней (МКБ-10) синдром Леннокса — Гасто кодируется как G40.4 Другие виды генерализованной эпилепсии и эпилептических синдромов.
Осложнения синдрома Леннокса — Гасто
Синдром Леннокса — Гасто часто приводит к развитию эпилептического статуса, из-за чего значительно ухудшаются умственные способности. Пациент может утратить необходимые для жизни навыки, и в дальнейшем ему потребуется постоянный уход.
Во время эпистатуса слабеют и беспорядочно подёргиваются мышцы лица и рук, снижается двигательная и психическая активность. Контакт с пациентом затруднён, он может лежать неподвижно или его движения становятся замедленными. Чаще всего приступы происходят по утрам. Эпистатус свидетельствует о неблагоприятном течении заболевания и может угрожать жизни.
У многих пациентов с синдромом Леннокса — Гасто развиваются экстрапирамидные и мозжечковые расстройства, которые проявляются слюнотечением, нарушением походки, координации, глотания и речи (дисфагией и диазартрией). При нарастающей дисфагии трудно есть и принимать лекарства, может потребоваться установка гастростомы [1] .
Нарушение походки и атонические приступы часто приводят к падениям и черепно-мозговым травмам. Нередко пациентам приходится пользоваться инвалидной коляской.
Также синдром Леннокса — Гасто может осложняться нарушением сна и психозом.
Диагностика синдрома Леннокса — Гасто
Диагностика синдрома Леннокса — Гасто включает сбор анамнеза, электроэнцефалографию (ЭЭГ), магнитно-резонансную или компьютерную томографию (МРТ или КТ). Может потребоваться генетическое тестирование.
Сбор жалоб и истории болезни
Для постановки диагноза важна подробная история болезни: как протекала беременность, роды и перинатальный период, какие симптомы беспокоят.
Неврологические нарушения при идиопатическом синдроме Леннокса — Гасто обычно отсутствуют. При симптоматических формах могут возникать нарушения со стороны нервной системы: паралич, неустойчивость при ходьбе, микроцефалия, нарушения речи, косоглазие. Также на синдром Леннокса — Гасто будет указывать задержка в развитии или потеря уже приобретённых навыков и устойчивость судорог к противоэпилептическим препаратам.
Магнитно-резонансная и компьютерная томография (МРТ и КТ)
Характер нарушений зависит от формы заболевания. Идиопатический вариант синдрома Леннокса — Гасто протекает без структурных изменений головного мозга. У многих пациентов на КТ и МРТ выявляют диффузную атрофию головного мозга, при которой отмирают нейроны и разрушаются нервные связи. При симптоматической форме наблюдаются очаговые поражения коры головного мозга.
Электроэнцефалография (ЭЭГ)
При диагностике синдрома Леннокса — Гасто проводится ЭЭГ с записью во время ночного сна и бодрствования. ЭЭГ позволяет выявить эпилептиформную активность — острые волны и пики. Для записи и оценки различных типов приступов может проводиться видео-ЭЭГ-мониторинг.

В самом начале заболевания на ЭЭГ заметно только изменение основной биоэлектрической активности. Кроме того, судороги и клинические симптомы развиваются с течением времени, отставание в развитии также не всегда проявляется в начале болезни. Поэтому часто диагноз устанавливают после нескольких лет наблюдения за пациентом [3] .
Синдром Леннокса — Гасто проявляется характерным паттерном ЭЭГ: медленными спайк-волновыми комплексами между приступами и генерализованной пароксизмальной быстрой активностью во сне (т. е. изменениями потенциала в форме острых волн, пиков и др.). Спайк-волна — это комплекс, который имеет высокую амплитуду и возникает при комбинации спайка с медленной волной.
Чтобы установить диагноз, необходимо наличие минимум двух типов генерализованных приступов и медленных спайк-волн на ЭЭГ в состоянии бодрствования.
![ЭЭГ при синдроме Леннокса — Гасто [15]](https://probolezny.ru/media/bolezny/sindrom-lennoksa-gasto/eeg-pri-sindrome-lennoksa-gasto-15_s.jpg)
Генетическое тестирование
Иногда проводится тестирование для выявления генетических нарушений: дефекта транспортёра глюкозы (SLC2A1), позднего детского нейронального липофусциноза цероидов (CLN2) и туберозного склероза (TSC 1,2). У детей с такими нарушениями часто наблюдаются эпилептические приступы.
Дифференциальная диагностика
Синдром Леннокса — Гасто следует отличать от других ранних эпилептических энцефалопатий:
- от синдромов Драве, Дуза, Ландау — Клеффнера, Веста и Ангельмана;
- атипичной эпилепсии детства с центро-темпоральными спайками;
- эпилептической энцефалопатии с продолженной спайк-волновой активностью во сне;
- синдрома псевдо-Леннокса (при такой эпилепсии во время засыпания или пробуждения быстро сокращаются мышцы лица и плеч, нарушается речь и усиливается потливость).
Также проводится дифференциальная диагностика с некоторыми наследственно-дегенеративными заболеваниями, например нейрофиброматозом 1-го типа [3] [12] .
Лечение синдрома Леннокса — Гасто
При синдроме Леннокса — Гасто применяют противосудорожные препараты, кортикостероды, кетогенную диету, стимуляцию блуждающего нерва и хирургическое лечение. Не доказано, что какой-то определённый препарат помогает лучше, чем другой. Поэтому терапию для каждого пациента подбирают индивидуально [8] .
Медикаментозное лечение
Судороги при синдроме Леннокса — Гасто устойчивы к лечению — полностью устранить их не получится, но можно уменьшить частоту.
Наиболее эффективны противосудорожные препараты широкого спектра: Вальпроат натрия, Клобазам, Ламотриджин, Топирамат и Зонисамид. Вальпроат натрия — это препарат первой линии в лечении синдрома Леннокса — Гасто. Часто сочетают два лекарства, например Вальпроат натрия с Ламотриджином или Клобазамом. Ламотриджин хорошо помогает при атонических припадках. Также могут быть эффективны Леветирацетам и Перампанел.
Если противосудорожные препараты не помогают, то назначают кортикостероиды: адренокортикотропный гормон, Преднизолон, Метилпреднизолон. При приёме кортикостероидов часто возникают побочные эффекты, например отёки, высокое давление, ожирение.
Для экстренного лечения частых атипичных абсансов, бессудорожного эпистатуса или других тяжёлых приступов обычно назначают бензодиазепины. При бессудорожном эпилептическом статусе может применяться короткий курс Клобазама или бензодиазепинов, высокие дозы кортикостероидов или вальпроевой кислоты. В некоторых случаях бензодиазепины могут увеличить частоту тонических судорог.
Кетогенная диета
Кетогенная диета показана, если лечение противосудорожными препаратами неэффективно. При такой диете потребляют много жиров, умеренное количество белков и мало углеводов. В рацион входят орехи, сыр, масло, жирная рыба. Исключают конфеты, картофель, выпечку, мёд, шоколад [13] .
Стимуляция блуждающего нерва
Стимуляцию блуждающего нерва проводят с помощью вживлённого электрода, подключённого к генератору импульсов. Этот метод рекомендован пациентам с атоническими приступами, которые могут привести к травмам и инвалидности. Стимуляция блуждающего нерва позволяет уменьшить частоту приступов.

Хирургическое лечение
При синдроме Леннокса — Гасто может проводиться мозолотомия и гемисферэктомия. Операции позволяют облегчить симптомы и в некоторых случаях уменьшить частоту приступов.
Мозолотомия — это рассечение мозолистого тела, т. е. пучка нервных волокон, соединяющих полушария мозга. После операции припадки не могут распространяться из одного полушария в другое.

Гемисферэктомия — это частичное или полное удаление полушария головного мозга. Проводится в редких случаях, когда остальные методы лечения неэффективны.
Прогноз. Профилактика
Судороги при синдроме Леннокса — Гасто часто не поддаются лечению. Они вызывают у пациентов страх перед физической травмой, при постоянных и плохо контролируемых приступах ухудшаются умственные способности. У пациентов часто возникают трудности в учёбе, социальной и личной жизни.
Практически во всех случаях судороги продолжаются и во взрослом возрасте. При этом заболевание сопровождается слюнотечением, нарушением походки, речи и глотания.
Прогноз лучше, если до начала приступов не было поражения мозга и отклонений в интеллектуальном развитии. Обычно если синдрому Леннокса — Гасто предшествовали инфантильные спазмы, то судороги тяжелее контролировать. Умственные способности у таких пациентов ухудшаются сильнее.
Как правило, частота припадков снижается в период полового созревания. Однако у 2/3 пациентов спустя десять лет после начала болезни приступы по-прежнему происходят ежедневно.
Атипичные абсансы иногда сменяются фокальными приступами: двигательными нарушениями, ощущением онемения и ударов током в руках, ногах или лице, покраснением кожи, дискомфортом в верхней части живота.
Гиперактивность, агрессия и аутистические черты могут сохраняться во взрослом возрасте, но чаще развивается медлительность и апатия. Большинство пациентов нуждаются в посторонней помощи, лишь немногие могут жить самостоятельно [7] .
Профилактика заключается в раннем выявлении заболевания, что позволит облегчить симптомы болезни [8] .
Баршоня — Тешендорфа синдром (Th. Barsony, 1887-1942, венгерский рентгенолог; W. Teschendorf, немецкий рентгенолог, XX в.; синонимы: дивертикулы пищевода множественные ложные, дивертикулы пищевода множественные функциональные, пищевод извитой, пищевод четкообразный, пищевод штопорообразный) — болезнь пищевода неизвестной этиологии, характеризующаяся спастическим сокращением его циркулярных мышц, что придает пищеводу четкообразный вид на рентгенограмме; клинически проявляется непостоянной дисфагией и загрудинными болями.
Бойса симптом (Boys) — появление булькающего звука или урчания при надавливании на боковую поверхность шеи; признак большого дивертикула шейного отдела пищевода.
Бушара болезнь (Ch.J. Bouchard, 1837-1915, французский патолог) — гастрэктазия (расширение полости желудка с растяжением его стенок при стенозе привратниковой части или двенадцатиперстной кишки).
Бэррета язва пищевода (N.R. Barret, род. в 1903 г., английский хирург) — язва пищевода, по клинико-морфологическим признакам напоминающая пептическую язву желудка или двенадцатиперстной кишки.
Кушинга эзофагит (Н.W. Cushing, 1869-1939, американский нейрохирург) — острый эзофагит, иногда развивающийся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кушинга язва (Н.W. Cushing) — язва желудка или двенадцатиперстной кишки, иногда развивающаяся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кэмерона синдром (A.J. Cameron) — язвенные или эрозивные повреждения слизистой оболочки грыжевого выпячивания при грыжах пищеводного отверстия диафрагмы, которые сопровождаются хроническим оккультным или явным кровотечением и железодефицитной анемией.
Ларрея грыжа (D.J. Larrey, 1766-1842, французский хирург). Левосторонняя парастернальная диафрагмальная грыжа, выходящая в средостение через грудино-реберный треугольник.
Лихтенштерна симптом (Lichtenstern, синоним дисфагия парадоксальная, d. paradoxalis) — дисфагия, при которой большие порции пищи легче проходят в желудок, чем малые; возможный признак грыжи пищеводного отверстия диафрагмы.
Лиана — Сигье — Вельти синдром (С.С. Lian, 1882-1969, французский врач; F. Siguier, 1909-1972, французский врач; Н.L. Welti, 1895-1970, французский хирург) — сочетание диафрагмальной грыжи, часто осложненной рефлюкс-эзофагитом, с повторными тромбозами и тромбофлебитами сосудов конечностей и нередко с гипохромной анемией; патогенез неясен.
Монтандона синдром (А. Montandon, швейцарский врач, XX век) — приступы длительной дисфагии вследствие миогенного стеноза шейной части пищевода (области пищеводноглоточного соединения), сопровождающиеся попаданием жидкой пищи в гортань, аспирации и приводящие к нарастающему истощению.
Морганьи грыжа (G.B. Morgagni, 1682-1771, итальянский анатом). Правосторонняя парастернальная грыжа.
Харрингтона грыжа (S. W. Harrington, 1889-1975, американский хирург). Антральная грыжа пищеводного отверстия диафрагмы (типы 1 и 2).
Ценкера дивертикул (F.A. Zenker, 1825-1898, немецкий патолог; синоним ценкеровский дивертикул) — мешковидный дивертикул глоточного конца пищевода, образующийся сначала на его задней стенке, а затем распространяющийся и на боковую. В начале заболевания — глоточные парестезии, перемежающаяся дисфагия, сухой кашель. С увеличением дивертикула в нем происходит задержка пищи, урчание при приеме воды. Мешок сдавливает пищевод, усиливается дисфагия, часты аспирационные пневмонии.
Эпонимные симптомы/синдромы, связанные с патологией кишечника
Алапи симптом (Alapy). Отсутствие или незначительное напряжение брюшной стенки при инвагинации кишечника.
Альвареса синдром (W.C. Alvarez, 1884-1978, американский врач, описан в 1949 г.; синонимы: псевдоилеус; истерическое вздутие живота). Преходящее вздутие живота нейрогенной природы. Чаще наблюдается у истеричных или психопатичных женщин.
Бувре признак (1) (L. Bouveret, 1850-1929, французский врач) — выпячивание в области проекции на переднюю брюшную стенку места перехода подвздошной кишки в слепую, наблюдаемое при непроходимости толстой кишки.
Бувре признак (2) (L. Bouveret; синоним Куссмауля симптом, Adolf Kußmaul, 1822-1902, немецкий терапевт) — периодическое выбухание брюшной стенки в надчревной области и левом подреберье при сужении привратника, обусловленное усиленной перистальтикой желудка.
Вербрайка синдром (J.R. Verbryke, род. в 1885 г., американский врач; синоним синдром печеночного перегиба ободочной кишки) — боль и чувство натяжения в подложечной области в сочетании с диспептическими явлениями при наличии сращений желчного пузыря с печеночным углом ободочной кишки.
Вильмса симптом падающей капли (М. Wilms, 1867-1918, немецкий хирург и онколог) — звук падающей капли жидкости, определяющийся аускультативно на фоне шумов перистальтики при непроходимости кишечника.
Гарднера синдром (Е.J. Gardner, 1909-1989, американский врач-генетик) — наследственная болезнь, характеризующаяся множественным полипозом прямой и ободочной кишок в сочетании с доброкачественными опухолями, чаще костей и кожи (остеомы, фибромы, липомы); наследуется по аутосомно-доминантному типу.
Гейбнер-гертеровская болезнь (O.J.L. Heubner, 1843-1926, немецкий педиатр; С.A. Herter, 1865-1910, американский врач и фармаколог; синоним Ги — Гертера — Гейбнера болезнь) — целиакия.
Данса симптом (J. В. H. Dance, 1797-1832, французский врач) — западение брюшной стенки в правой подвздошной области при илеоцекальной инвагинации.
Жанбона синдром (М. Janbon, современный французский терапевт; синоним syndromus choleriformis, enteritis choleriformis). Описан как гастроинтестинальная симптоматика после лечения окситетрациклином. Патогенез заключается в уничтожении антибиотиками физиологической бактериальной флоры кишечника, что способствует развитию патогенной, устойчивой к антибиотикам.
Ирасека — Цюльцера — Уилсона синдром (A. Jirasek, 1880-1960, чехословацкий хирург; W.W. Zuelzer, 1909-1987, американский педиатр; J.L. Wilson, американский педиатр) — аганглиоз толстой кишки врожденный сегментарный. Патоморфологическая форма болезни Гиршспрунга, при которой в толстой кишке имеются одна или две аганглионарные зоны с нормальным участком кишки между ними; при этом аганглиоз не распространяется на прямую кишку.
Кантора симптом (М.О. Cantor, род. в 1907 г., американский хирург) — нитевидные тени в дефектах наполнения кишки; рентгенологический признак колита и регионарного илеита.
Карно симптом (M.O. Carnot) — боль в эпигастральной области, возникающая при резком разгибании туловища. Бывает при спаечной болезни.
Кенига синдром (F. Konig, 1832-1910, немецкий хирург) — сочетание приступов коликообразных болей в животе, метеоризма, чередования запоров и поносов, урчания при пальпации правой подвздошной ямки, наблюдающееся при хронической непроходимости кишечника в области перехода подвздошной кишки в слепую.
Клойбера симптом (Н. Kloiber, немецкий рентгенолог; синоним Клойбера чаши) — наличие на рентгенограмме живота (при вертикальном положении больного) теней, напоминающих чаши с жидкостью; признак скопления жидкости и газа в кишечнике при его непроходимости.
Кронкхайта — Канада синдром (L.М. Cronkhite, род. в 1919 г., американский врач; W.J. Canada, современный американский рентгенолог) — облысение, атрофия ногтей и гиперпигментация кожи при семейном полипозе органов пищеварительного тракта.
Кюсса синдром (G.Е. Kuss, 1877-1967, французский хирург) — хроническая частичная непроходимость кишечника, обусловленная наличием спаек в области малого таза, например при хроническом сальпингите.
Лобри — Сулля синдром (Ch. Laubry, 1872-1941, французский врач; P.L.J. Soulle, 1903-1960, франц. врач) — сочетание избыточного содержания газа в желудке, метеоризма и высокого стояния правого купола диафрагмы при ишемической болезни сердца, обусловленное рефлекторной гипокинезией пищеварительного тракта.
Мачеллы — Дворкена — Биля симптом (T.E. Machella, F.J. Dworken, H.J. Biel; 1952). Синоним: симптом селезеночного угла. Сильная боль в левой половине живота, вызванная растяжением газами селезеночного угла толстой кишки. Облегчение наступает после опорожнения кишечника и отхождения газов. См. Пайра болезнь, синдром.
Матье симптом (A. Mathieu, 1855-1917, французский врач) — шум плеска в пупочной области при толчкообразной пальпации; признак непроходимости кишечника.
Меккеля дивертикул (J.F. Meckel junior, 1781-1833, немецкий анатом). Врожденный дивертикул подвздошной кишки. Правило двоек: 2 дюйма длиной, в 2 футах от илеоцекального клапана (баугиниевой заслонки), у 2% населения.
Образцова признак (В.П. Образцов, 1849-1920, отечественный терапевт) — шум плеска при пальпации слепой кишки; признак хронического колита.
Обуховской больницы симптом — баллонообразное расширение ампулы прямой кишки, определяемое пальцевым исследованием; признак заворота сигмовидной кишки.
Пайра болезнь, синдром (Erwin Payr, австрогерманский хирург, 1871-1946; синонимы: синдром селезеночного изгиба, flexura lienalis syndrome, splenic flexure syndrome). Первично описана как запоры, обусловленные перегибом и сращением поперечной и нисходящей ободочной кишок («двустволка»). Разнообразная симптоматика (в т. ч. кардиалгии) связана с скоплением газа в области изгиба. Одни считают вариантом СРК (облегчение после опорожнения кишечника и отхождения газов), другие - самостоятельной патологией. Иногда называют также синдромом Мачеллы-Дворкена-Била.
Пиулахса — Хедериха (Пиулакса - Эдерика) болезнь, синдром (испанские врачи P. Piulachs, H. Hederich; синоним: тимпанит при синдроме долихомегаколона, tympanites in dolichomegacolon syndrome). Острое паралитическое расширение толстой кишки (без механической непроходимости). Симптомы: резкий метеоризм и боль в животе. Рентгенологически обнаруживают долихомегаколон.
Протопопова синдром (В.П. Протопопов, 1880-1957, отечественный психиатр; синоним Протопопова триада) — сочетание тахикардии, мидриаза (расширения зрачка) и спастического запора, наблюдающееся при депрессиях.
Рапунцель синдром (описан E.D. Vaughan, J.L. Sawyers и H.W. Scott в 1968 г.). Закупорка кишок, вызванная систематическим проглатыванием волос. В кишках больного образуются трихобезоары. Может потребовать хирургического вмешательства. Наблюдается при психопатиях, шизофрении, эпилепсии, олигофрении, преимущественно в детском возрасте. Рапунцель (Rapunzel) - персонаж одноименной сказки братьев Гримм, девушка с длинными косами.
Сейнта синдром (Ch.F.М. Saint, совр. южноафриканский патолог; синонимы: Сейнта триада, Сена синдром — нрк*, Сента синдром — нрк*) — сочетание грыжи пищеводного отверстия диафрагмы, желчнокаменной болезни и дивертикулеза толстой кишки.
Тавеля болезнь (Е. Tavevl, швейцарский хирург) — периколит после аппендэктомии, характеризующийся образованием спаек и сужением толстой кишки, лихорадкой и расстройствами функции кишечника.
Miserere! (помилосердствуй!) от начала католической молитвы "miserere mei" - помилуй меня (Боже), так по-старому называлась каловая рвота (copremesis) при непроходимости кишечника.
Синдром Уотерхауса — Фридериксена (аддисонический криз) - симптомы и лечение
Что такое синдром Уотерхауса — Фридериксена (аддисонический криз)? Причины возникновения, диагностику и методы лечения разберем в статье доктора Троицкой Ирины Николаевны, эндокринолога со стажем в 8 лет.
Над статьей доктора Троицкой Ирины Николаевны работали литературный редактор Вера Васина , научный редактор Екатерина Шрёдер и шеф-редактор Маргарита Тихонова
Синдром Уотерхауса — Фридериксена (Waterhouse — Friderichsen syndrome) — это острая надпочечниковая недостаточность, которая развивается при двустороннем кровоизлиянии в надпочечники. Может возникать на фоне сепсиса. Проявляется слабостью, головокружением, рвотой, диареей, низким давлением, обмороками и судорогами [1] .

Заболевание ещё называют гипоадреналовым кризом, острым гипокортицизмом, кризом надпочечниковой недостаточности и аддисоническим кризом [1] .
Синдром Уотерхауса — Фридериксена — это редкое состояние, которое чаще развивается у детей. Среди всех заболевших 90 % — дети до 9 лет, 70 % — до 2 лет [24] .
Причины синдрома Уотерхауса — Фридериксена
- Бактериальная инфекция. Менингококковая инфекция — это самая распространённая причина кровоизлияния в надпочечники, но к этому состоянию могут привести и другие бактерии: стрептококки, которые вызывают пневмонию, гемофильная и кишечная палочки, золотистый стафилококк, гемолитический стрептококк группы А, гонококк, бледная трепонема и другие микробы [6][8][13] .
- Вирусы: цитомегаловирус, парвовирус, вирус ветряной оспы и Эпштейна — Барр [15][16] .
- Приём антикоагулянтов — препаратов, которые препятствуют свёртыванию крови [17] .
- Генетические заболевания: антифосфолипидный синдром, гемофилия, болезнь Шенлейн — Геноха, системная красная волчанка, узелковый периартериит[1][12] .
- Травма надпочечников [3] .
- Ожоги [1] .
- Адреналэктомия — удаление надпочечников [1] .
- Операция — осложнение после хирургического лечения, например эндопротезирования [3][18] .
Симптомы синдрома Уотерхауса — Фридериксена
К первым проявлениям синдрома Уотерхауса — Фридериксена относятся:
- слабость и упадок сил;
- головокружение;
- ухудшение аппетита;
- снижение артериального давления;
- чаще увеличение и реже замедление частоты сердечных сокращений;
- угнетение рефлексов;
- уменьшение объёма мочи [23] .
При заболевании резко снижается давление, кожа становится бледной и синюшной. Уменьшение уровня кортизола и глюкозы проявляется потерей сознания и судорогами [1] . Присоединяются рвота и диарея, приводящие к обезвоживанию и коме [23] .
Если синдром Уотерхауса — Фридериксена развился на фоне менингококковой инфекции, на туловище, ногах и слизистых оболочках может появиться петехиальная сыпь. Петехии — это красные пятна размером меньше 2 мм. Они могут объединяться в кровоизлияния покрупнее: пурпуру и экхимозы (больше 2 и 3 мм). Чем ниже уровень тромбоцитов, тем чаще возникают крупные кровоизлияния [14] .
![Петехиальная сыпь [25]](https://probolezny.ru/media/bolezny/sindrom-uoterhausa-frideriksena/petehialnaya-syp-25_s.jpg)
Патогенез синдрома Уотерхауса — Фридериксена
К развитию синдрома Уотерхауса — Фридериксена приводят:
- нарушение работы надпочечников;
- интоксикация из-за острой инфекции.
Эти процессы отягощают друг друга. Из-за прекращения работы надпочечников нарушается выработка глюко- и минералокортикоидных гормонов, которые участвуют в белковом, углеводном, жировом и водно-электролитном обмене. При нехватке этих гормонов нарушается обмен веществ, что приводит к сбою в работе всех клеток . Ионы натрия и хлориды при этом выводятся с мочой, хуже всасываются в кишечнике, что приводит к обезвоживанию — так возникает порочный круг заболевания.
Нарушения электролитного баланса проявляются сильной рвотой и частым жидким стулом, у новорождённых — срыгиваниями [1] . Уменьшение объёма крови из-за резкой потери жидкости приводит к шоку.
Также в организме нарушается обмена калия: он плохо выводится с мочой, повышается его уровень в крови, клетках и жидкости между ними. Из-за избытка калия в миокарде сердце начинает хуже сокращаться и не может адекватно реагировать на повышенную нагрузку.
При синдроме Уотерхауса — Фридериксена также нарушается углеводный обмен, в частности снижается уровень глюкозы в крови. Если это происходит резко, то развивается гипогликемическая кома [19] .
Классификация и стадии развития синдрома Уотерхауса — Фридериксена
Общепринятой классификации синдрома Уотерхауса — Фридериксена не существует.
В Международной классификации болезней (МКБ-10) он кодируется как:
- E35.1 — синдром Уотерхауса — Фридериксена;
- А39.1 — менингококковая инфекция [10] .
Осложнения синдрома Уотерхауса — Фридериксена
Синдром Уотерхауса — Фридериксена сам по себе является осложнением инфекций и ожогов, следствием удаления надпочечников, побочным эффектом приёма антикоагулянтов и осложнением операций.
При заболевании резко ухудшается работа надпочечников и развивается сосудистый коллапс. Без своевременной медицинской помощи наступает кома и смерть [1] .
Диагностика синдрома Уотерхауса — Фридериксена
При подозрении на синдром Уотерхауса — Фридериксена собирают анамнез, проводят осмотр, применяют лабораторные и инструментальные методы диагностики.
Сбор анамнеза
По возможности врач постарается выяснить историю болезни: наличие заболеваний надпочечников, ДВС-синдрома (нарушения свёртываемости крови), острой инфекции, ожогов и перенесённой операции.
При синдроме Уотерхауса — Фридериксена пациенты часто жалуются на головные боли, тошноту, боли в животе, диарею и сильную рвоту, возможно с кровью.
Осмотр
Состояние обычно тяжёлое, возникает заторможенность, ступор и бред.
При осмотре врач обратит внимание на положительные менингеальные симптомы, характерные для синдрома:
- поза «легавой собаки» — больной лежит на боку, ноги согнуты в коленях и подтянуты к животу, голова запрокинута назад;
- ригидность затылочных мышц — при попытке пригнуть голову к груди мышцы сопротивляются и не дают этого сделать;
- симптом Кернига — ногу, согнутую под прямым углом в коленном и тазобедренном суставе, невозможно разогнуть в колене [22] .

Тоны сердца глухие, пульс едва прощупывается. Развиваются судороги и очаговые неврологические симптомы. Кожа бледная, может появиться сыпь, руки и ноги холодные, давление сильно снижено, вплоть до коллапса, сердцебиение учащается, может прекратиться выделение мочи [1] .
Лабораторная диагностика
Проводится анализ на базальные уровни гормонов коры надпочечников. Кровь нужно сдавать с 6 до 9 утра, так как именно в это время вырабатывается больше всего гормонов. Анализ назначают строго до начала лечения глюкокортикоидами: Гидрокортизоном, Дексаметазоном, Преднизолоном [23] .
Также проводятся другие анализы:
- Общий анализ крови — при синдроме Уотерхауса — Фридериксена сильно увеличены лейкоциты со сдвигом лейкоцитарной формулы влево, повышены эозинофилы.
- Коагулограмма (исследование системы свёртывания крови) — снижен протромбиновый индекс, повышено активированное частичное тромбопластиновое время и международное нормализованное отношение (АЧТВ и МНО).
- Анализ крови на электролиты — калий повышен, натрий снижен.
- Анализ на глюкозу — уровень глюкозы в крови понижен [23] .
Инструментальная диагностика
- Электрокардиография (ЭКГ) — на кардиограмме видны изменения, характерные для высокого уровня калия в крови.
- Рентген органов грудной клетки — видны очагово-инфильтративные изменения в лёгких.
- Ультразвуковое исследование почек и надпочечников (УЗИ) — наличие кровоизлияний в надпочечники.
- Компьютерная томография или магнитно-резонансная томография (КТ или МРТ) — может проводиться при стабильном состоянии, чтобы выявить причину кровоизлияния в надпочечники, например очаг инфекции в головном мозге, грудной клетке или брюшной полости, туберкулёз, опухоли и метастазы. КТ надпочечников при инфильтративном процессе не всегда информативна [1][21] .
Синдром Уотерхауса — Фридериксена следует отличать от септического шока. Эту форму сепсиса можно предположить, если давление остаётся низким, несмотря на введение вазопрессоров [19] .
Также симптомы острой надпочечниковой недостаточности бывают схожи с гиповолемическим шоком. Эти два состояния легко спутать, особенно если нет бактериальной инфекции.
Лечение синдрома Уотерхауса — Фридериксена
При подозрении на синдром Уотерхауса — Фридериксена больного нужно немедленно госпитализировать в реанимационное отделение.
Лечение начинают сразу, не дожидаясь результатов анализов. Если пациент находится в бессознательном состоянии, устанавливают катетер для введения лекарств, мочевой катетер для выведения мочи и желудочный зонд для промывания желудка.
При острой надпочечниковой недостаточности внутривенно вводят 0,9 % -й раствор натрия хлорида или 5 % -й раствор глюкозы. В первые сутки потребуется не меньше четырёх литров жидкости. Восполнение потерянной при рвоте и диарее жидкости поможет предотвратить развитие гиповолемического шока [1] .
Лечение проводят под контролем артериального и центрального венозного давления, объёма мочи и хрипов в лёгких. Также каждые два часа проверяют уровень калия, натрия и глюкозы.
При неукротимой рвоте в начале лечения и повторно при очень низком артериальном давлении внутривенно вводят 10 % -й раствор хлорида натрия. При необходимости используют плазму крови и её заменители.
Из-за нарушенной работы надпочечников в крови повышен калий и снижен натрий. Поэтому нельзя применять диуретики, калийсодержащие и гипотонические растворы.
Чтобы заместить выработку гормонов надпочечников, вводят Гидрокортизон. Препарат применяют немедленно, а затем через каждые шесть часов. При острой надпочечниковой недостаточности используют Гидрокортизон натрия гемисукцинат, его можно вводить внутривенно и внутримышечно. Другой препарат — Гидрокортизон ацетат — действует медленнее, поэтому не подходит для неотложной помощи [1] .
Если нет Гидрокортизона, например при транспортировке пациента в специализированный стационар, можно использовать Дексаметазон или Преднизолон. Но это временное лечение, затем нужно перейти на Гидрокортизон [1] .
Гидрокортизон применяют до тех пор, пока пациент не будет выведен из коллапса и систолическое артериальное давление не повысится до 100 мм рт. ст. На 2-3-и сутки лечения при стабилизации состояния дозировку постепенно снижают.
При уменьшении дозы Гидрокортизона добавляют Флудрокортизон (Кортинефф): при низких дозах Гидрокортизон перестаёт оказывать минералокортикоидный эффект, т. е. замедлять выделение натрия и воды из организма, а также усиленно выводить калий с мочой.
При лихорадке применяют антибиотики. Исключение — лихорадка на фоне обезвоживания [1] .
Прогноз зависит от тяжести заболевания. При двухстороннем поражении надпочечников около 15 % пациентов погибает, но при своевременной диагностике и адекватном лечении даже с таким диагнозом можно выздороветь. Около половины смертей связаны с поздним обращением за медицинской помощью [9] .
Профилактика синдрому Уотерхауса — Фридериксена
Всем пациентам с заболеваниями надпочечников рекомендуется носить идентификационный браслет или кулон, где будет указан диагноз и препараты, которые нужно ввести. Также желательно иметь под рукой инъекции глюкокортикостероидов для неотложных ситуаций.
Пациентам с болезнями надпочечников нужно не реже раза в год посещать эндокринолога. Это важно для оценки адекватности дозировки заместительной гормональной терапии [20] .
Синдром Форсиуса-Эрикссона (Forsius-Eriksson) - синонимы, авторы, клиника
Альбинизм — клинически и генетически гетерогенная группа наследственных заболеваний, в основе патогенеза которых лежит нарушение биосинтеза меланина, приводящее к полному или частичному его отсутствию. Клинически снижение количества меланина проявляется в гипопигментации кожи, волос и глаз. Поражение глаз включает гипопигментацию или отсутствие пигмента глазного дна и радужной оболочки, гипоплазию фовеа, снижение остроты зрения, нистагм и косоглазие, фотофобию, трансиллюминацию радужной оболочки и асимметричное пересечение зрительных нервов в хиазме. Однако альбинизм может выступать частью симптомокомплекса более сложных генетических синдромов, таких как синдромы Германски — Пудлака и Чедиака — Хигаси, которые требуют как можно более раннего выявления и начала терапии из-за риска развития жизнеугрожающих состояний. Также встречаются случаи частичного альбинизма с гипопигментацией структур глаз, кожи или волос, но не связанные с меланогенезом, такие как синдром Грисцелли, синдром Ваарденбурга, синдром Титца, болезнь Аландских островов. При исследовании каждого случая альбинизма необходимо установить точный молекулярно-генетический диагноз. Это обеспечит персонализированный подход к лечению, позволит корректно прогнозировать жизнь и здоровье пациентов, а также планировать деторождение.
Ключевые слова: альбинизм, синдром Германски — Пудлака, синдром Чедиака — Хигаси, гипопигментация, клинический полиморфизм, генетическая гетерогенность.
V.V. Kadyshev, S.A. Ryazhskaya, O.V. Khalanskaya, N.V. Zhurkova, R.A. Zinchenko
Research Center for Medical Genetics, Moscow, Russian Federation
Albinism is a clinically and genetically heterogeneous group of hereditary diseases whose pathogenesis is mediated by impaired synthesis of melanin which results in its partial or total loss. Reduced melatonin level clinically manifests as skin, hair, and ocular hypopigmentation. Ocular presentations include hypopigmentation/lack of pigmentation of eye fundus and iris, foveal hypoplasia, low vision, nystagmus and strabismus, photophobia, iris transillumination, and asymmetrical decussation of nerve fibers at the optic chiasm. However, albinism can be a part of more complex genetic syndromes, e.g., Hermansky-Pudlak syndrome or Chediak-Higashi syndrome. These disorders should be identified as early as possible to start therapy to prevent life-threatening conditions. Partial albinism with ocular, skin or hair hypopigmentation not associated with melanogenesis (e.g., Griscelli syndrome, Waardenburg syndrome, Aland Island eye disease, etc.) also occurs. Each case of albinism requir es an accurate molecular genetic diagnosis to provide a personalized treatment approach, predict life expectancy and health status, and plan pregnancy.
Keywords: albinism, Hermansky-Pudlak syndrome, Chediak-Higashi syndrome, hypopigmentation, clinical polymorphism, genetic heterogeneity.
For citation: Kadyshev V.V., Ryazhskaya S.A., Khalanskaya O.V. et al. Clinical and genetic aspects of albinism. Russian Journal of Clinical Ophthalmology. 2021;21(3):175-180 (in Russ.). DOI: 10.32364/2311-7729-2021-21-3-175-180.
Для цитирования: Клинико-генетические аспекты альбинизма. Клиническая офтальмология. 2021;21(3):175-180. DOI: 10.32364/2311-7729-2021-21-3-175-180.

Альбинизм — это клинически и генетически гетерогенная группа наследственных заболеваний, в основе патогенеза которых лежит нарушение биосинтеза меланина, приводящее к полному или частичному его отсутствию. Клинически снижение количества меланина проявляется в гипопигментации кожи, волос и глаз. Поражение глаз включает гипопигментацию или отсутствие пигмента глазного дна и радужной оболочки, гипоплазию фовеа, снижение остроты зрения, нистагм, косоглазие, фотофобию и трансиллюминацию радужки. Кроме того, у пациентов с альбинизмом при исследовании зрительных вызванных потенциалов, МРТ или трактографии выявляется неправильная маршрутизация волокон зрительных нервов в хиазме (перекрестная асимметрия) — чрезмерный переход височной доли волокон на противоположную сторону [1, 2].
Средняя частота альбинизма в мире составляет 1:17000, частота носительства — 1:70 (варьирует в зависимости от региона) [3].
Глазной альбинизм
Глазо-кожный альбинизм
Глазо-кожный альбинизм объединяет группу заболеваний с аутосомно-рецессивным типом наследования, для которых, в отличие от изолированного глазного альбинизма, дополнительным характерным признаком является гипопигментация кожи и волос. Воздействие солнечных лучей на гипопигментированную кожу приводит к пахидермии, актиническому дерматиту и раку кожи, базальноклеточной и плоскоклеточной карциноме и, реже, меланоме, что может встречаться даже у гетерозиготных носителей [6].
Молекулярно-генетический дефект выявляется примерно у 60% пациентов с клиническими проявлениями альбинизма. Значительное число из оставшихся 40% являются гетерозиготными носителями патогенных мутаций в генах TYR или OCA2: описаны случаи развития заболевания при наличии патогенной мутации на одной аллели и двух полиморфных вариантов — на другой [7, 8].
Глазо-кожный альбинизм 1 типа (OCA1) ассоциирован с геном TYR, расположенным на хромосоме 11 в положении 11q14.3, и разделяется на два клинических подтипа: глазо-кожный альбинизм 1A и 1B типов. Ген TYR кодирует ключевой фермент меланогенеза — тирозиназу, участвующую в следующих этапах меланогенеза: гидроксилирование тирозина до диоксифенилаланина (ДОФА) и окисление ДОФА до L-ДОФА. На сегодняшний день в базе Human Gene Mutation Database описано более 350 патогенных вариантов в гене TYR [9], подавляющее большинство которых продуцируют неактивный фермент и приводят к фенотипу глазо-кожного альбинизма 1A типа, а около 10 вариантов продуцируют фермент с остаточной активностью (гипоморфные варианты) и, соответственно, приводят к фенотипу глазо-кожного альбинизма 1B типа. Глазо-кожный альбинизм 1 типа составляет около 40% от всех случаев альбинизма [10], его частота в мире — 1-9:100000 [3].
Глазо-кожный альбинизм 8 типа (OCA8) ассоциирован с геном DCT (TYRP2) и описан Pennamen et al. в 2020 г. в двух неродственных семьях. Ген DCT кодирует фермент допахромтаутомеразу, который катализирует превращение допахрома в дигидроксииндол-2-карбоновую кислоту. Фенотипические проявления 8 типа глазо-кожного альбинизма: легкая гипопигментация кожи и волос и умеренные поражения глаз, типичные для альбинизма (нистагм, светобоязнь, трансиллюминация радужки, гипопигментация сетчатки и глазного дна, гипоплазия фовеа 0-1-й степени, снижение остроты зрения до 0,4-0,5) [18, 19].
Редкие наследственные синдромы в сочетании с альбинизмом
Кроме изолированного глазного и глазо-кожного типов существуют редкие наследственные синдромы альбинизма, связанные с системными поражениями. К ним относятся синдром Германски — Пудлака и синдром Чедиака — Хигаси.
Синдром Германски — Пудлака
Синдром Германски — Пудлака (Hermansky — Pudlak syndrome, HPS) впервые описан в 1959 г. в двух неродственных семьях у пациентов с альбинизмом, геморрагическим диатезом и пигментированными ретикулярными клетками в костном мозге, а также в биоптатах лимфатических узлов и печени.
Синдром Германски — Пудлака относится к орфанным заболеваниям: его частота в мире составляет от 1:50000 до 1:1000000, однако некоторые типы распространены в Пуэрто-Рико (так, частота 1 типа в этой популяции составляет 1:1800, 3 типа — 1:16000) [10].
В настоящее время синдром Германски — Пудлака включает 11 типов, ассоциированных с 11 генами. Главным патогенетическим звеном выступает нарушение адапторного белкового комплекса-3 (AP3) или системы биогенеза комплекса связанных с лизосомами органелл (BLOC 1-3), являющихся важными компонентами мембран цитоплазматических органелл и участниками транспортировки везикул. Дефекты белков, составляющих эти комплексы, приводят к патологическим изменениям в структуре и функции лизосом, меланосом, плотных гранул тромбоцитов.
Основные клинические проявления синдрома Германски — Пудлака: альбинизм, отложение пигмента в клетках ретикуло-эндотелиальной системы и нарушение агрегации тромбоцитов (за счет отсутствия плотных гранул, содержащих серотонин, адениновые нуклеотиды, Са2 + , фибриноген, адреналин, фактор Виллебранда, антигепариновый фактор). Геморрагический диатез может проявляться частыми носовыми, десневыми, интра- и послеоперационными кровотечениями, меноррагией у женщин. В лабораторных показателях отмечается отсутствие плотных гранул тромбоцитов, выявляемое при электронной микроскопии, при нормальном количестве тромбоцитов и нормальном протромбиновом времени. Кроме того, с синдромом Германски — Пудлака связаны гранулематозный колит, легочный фиброз и кардиомиопатия (характерные не для всех типов), возникающие из-за цероидоподобных отложений в тканях [20, 21]. Краткая клиническая характеристика всех известных типов синдрома Германски — Пудлака приведена в таблице 1 33.
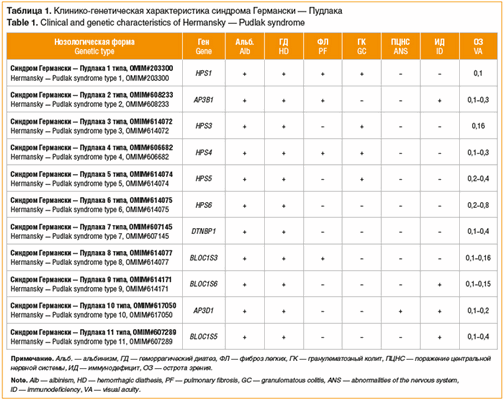
Альбинизм и геморрагический диатез проявляются при всех типах синдрома Германски — Пудлака. Основными различиями являются системные проявления: так, фиброз легких развивается при 1, 2, 4 и 8 типах, высокий риск развития гранулематозного колита имеется при 1, 3, 4 и 5 типах. Клеточный иммунодефицит с повышенным риском развития гемофагоцитарного лимфогистиоцитоза чаще встречается у пациентов с 2, 9, 10 и 11 типами синдрома. При 10 типе также описано поражение ЦНС, включающее задержку моторного развития, мышечную гипотонию и эпилепсию.
Синдром Чедиака — Хигаси
Впервые синдром Чедиака — Хигаси описан Beguez — Cesar в 1943 г., впоследствии Чедиак (в 1952 г.) и Хигаси (в 1954 г.) дополнили фенотипическое описание синдрома. В настоящее время в мире описано около 500 случаев этого заболевания, однако точное количество пациентов невозможно установить в связи с существованием аномальных мягких форм, при которых диагноз подтверждается в 30 лет и позже [38], либо из-за повторных описаний пациентов в течение жизни [39].
Фенотипическими проявлениями синдрома Чедиака — Хигаси являются глазо-кожный альбинизм, геморрагический диатез, неврологические нарушения, а также первичный иммунодефицит и повторные инфекционные заболевания. Зрительные нарушения включают горизонтальный нистагм, астигматизм, гипопигментацию радужки, гипоплазию макулы и фовеа. Острота зрения варьирует от 0,3 до нормальных показателей [40]. Поражение нервной системы проявляется задержкой моторного и психоречевого развития, периферической нейропатией, атаксией, паркинсонизмом и судорогами; при обследовании выявляется снижение глубоких сухожильных рефлексов, снижение скорости нервной проводимости, на МРТ обнаруживается диффузная атрофия головного и спинного мозга, выявляются также гигантские гранулы в шванновских клетках при биопсии [41, 42]. При лабораторном исследовании крови отмечаются: гигантские включения в полиморфноядерных нейтрофилах и, в меньшей степени, в лимфоцитах; нормальное или пониженное количество естественных клеток-киллеров с аномально сниженной функцией; нейтропения и нарушение функции нейтрофилов, таких как хемотаксис и внутриклеточная бактерицидная активность; отсутствие или пониженное количество и неправильная морфология плотных гранул тромбоцитов [39].
У 65-85% пациентов с синдромом Чедиака — Хигаси в течение первого десятилетия жизни на фоне инфекционного процесса развивается фаза «акселерации» с быстропрогрессирующей массивной лимфопролиферативной реакцией, «цитокиновым штормом» и риском развития гемофагоцитарного лимфогистиоцитоза [43].
Редкие наследственные синдромы с частичным альбинизмом
Гено-фенотипические корреляции при разных типах альбинизма до сих пор не установлены: даже пациенты, заболевание у которых обусловлено одинаковыми мутациями, могут обладать различными по степени выраженности клиническими проявлениями, в том числе в пределах одной семьи. Кроме того, существуют наследственные заболевания, связанные с гипопигментацией структур глаз, кожи и волос, но не имеющие ключевых признаков альбинизма, таких как гипоплазия фовеа, гипопигментация глазного дна, неправильный перекрест зрительных нервов и нистагм, — такие синдромы входят в группу частичного, или парциального, альбинизма. К этим состояниям относятся синдром Грисцелли, синдром Ваарденбурга, синдром Титца, болезнь Аландских островов и синдром FHONDA. Кроме того, необходимо дифференцировать глазной альбинизм и группу заболеваний с Х-сцепленным врожденным нистагмом.
Синдром Ваарденбурга обладает генетической и фенотипической гетерогенностью, включая тип наследования — большинство типов наследуются аутосомно-доминантно с неполной пенетрантностью и вариабельной экспрессивностью, отдельные типы наследуются и аутосомно-рецессивно. Известны все гены, ассоциированные с синдромом Ваарденбурга: 1 и 3 типа — PAX3, 2А типа — MITF, 2D типа — SNAI2, 2Е и 4С типа — SOX10, 4A типа (вариабельное наследование в зависимости от патогенного варианта — аутосомно-доминантный (AD) либо аутосомно-рецессивный (AR)) — EDNRB, 4B типа (также AD- либо AR-наследование) — EDN3. Основными признаками этой группы заболеваний являются нейросенсорная тугоухость, лицевые дизморфии (характерны для отдельных типов), нарушение пигментации пряди волос в лобной области (пьебалдизм), полная или частичная гетерохромия радужек, дистопия медиальной спайки век, гипопигментация радужки и глазного дна с нистагмом (описаны не при всех типах).
Заключение
Альбинизм — редкое, клинически и генетически гетерогенное состояние с множеством форм, но, несмотря на длительность и большое количество проводимых исследований, не до конца изученное. В настоящее время продолжается поиск генов, мутации в которых ответственны за глазо-кожный альбинизм или редкие синдромальные формы альбинизма: в 2020 г. описаны два новых заболевания — глазо-кожный альбинизм 8 типа и альбинизм 11 типа (синдром Германски — Пудлака).
Пациенты с альбинизмом должны наблюдаться офтальмологами и дерматологами, однако при выявлении синдромальной формы необходимо подключать к наблюдению и врачей других специальностей — пульмонологов, гематологов, неврологов и гастроэнтерологов — для решения вопроса о начале терапии и профилактике осложнений. Кроме того, необходимо выявление корреляции между генотипом, клиническими проявлениями и тяжестью течения заболевания для прогнозирования жизни пациентов, а также определение популяционной частоты заболеваний и носительства гетерозиготных мутаций в генах и выявление других типов наследования — для планирования деторождения.
Сведения об авторах:
Кадышев Виталий Викторович — к.м.н., ст. научный сотрудник лаборатории генетической эпидемиологии, заведующий кафедрой офтальмогенетики Института ВиДПО, врач-генетик, офтальмолог; ORCID iD 0000-0001-7765-3307.
Ряжская Светлана Андреевна — клинический ординатор; ORCID iD 0000-0002-5224-5726.
Журкова Наталия Вячеславовна — к.м.н., ст. научный сотрудник лаборатории генетической эпидемиологии; ORCID iD 0000-0001-6614-6115.
Зинченко Рена Абульфазовна — д.м.н., профессор, заместитель директора по научно-клинической работе, заведующая лабораторией генетической эпидемиологии; ORCID iD 0000-0003-3586-3458.
ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова». 115522, Россия, г. Москва, ул. Москворечье, д. 1.
Источник финансирования: работа выполнена при финансовой поддержке РНФ, проект № 17-15-01051, и государственного задания Министерства науки и высшего образования РФ.
Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах.
Конфликт интересов отсутствует.
Статья поступила 16.02.2021.
About the authors:
Vitaly V. Kadyshev — C. Sc. (Med.), senior researcher of the Laboratory of Genetic Epidemiology, Head of the Department of Ophthalmogenetics of the Institute of Higher and Additional Vocational Education, geneticist, ophthalmologist; ORCID iD 0000-0001-7765-3307.
Svetlana A. Ryazhskaya — clinical resident; ORCID iD 0000-0002-5224-5726.
Nataliya V. Zhurkova — C. Sc. (Med.), senior researcher of the Laboratory of Genetic Epidemiology; ORCID iD 0000-0001-6614-6115.
Rena A. Zinchenko — Dr. Sc. (Med.), Professor, Deputy Director for Scientific Clinical Work, Head of the Laboratory of Genetic Epidemiology; ORCID 0000-0003-3586-3458.
Research Center for Medical Genetics. 1, Moskvorechye str., Moscow, 115522, Russian Federation.
There is no conflict of interests.
Received 16.02.2021.
Список литературы Свернуть Развернуть
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Читайте также: