Синдром Гельмгольца-Харрингтона (Helmholz-Harrington) - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 21.01.2026
«Проезжая по лесной дороге. мы с отцом повстречали двух женщин, мать и дочь, которые были высокими и полными, мертвенно-бледными, и которые беспрестанно раскачивались, присвистывали и гримасничали. Я застыл не столько в изумлении, сколько в страхе. Что бы это могло означать? Мой отец остановился поговорить с ними, и затем мы продолжили путь.
С тех пор как я начал изучать медицину, мой интерес к этому заболеванию стал неисчерпаем.»
Мальчику, наблюдавшему этот эпизод в 1858 году, было на тот момент всего 8 лет. Сегодня мы знаем его как доктора Джорджа Хантингтона.
- Изабелла Ивановна, сегодня, в день рождения Джорджа Хантингтона - американского врача, ставшего известным благодаря первому в истории медицины классическому описанию заболевания, названного в его честь, поговорим о болезни или хорее Хантингтона. Что это такое?
Болезнь Гентингтона (а также все ее синимы - болезнь/хорея Хантингтона, хорея наследственная/дегенеративная и т.д.) - это тяжелая, наследственная патология нервной системы, проявляющаяся двигательными нарушениями, психическими изменениями и деменцией (слабоумием).
Несмотря на то, что честь первой публикации об этом заболевании принадлежит Хантингтону, до него этот недуг изучал норвежский врач Йохан Кристиан Лунд. Однако его труды были переведены на английский язык лишь в 50-х годах прошлого столетия, в связи с чем большая часть научного мира узнала о хорее от Хантингтона. Тем не менее, отдавая должное заслуге Лунда, в литературе заболевание иногда называют болезнью Лунда-Хантингтона.
- Это заболевание неврологическое или психиатрическое?
Хорея Гентингтона - болезнь, лечением которой занимаются разные специалисты. Всё зависит от того, какие проявления отмечаются или преобладают у пациента в тот или иной период развития заболевания.
Среди начальных проявлений болезни иногда отмечаются: нарушения поведения, снижение сексуального влечения, игромания, склонность к чрезмерному выражению эмоций, появление бреда, галлюцинаций. Это - сфера компетенции психиатров.
В других случаях хорея дебютирует именно с двигательных расстройств - так называемых гиперкинезов. Это непроизвольные движения в различных частях тела - лице, конечностях. Лечением таких проявлений занимаются неврологи.
- Болезнь Гентингтона как-то отражена в МКБ-10?
Да, соответственно в двух разделах: в болезнях нервной системы под кодом G10, а в психических расстройствах - под кодом F02.2.
- Насколько часто встречается это заболевание?
Достаточно редко. У европеоидов частота встречаемости колеблется от 3 до 10 случаев на 100 000 человек. У представителей других рас частота ниже - 1 на 1000 000. Вместе с тем нужно не сбрасывать со счетов то, что европейцы могут просто чаще обследоваться, в связи с чем выявляемость болезни Гентингтона у них выше.
- Что является причиной развития хореи Хантингтона?
Генетические изменения. Ген HTT, ответственный за развитие болезни, находится в 4-й хромосоме. Благодаря данному гену, кодируется особый белок - гентингтин (хантингтин). Функция его нормальной формы неизвестна. Измененный генетической мутацией белок приобретает иные свойства, что приводит к развитию болезни Гентингтона.
- Что говорит наука о вероятности наследования этого заболевания?
Оно передается по наследству от человека-носителя генетической мутации. Если один из родителей болен, вероятность наследования - 50%. Болеть могут представители обоих полов, несколько чаще оно отмечается у мужчин. Если мутация передается от отца, тяжесть мутации может увеличиваться, что означает, что болезнь проявится у детей в более раннем по сравнению с отцом возрасте. В случае если мутация не передалась следующему поколению и все его представители не являются ее носителями, передача болезни Хантингтона по наследству прекращается.
- Болезнь Гентигтона передаётся по мужской или женской линии?
- Как по внешним признакам можно определить, что у человека - болезнь Гентигтона?
Проявления могут проявиться как во взрослом возрасте (чаще примерно в 40-50 лет), так и в детском (чаще всего в 7-10 лет).
Симптомы хореи Гентингтона достаточно разнообразны и включают в себя двигательные проявления, психические нарушения, деменцию.
Для взрослого человека типичны двигательные нарушения, выражающиеся быстрыми, непроизвольными, иногда вычурными движениями в любых мышечных группах. Контролировать их больной не может.
Зачастую они начинаются с лицевой мускулатуры. Могут отмечаться гримасы, с высовыванием языка, поднятием щек, сворачиванием губ «трубочкой», подмигиванием, нахмуриванием бровей.
По мере прогрессирования процесса патологические движения появляются в других мышечных группах. Это быстрые сгибания-разгибания в пальцах, кистях, одно- или двусторонние. Затем добавляются другие отделы рук, ног.
Какой болезнью страдала Маргарет Тэтчер? Читать далее
В некоторых случаях такие движения появляются сразу в нескольких частях тела.
Психические нарушения проявляются появлением и постепенным снижение когнитивных способностей, может отмечаться подавленность, апатия, раздражительность, тревожность, нарушения памяти, ослабление критического отношения к происходящему. Возможно развитие бреда, галлюцинаций. На фоне развития депрессии возможны попытки суицида.
У детей в двигательной сфере на первых этапах может, напротив, отмечаться замедление, вялость движений. Также сравнительно рано страдает речь: ее восприятие, произношение звуков, скорость речи, ритмичность.
Для хореи Хантингтона в детском возрасте также характерны изменения со стороны глаз. Это колебания глазного яблока вверх-вниз, нарушение способности переводить взгляд, точность фиксации на чем-либо. Иногда у детей могут отмечаться судороги. Изменения в психике выражаются дурашливостью, повышенной сексуальностью, склонность к употреблению алкоголя, игроманией. Так как страдает критика, ребенок не осознает, что его действия неправильны.
- Как проводится диагностика хореи Хантигтона?
Ввиду особенностей заболевания очень желательно, чтобы во время осмотра больного присутствовали его близкие, которые могут предоставить ценную для постановки диагноза информацию. Оцениваются неврологическая, психосоматическая сферы.
Для уточнения и подтверждения наличия болезни Гентингтона выполняется МРТ, обязательно - генетическое обследование.
Зачем проходить МРТ, когда ничего не болит? Рассказывает исполнительный директор «Клиника Эксперт Оренбург» Юрий Андреевич Подлевских
- Что происходит с головным мозгом человека, поражённого хореей Хантингтона?
На МРТ можно видеть уменьшение плотности вещества головного мозга, расширение боковых желудочков, атрофию головок хвостатых ядер.
О каких заболеваниях расскажет МРТ головного мозга? Здесь собраны вопросы, касающиеся МРТ головного мозга и его сосудов, которые пациенты задают нам наиболее часто
При патологоанатомическом исследовании отмечается уменьшение массы определенных анатомических образований в головном мозге, появление своеобразных «рубцов» в разных его отделах.
- Если человек является носителем мутации болезнь Гентингтона, то заболевание можно предотвратить?
На данный момент нет.
- Хорею Хантингтона возможно вылечить? Каков прогноз при этой болезни?
Лечение симптоматическое, т.е. направление на устранение двигательных проявлений, депрессии и т.д. Коррекция симптомов позволяет улучшить качество жизни и пациента, и его близких. Огромную помощь оказывают больному те, кто находится рядом с ним - родные, друзья и т.д.
Исследования по поиску специфического лечения ведутся непрерывно. Значительную роль в этой области играет Европейская ассоциация по изучению болезни Гентингтона (EHDN). В нее уже входит несколько российских исследовательских центров.
Дополнительную информацию можно получить на сайте организации
Возможно, вас заинтересуют:
Капралова Изабелла Ивановна
Выпускница Российского государственного медицинского университета Федерального агентства по здравоохранению и социальному развитию. Закончила университет в 2009 году.
В 2009-2011 годах прошла клиническую ординатуру ГОУ ВПО «РГМУ Росздрава» на кафедре неврологии и нейрохирургии.
С 2011 года работала врачом-неврологом в лечебных учреждениях Москвы и Тульской области.
В настоящее время занимает должность врача-невролога в ООО «Клиника Эксперт Тула». Принимает по адресу: ул. Болдина, д. 74
Другие статьи по теме
С диагнозом «киста» сталкивается немало пациентов, среди них большинство - женщины. Однако. Что такое дермоидная киста?
По данным ВОЗ, врождённые пороки развития становятся причиной практически каждого пятого случая. Выявление врожденных пороков в антенатальном периоде
Внедрение в практику невролога МРТ позволило расширить прижизненную диагностику аномалий развития головного. МРТ в диагностике изменений головного мозга при наследственных заболеваниях
Ставинский Юрий Алексеевич
Для жителей с Востока Украины и бойцов АТО в Киеве откроют Отделение травматических повреждений опорно-двигательного аппарата и проблем остеосинтеза
Для жителей с Востока Украины и бойцов АТО в Киеве откроют Отделение травматических повреждений опорно-двигательного аппарата и проблем остеосинтеза
The algorithm of proximal femoral nail
The algorithm of proximal femoral nail use for surgical treatment of trochanteric fractures
Фиксации при переломах костей голени
Фиксации при переломах костей голени
Мультфильм ортопеды Киева, Украина
Мультфильм ортопеды Киева, Украина
Видео ортопеды Киева, Украина
Видео ортопеды Киева, Украина
- 2016.02.05 Фиксации при переломах костей голени 2015.07.02 Мультфильм ортопеды Киева, Украина 2015.03.11 The algorithm of proximal femoral nail 2014.01.20 Артроскопия кистевого сустава Киев 2014.01.20 Видео ортопеды Киева, Украина 2014.01.15 Анкилозирующий спондилит 2014.01.15 Болезнь Бехтерева 2014.01.15 Эндопротезирование коленного сустава Киев 2014.01.15 Операции голеностопный сустав Киев 2014.01.15 Операции локтевой сустав Киев
Синдром Гельмгольца - Харрингтона, s. Helmholz - Harrington Главная » Справочник ортопеда » С » Синдром Гельмгольца - Харрингтона
Синдром Гельмгольца - Харрингтона (s. Helmholz - Harrington), синдром помутнения роговицы при дизостозе скелета. Описан совместно американскими врачами Helmholz H. F. и Harrington Е. в 1931 г.
Этиология, патогенез и симптомы заболевания соответствуют синдрому Пфаундлера - Гурлера, в отличие от которого в роговице отмечается только ограниченное помутнение.
Информационно-консультативный партнер проекта Ортопеды Киева : ГУ "Институт травматологии и ортопедии" АМН Украины, отдел повреждений опорно — двигательного аппарата и проблем остеосинтеза
Дни консультаций : понедельник - пятница с 9 до 18 (возможна предварительная договоренность по телефону)
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
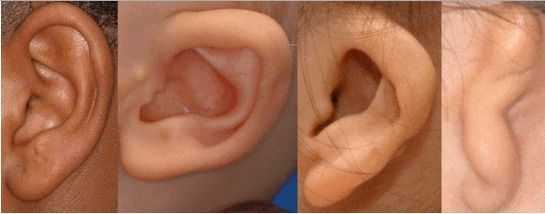
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
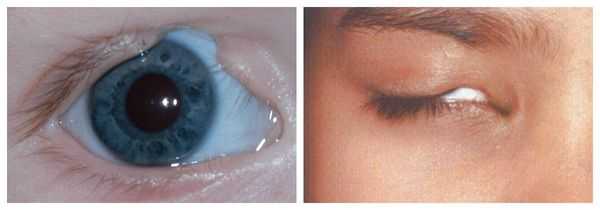
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
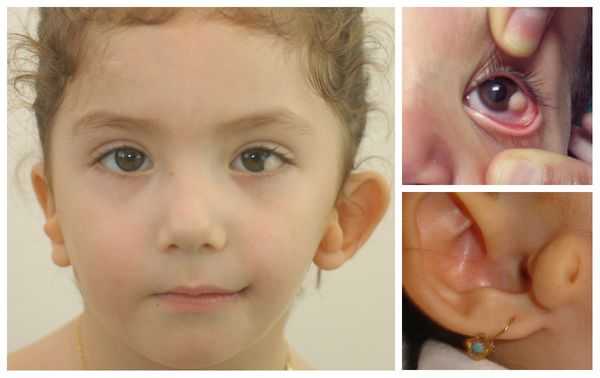
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
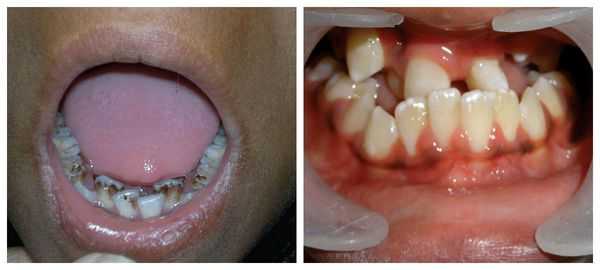
Патогенез синдрома Гольденхара
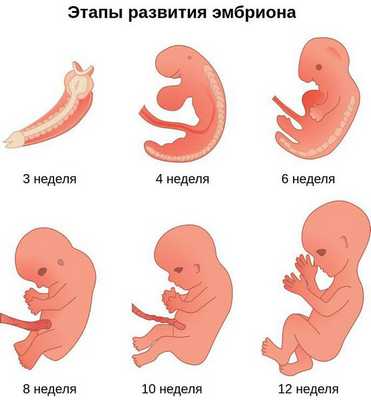
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
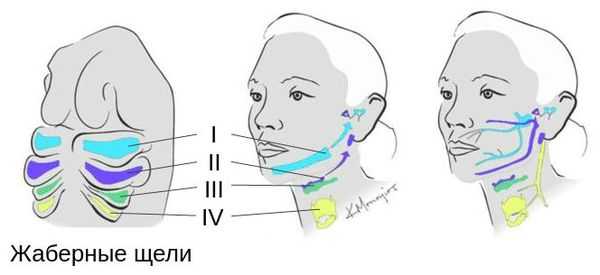
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
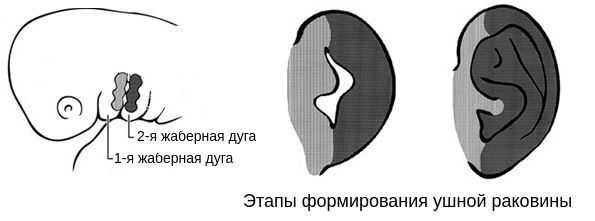
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
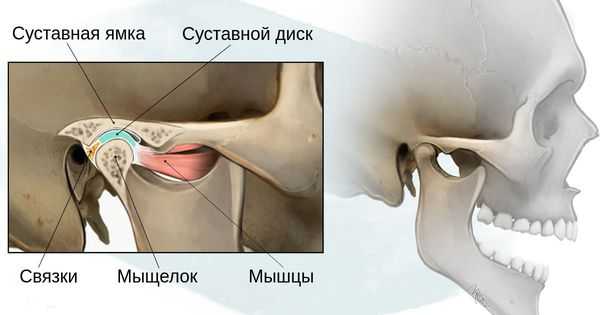
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
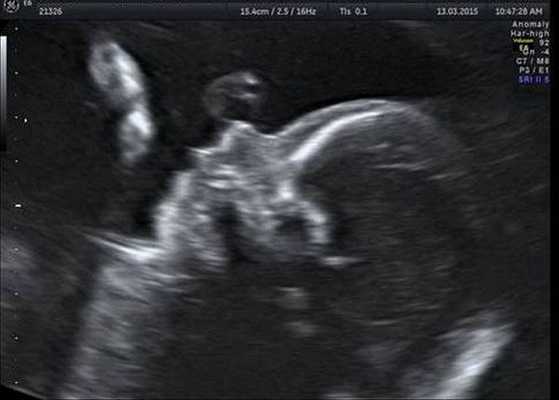
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
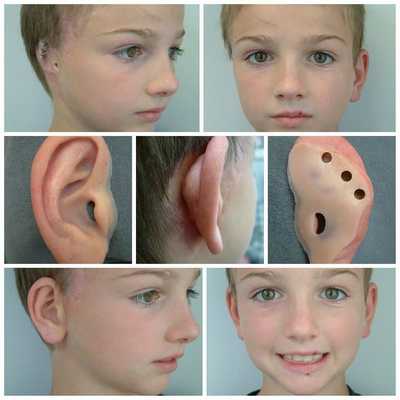
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
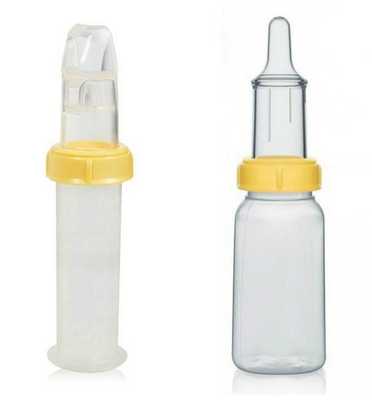
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Синдром Гольденхара ( Окуло-аурикуло-вертебральная дисплазия )
Синдром Гольденхара — это редкое врожденное заболевание, которое проявляется множественными пороками развития, выраженным клиническим полиморфизмом. Возникает вследствие мутации генов, локализованных в 5, 14, 20 хромосомах. Для синдрома характерны разнообразные аномалии лицевого скелета, патологии органов чувств, зачастую болезнь сопровождается задержкой психического развития. План обследования включает генетическое тестирование для верификации диагноза, лабораторно-инструментальные методы с учетом ведущих клинических признаков. Лечение поддерживающее, многим больным требуется слухопротезирование, комплексная нейрореабилитация.
МКБ-10
Общие сведения
Синдром назван в честь американского ученого Мориса Гольденхара, который описал его типичные клинические проявления в 1952 г. В 1963 г. Р.Дж. Горлин и его коллеги сообщили о собственных наблюдениях пациентов, дав болезни второе название «окуло-аурикуло-вертебральная дисплазия». Состояние выявляется с частотой от 1:3500 до 1:7000 живорожденных новорожденных, соотношение мальчиков и девочек составляет 3:2. Если в семье есть больной ребенок, то вероятность появления синдрома Гольденхара у следующих детей — не более 3%.
Причины
Заболевание возникает при нарушении дифференцировки 1-2-й жаберных дуг, что провоцируется мутацией в генах GSC и TCOF1. В 98% случаев синдрома роль наследственности не удается проследить. Однако около 2% больных имеют родственников с аналогичной клинической симптоматикой. В литературе описаны случаи аутосомно-доминантного и аутосомно-рецессивного наследования.
Основными предрасполагающими факторами считаются:
- воздействие тератогенных факторов на ранних сроках гестации;
- наличие у матери сахарного диабета, избыточного веса;
- предшествующие искусственные прерывания беременности и выкидыши.
Патогенез
Как следствие, лобный, нижне- и верхнечелюстные эктодермальные отростки, происходящие из первой жаберной дуги, и ушные раковины, образованные 1 и 2 жаберными дугами, развиваются асимметрично. После рождения ребенка это проявляется специфическими изменениями верхней и нижней челюстей, глаз и глазниц, мимической и жевательной мускулатуры, структур наружного и среднего уха, аномалиями прикуса, дефицитом мягких тканей.
Симптомы
Основные признаки синдрома Гольденхара — аномалии строения лица, которые в 70% случаев являются правосторонними. У всех пациентов наблюдается асимметрия, недоразвитие нижней челюсти, уменьшение в размерах, деформация или отсутствие ушных раковин. Патологии органа слуха чаще бывают односторонними, сопровождаются атрезией слухового прохода, преаурикулярными выростами. Иногда формируется дополнительная рудиментарная ушная раковина.
Также характерно уменьшение размера глазных яблок (микрофтальмия), косоглазие, атрезия радужки, катаракта. Около 50% пациентов с болезнью Гольденхара имеют высокое готическое небо, широкий рот (макростомию), расщепление языка, аномальный прикус, отсутствие части зубов. В 40% случаев отмечаются косолапость, аномалии позвоночника (сколиоз, spina bifida), искривление ребер. У 30% больных возникают врожденные патологии внутренних органов: пороки сердца, гипоплазия легких, дисплазия почек.
Осложнения
Самым опасным последствием синдрома Гольденхара считается умственная отсталость, которая обусловлена двусторонней тугоухостью, снижением остроты зрения. При этом у большинства детей наблюдается нормальное функционирование нервной системы, однако сенсорная депривация создает затруднения при речевом и психомоторном развитии. Осложненное течение синдрома встречается при тяжелых врожденных пороках сердца, легких, почек.
Диагностика
В большинстве случаев предварительный диагноз устанавливается на основании характерных фенотипических признаков: деформации ушей, асимметрии лица, патологии развития нижней челюсти. Для подтверждения синдрома Гольденхара обязательно назначается консультация генетика, проводится комплекс диагностических мероприятий:
- Генетический анализ. Тестирование на специфическую мутацию генов 14q32, 5p15, MYT1 позволяет 100% подтвердить диагноз. Исследование выполняется в специальных генетических лабораториях с использованием методов секвенирования экзона, флуоресцентной гибридизации.
- Аудиометрия. Оценка слуха в динамике необходима всем пациентам, чтобы вовремя выявить кондуктивную тугоухость, обеспечить мероприятия по ее коррекции. Расширенное обследование у отоларинголога может включать импедансометрию, оценку рефлекса стапедиальной мышцы.
- Неврологический осмотр. Консультация детского невролога требуется для определения причин задержки психомоторного развития, оценки функционирования центральной и периферической нервной системы. При наличии показаний больного направляют на дообследование к психиатру.
- Инструментальная визуализация. Чтобы подтвердить или исключить соматические пороки, при синдроме Гольденхара проводятся рентгенография грудной клетки, эхокардиография, УЗИ органов брюшной полости. Для исследования ЦНС применяется КТ или МРТ головного мозга.
Пациенты, у которых диагностирована болезнь Гольденхара, подлежат пожизненному динамическому наблюдению. Им назначаются регулярные обследования у офтальмолога, отоларинголога, невролога и других специалистов, чтобы контролировать состояние здоровья. Курсы поддерживающего лечения включают несколько направлений помощи, основными из которых являются:
- Метаболическая терапия. Для правильного развития ЦНС, улучшения когнитивных навыков используются ноотропы, витамины группы В.
- Нейрореабилитация. Чтобы улучшить речевое развитие, рекомендованы продолжительные курсы занятий с логопедами, сурдологами. По показаниям проводится психологическая коррекция.
- Специальное обучение. Пациентам с признаками ЗПР необходимо продолжать учебу в коррекционных классах с дефектологами, сурдопедагогами.
Прогноз и профилактика
При отсутствии у больного врожденных соматических пороков и достаточном уровне интеллекта прогноз благоприятный, большинство пациентов доживают до зрелого и пожилого возраста. Вызывают опасения случаи синдрома Гольденхара, которые сопровождаются кардиологическими, пульмонологическими или нефрологическими пороками, также неблагоприятный прогноз устанавливают при тяжелой умственной отсталости.
Основу профилактики синдрома составляет антенатальная охрана плода. Критическим периодом является первый триместр, поскольку именно в это время возникают характерные костные аномалии. Беременным женщинам рекомендовано избегать контакта с химикатами, рентгеновским излучением, а также соблюдать противоэпидемические меры для защиты от вирусных инфекций.
1. Клинический случай синдрома Гольденхара в психиатрической практике/ А.В. Ковалева// Acta Biomedica Scientifica. — 2020. — №3.
2. Клинический случай синдрома Гольденхара у новорожденного/ Л.Г. Киселева, Л.П. Мокеева, Ю.С. Тишкова, Н.В. Павловский// Вятский медицинский вестник. — 2015. — №46.
3. Диагностика синдрома Гольденхара в периоде новорожденности/ С.И. Мизинцева// Бюллетень Северного государственного медицинского университета. — 2012.
4. Особенности общеклинических проявлений синдрома Гольденхара/ И.А. Карякина// Системная интеграция в здравоохранении. — 2010. — №2.
Баршоня — Тешендорфа синдром (Th. Barsony, 1887-1942, венгерский рентгенолог; W. Teschendorf, немецкий рентгенолог, XX в.; синонимы: дивертикулы пищевода множественные ложные, дивертикулы пищевода множественные функциональные, пищевод извитой, пищевод четкообразный, пищевод штопорообразный) — болезнь пищевода неизвестной этиологии, характеризующаяся спастическим сокращением его циркулярных мышц, что придает пищеводу четкообразный вид на рентгенограмме; клинически проявляется непостоянной дисфагией и загрудинными болями.
Бойса симптом (Boys) — появление булькающего звука или урчания при надавливании на боковую поверхность шеи; признак большого дивертикула шейного отдела пищевода.
Бушара болезнь (Ch.J. Bouchard, 1837-1915, французский патолог) — гастрэктазия (расширение полости желудка с растяжением его стенок при стенозе привратниковой части или двенадцатиперстной кишки).
Бэррета язва пищевода (N.R. Barret, род. в 1903 г., английский хирург) — язва пищевода, по клинико-морфологическим признакам напоминающая пептическую язву желудка или двенадцатиперстной кишки.
Кушинга эзофагит (Н.W. Cushing, 1869-1939, американский нейрохирург) — острый эзофагит, иногда развивающийся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кушинга язва (Н.W. Cushing) — язва желудка или двенадцатиперстной кишки, иногда развивающаяся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кэмерона синдром (A.J. Cameron) — язвенные или эрозивные повреждения слизистой оболочки грыжевого выпячивания при грыжах пищеводного отверстия диафрагмы, которые сопровождаются хроническим оккультным или явным кровотечением и железодефицитной анемией.
Ларрея грыжа (D.J. Larrey, 1766-1842, французский хирург). Левосторонняя парастернальная диафрагмальная грыжа, выходящая в средостение через грудино-реберный треугольник.
Лихтенштерна симптом (Lichtenstern, синоним дисфагия парадоксальная, d. paradoxalis) — дисфагия, при которой большие порции пищи легче проходят в желудок, чем малые; возможный признак грыжи пищеводного отверстия диафрагмы.
Лиана — Сигье — Вельти синдром (С.С. Lian, 1882-1969, французский врач; F. Siguier, 1909-1972, французский врач; Н.L. Welti, 1895-1970, французский хирург) — сочетание диафрагмальной грыжи, часто осложненной рефлюкс-эзофагитом, с повторными тромбозами и тромбофлебитами сосудов конечностей и нередко с гипохромной анемией; патогенез неясен.
Монтандона синдром (А. Montandon, швейцарский врач, XX век) — приступы длительной дисфагии вследствие миогенного стеноза шейной части пищевода (области пищеводноглоточного соединения), сопровождающиеся попаданием жидкой пищи в гортань, аспирации и приводящие к нарастающему истощению.
Морганьи грыжа (G.B. Morgagni, 1682-1771, итальянский анатом). Правосторонняя парастернальная грыжа.
Харрингтона грыжа (S. W. Harrington, 1889-1975, американский хирург). Антральная грыжа пищеводного отверстия диафрагмы (типы 1 и 2).
Ценкера дивертикул (F.A. Zenker, 1825-1898, немецкий патолог; синоним ценкеровский дивертикул) — мешковидный дивертикул глоточного конца пищевода, образующийся сначала на его задней стенке, а затем распространяющийся и на боковую. В начале заболевания — глоточные парестезии, перемежающаяся дисфагия, сухой кашель. С увеличением дивертикула в нем происходит задержка пищи, урчание при приеме воды. Мешок сдавливает пищевод, усиливается дисфагия, часты аспирационные пневмонии.
Эпонимные симптомы/синдромы, связанные с патологией кишечника
Алапи симптом (Alapy). Отсутствие или незначительное напряжение брюшной стенки при инвагинации кишечника.
Альвареса синдром (W.C. Alvarez, 1884-1978, американский врач, описан в 1949 г.; синонимы: псевдоилеус; истерическое вздутие живота). Преходящее вздутие живота нейрогенной природы. Чаще наблюдается у истеричных или психопатичных женщин.
Бувре признак (1) (L. Bouveret, 1850-1929, французский врач) — выпячивание в области проекции на переднюю брюшную стенку места перехода подвздошной кишки в слепую, наблюдаемое при непроходимости толстой кишки.
Бувре признак (2) (L. Bouveret; синоним Куссмауля симптом, Adolf Kußmaul, 1822-1902, немецкий терапевт) — периодическое выбухание брюшной стенки в надчревной области и левом подреберье при сужении привратника, обусловленное усиленной перистальтикой желудка.
Вербрайка синдром (J.R. Verbryke, род. в 1885 г., американский врач; синоним синдром печеночного перегиба ободочной кишки) — боль и чувство натяжения в подложечной области в сочетании с диспептическими явлениями при наличии сращений желчного пузыря с печеночным углом ободочной кишки.
Вильмса симптом падающей капли (М. Wilms, 1867-1918, немецкий хирург и онколог) — звук падающей капли жидкости, определяющийся аускультативно на фоне шумов перистальтики при непроходимости кишечника.
Гарднера синдром (Е.J. Gardner, 1909-1989, американский врач-генетик) — наследственная болезнь, характеризующаяся множественным полипозом прямой и ободочной кишок в сочетании с доброкачественными опухолями, чаще костей и кожи (остеомы, фибромы, липомы); наследуется по аутосомно-доминантному типу.
Гейбнер-гертеровская болезнь (O.J.L. Heubner, 1843-1926, немецкий педиатр; С.A. Herter, 1865-1910, американский врач и фармаколог; синоним Ги — Гертера — Гейбнера болезнь) — целиакия.
Данса симптом (J. В. H. Dance, 1797-1832, французский врач) — западение брюшной стенки в правой подвздошной области при илеоцекальной инвагинации.
Жанбона синдром (М. Janbon, современный французский терапевт; синоним syndromus choleriformis, enteritis choleriformis). Описан как гастроинтестинальная симптоматика после лечения окситетрациклином. Патогенез заключается в уничтожении антибиотиками физиологической бактериальной флоры кишечника, что способствует развитию патогенной, устойчивой к антибиотикам.
Ирасека — Цюльцера — Уилсона синдром (A. Jirasek, 1880-1960, чехословацкий хирург; W.W. Zuelzer, 1909-1987, американский педиатр; J.L. Wilson, американский педиатр) — аганглиоз толстой кишки врожденный сегментарный. Патоморфологическая форма болезни Гиршспрунга, при которой в толстой кишке имеются одна или две аганглионарные зоны с нормальным участком кишки между ними; при этом аганглиоз не распространяется на прямую кишку.
Кантора симптом (М.О. Cantor, род. в 1907 г., американский хирург) — нитевидные тени в дефектах наполнения кишки; рентгенологический признак колита и регионарного илеита.
Карно симптом (M.O. Carnot) — боль в эпигастральной области, возникающая при резком разгибании туловища. Бывает при спаечной болезни.
Кенига синдром (F. Konig, 1832-1910, немецкий хирург) — сочетание приступов коликообразных болей в животе, метеоризма, чередования запоров и поносов, урчания при пальпации правой подвздошной ямки, наблюдающееся при хронической непроходимости кишечника в области перехода подвздошной кишки в слепую.
Клойбера симптом (Н. Kloiber, немецкий рентгенолог; синоним Клойбера чаши) — наличие на рентгенограмме живота (при вертикальном положении больного) теней, напоминающих чаши с жидкостью; признак скопления жидкости и газа в кишечнике при его непроходимости.
Кронкхайта — Канада синдром (L.М. Cronkhite, род. в 1919 г., американский врач; W.J. Canada, современный американский рентгенолог) — облысение, атрофия ногтей и гиперпигментация кожи при семейном полипозе органов пищеварительного тракта.
Кюсса синдром (G.Е. Kuss, 1877-1967, французский хирург) — хроническая частичная непроходимость кишечника, обусловленная наличием спаек в области малого таза, например при хроническом сальпингите.
Лобри — Сулля синдром (Ch. Laubry, 1872-1941, французский врач; P.L.J. Soulle, 1903-1960, франц. врач) — сочетание избыточного содержания газа в желудке, метеоризма и высокого стояния правого купола диафрагмы при ишемической болезни сердца, обусловленное рефлекторной гипокинезией пищеварительного тракта.
Мачеллы — Дворкена — Биля симптом (T.E. Machella, F.J. Dworken, H.J. Biel; 1952). Синоним: симптом селезеночного угла. Сильная боль в левой половине живота, вызванная растяжением газами селезеночного угла толстой кишки. Облегчение наступает после опорожнения кишечника и отхождения газов. См. Пайра болезнь, синдром.
Матье симптом (A. Mathieu, 1855-1917, французский врач) — шум плеска в пупочной области при толчкообразной пальпации; признак непроходимости кишечника.
Меккеля дивертикул (J.F. Meckel junior, 1781-1833, немецкий анатом). Врожденный дивертикул подвздошной кишки. Правило двоек: 2 дюйма длиной, в 2 футах от илеоцекального клапана (баугиниевой заслонки), у 2% населения.
Образцова признак (В.П. Образцов, 1849-1920, отечественный терапевт) — шум плеска при пальпации слепой кишки; признак хронического колита.
Обуховской больницы симптом — баллонообразное расширение ампулы прямой кишки, определяемое пальцевым исследованием; признак заворота сигмовидной кишки.
Пайра болезнь, синдром (Erwin Payr, австрогерманский хирург, 1871-1946; синонимы: синдром селезеночного изгиба, flexura lienalis syndrome, splenic flexure syndrome). Первично описана как запоры, обусловленные перегибом и сращением поперечной и нисходящей ободочной кишок («двустволка»). Разнообразная симптоматика (в т. ч. кардиалгии) связана с скоплением газа в области изгиба. Одни считают вариантом СРК (облегчение после опорожнения кишечника и отхождения газов), другие - самостоятельной патологией. Иногда называют также синдромом Мачеллы-Дворкена-Била.
Пиулахса — Хедериха (Пиулакса - Эдерика) болезнь, синдром (испанские врачи P. Piulachs, H. Hederich; синоним: тимпанит при синдроме долихомегаколона, tympanites in dolichomegacolon syndrome). Острое паралитическое расширение толстой кишки (без механической непроходимости). Симптомы: резкий метеоризм и боль в животе. Рентгенологически обнаруживают долихомегаколон.
Протопопова синдром (В.П. Протопопов, 1880-1957, отечественный психиатр; синоним Протопопова триада) — сочетание тахикардии, мидриаза (расширения зрачка) и спастического запора, наблюдающееся при депрессиях.
Рапунцель синдром (описан E.D. Vaughan, J.L. Sawyers и H.W. Scott в 1968 г.). Закупорка кишок, вызванная систематическим проглатыванием волос. В кишках больного образуются трихобезоары. Может потребовать хирургического вмешательства. Наблюдается при психопатиях, шизофрении, эпилепсии, олигофрении, преимущественно в детском возрасте. Рапунцель (Rapunzel) - персонаж одноименной сказки братьев Гримм, девушка с длинными косами.
Сейнта синдром (Ch.F.М. Saint, совр. южноафриканский патолог; синонимы: Сейнта триада, Сена синдром — нрк*, Сента синдром — нрк*) — сочетание грыжи пищеводного отверстия диафрагмы, желчнокаменной болезни и дивертикулеза толстой кишки.
Тавеля болезнь (Е. Tavevl, швейцарский хирург) — периколит после аппендэктомии, характеризующийся образованием спаек и сужением толстой кишки, лихорадкой и расстройствами функции кишечника.
Miserere! (помилосердствуй!) от начала католической молитвы "miserere mei" - помилуй меня (Боже), так по-старому называлась каловая рвота (copremesis) при непроходимости кишечника.
Читайте также: