Синдром Гийена-Барре (Guillain-Barre) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 21.01.2026
Порохина Ирина Дмитриевна 1 , Кузьмина Светлана Валентиновна 2
1 Первый Санкт-Петербургский Государственный Медицинский университет им. акад. И.П. Павлова, студентка 6 курса
2 Первый Санкт-Петербургский Государственный Медицинский университет им. акад. И.П. Павлова, кандидат медицинских наук, ассистент кафедры неврологии и нейрохирургии с клиникой
Аннотация
Данная статья посвящена обзору основных заболеваний, требующих проведения дифференциальной диагностики с синдромом Гийена-Барре, который в настоящее время является основной причиной возникновения тяжелых острых прогрессирующих периферических параличей и приводит к формированию стойкого неврологического дефицита.
Porokhina Irina Dmitrievna 1 , Kuzmina Svetlana Valentinovna 2
1 Pavlov First Saint Petersburg State Medical University, 6th year student
2 Pavlov First Saint Petersburg State Medical University, Candidate of Medical Science, Assistant professor of the Neurology and Neurosurgery Department
Abstract
This article is concerned with the most common diseases from which Guillain-Barre syndrome must be differentiated by the doctor. Nowadays Guillain-Barre syndrome is the main cause of severe acute progressive flaccid paralyses and it results in persistent neurological deficits.
В последние десятилетия по данным российской и зарубежной литературы роль синдрома Гийена-Барре в возникновении у пациентов тяжелых острых прогрессирующих периферических парезов рассматривается как ведущая, но при этом для данного синдрома характерен однофазный вариант течения с высокой вероятностью полного восстановления вне зависимости от тяжести клинических проявлений развернутой стадии заболевания при своевременной диагностике и адекватной патогенетической терапии [1, с.20].
Средний возраст пациентов на момент начала заболевания составляет 40 лет (диапазон возраста заболевших от 3 недель до 95 лет). Характерно наличие двух возрастных пиков: первый - в 20 - 29 лет, второй - в 50 и более лет. [2, с. 23 - 24]
У 1/3 пациентов развиваются дыхательные нарушения, требующие проведения ИВЛ.
5- 87% - полное выздоровление;
5 - 22% - выздоровление с дефектом;
5 - 33% - летальность [2, c.28].
Заболеваемость синдромом Гийена-Барре в мире составляет 1 - 2 случая на 100 тыс. населения в год. Заболеваемость в отдельных субъектах Российской Федерации по данным исследований от 2010 года составила от 0,34 до 2,5 на 100 тыс. населения [1, с.22].
По современным представлениям синдром Гийена - Барре является приобретенной иммуноопосредованной невропатией, развитие которой происходит вследствие иммунной реакции на предшествующее иммуноактивирующее событие (вирусная инфекция, оперативное вмешательство и др.) [3, c.23].
Установлено, что более чем 60% пациентов переносят респираторную или желудочно-кишечную инфекцию за 1 - 6 недель до появления первых симптомов заболевания. По данным серологических исследований эта инфекция может быть вирусной (CMV, EBV, HSV1, HSV2, VZ, вирус гриппа, вирусы Коксаки и ECHO и др.) или бактериальной (Mycoplasma pneumoniae, Campylobacter jejuni) [4, c.97].
Среди бактериальных триггеров ведущая роль принадлежит С. jejuni. Молекулярная мимикрия между ганглиозидами (эпитопы GM1, GM1b, GD1a, GQ1b, GalNAc-GD1a) и липосахаридами С. jejuni способствует выработке антиганглиозидных антител.
Среди триггеров вирусной природы ведущая роль принадлежит цитомегаловирусу (10 - 22% наблюдений). У пациентов с ЦМВ-ассоциированным синдромом Гийена-Барре выявляются анти-GM2-ганглиозидные антитела, при этом в клинической картине доминирует поражение чувствительных волокон, особенно черепных нервов [3, c.23], [6, c. 187, 188].
Клинические формы синдрома Гийена-Барре
А) Варианты с типичной клинической картиной:
1) Классическая форма - острая воспалительная демиелинизирующая полирадикулоневропатия (ОВДП) - 85 и >%
- Парапаретическая
- Фарингоцервикобрахиальная
2) Острая моторная аксональная полиневропатия (ОМАН) - 3%
3) Острая моторно-сенсорная аксональная полиневропатия (ОМСАН) -
Б) Варианты с атипичной клинической картиной:
1) Синдром Миллера Фишера - 5%
2) Острая вегетативная полиневропатия (острая пандизавтономия) -
3) Острая сенсорная полиневропатия - 1%
4) Острая краниальная полиневропатия -
5) Фарингоцервикокраниальная невропатия - 3% [2, c. 12], [4, c. 106 - 107].
Классическая форма (ОВДП) :
1 этап. Представление макрофагами перекрестно-реагирующего антигена наивным Т-лимфоцитам, их активация, циркуляция по кровотоку и фиксация на эндотелии венул периферических нервов.
2 этап. Активация продукции В-лимфоцитами антиганглиозидных антител (в основном, анти-GM1).
3 этап. Преодоление макрофагами, Т-лимфоцитами и антиганглиозидными антителами гематоэнцефалического барьера, проникновение их через эндотелий в периваскулярную зону и миграция к эндоневрию.
4 этап. Вхождение макрофагов Т-лимфоцитов и аутоантител в эндоневрий, идентификация аутоантигенов на леммоцитах, что приводит к комплемент-опосредованному повреждению их мембран [3, c.23 - 24], [5, c.2 - 7].
В настоящее время ведется поиск новых патогенетических механизмов развития синдрома Гийена - Барре. По данным публикации Y.Z. Wang с соавторами от 2012 года обнаружено повышение экспрессии Toll-подобных рецепторов 2,4 и 9 типов (TLR2, TLR4 и TLR9) у пациентов с синдромом Гийена - Барре по сравнению со здоровыми, а также установлена достоверная положительная корреляция между высокими уровнями этих показателей и выраженностью неврологического дефицита [3, c.26].
Аксональные формы (ОМАН, ОМСАН) :
Первоначальное поражение локализовано не в леммоцитах, а в области перехватов Ранвье и вентральных корешков двигательных (ОМАН) или двигательных и чувствительных нервов и дорсальных корешков (ОМСАН).
При взаимодействии анти-GM1-ганглиозидных антител с перехватами Ранвье происходит блокада ионных К/Na каналов и, как следствие, нарушение проведения нервного импульса. Это приводит к аксональной дегенерации с почти полным отсутствием или минимальной выраженностью лимфоцитарной воспалительной реакции и периаксональной макрофагальной инфильтрации [2, c.39 - 41, 138].
Для ОМАН и ОМСАН характерно развитие вторичной демиелинизации нервных волокон. Течение ОМАН, как правило, более благоприятное, что связывают с поражением только синаптических окончаний двигательных волокон. Если же в патологический процесс вовлекаются аксоны на значительном протяжении (как при ОМСАН), скорость их регенерации значительно снижается, а иногда и полностью прекращается вследствие гибели тел нейронов. Клинически ОМАН и ОМСАН практически неотличимы от классической формы синдрома Гийена-Барре [2, c. 138 - 140],[7, c.1323, 1326].
Чаще всего в дебюте заболевания у пациентов появляются двигательные и/или чувствительные расстройства (гипестезии, гиперестезии, парестезии) в нижних и/или верхних конечностях. Реже заболевание дебютирует с изолированного болевого синдрома в мышцах плечевого, тазового пояса и нижних конечностей или с изолированной слабости бульбарных мышц и наружных мышц глаз. Очень редко (при тяжелом течении) - с нарушения функций тазовых органов. [2, c. 87 - 88], [4, c.98 - 99].
Как правило, двигательные и чувствительные нарушения первоначально появляются в нижних конечностях и через несколько часов или дней распространяются на верхние конечности, туловище, шею, краниальную мускулатуру (восходящий паралич Ландри). Примерно у трети пациентов заболевание манифестирует с одновременным поражением верхних и нижних конечностей. [7, c. 1323].
Расстройства поверхностной (болевой и температурной) чувствительности выражены не резко, по типу полиневропатии «носки-перчатки», тогда как расстройства вибрационной и тактильной чувствительности ярко выражены.
Мышечная слабость появляется сначала в проксимальных отделах конечностей, симметрична, характерно резкое снижение, а затем выпадение сухожильных рефлексов [2, c.87 - 88], [4, c. 99].
Парезы различной степени выраженности / параличи конечностей, чаще симметричные. Вовлечение краниальной мускулатуры и мышц шеи свидетельствует о тяжелом течении заболевания и обычно является предвестником развития слабости межреберных мышц и диафрагмы и, как следствие, дыхательных расстройств (у 25 - 30% больных возникает потребность в ИВЛ).
Характерны резкое угнетение глубоких рефлексов (причем степень угнетения рефлексов не соответствует тяжести паралича), гипотония с последующей атрофией мышц [2, c. 89 - 90, 94], [4, c.99].
Характерно нерезкое нарушение поверхностной чувствительности (гипестезии, гиперестезии, парестезии) по типу полиневропатии «носки-перчатки». Могут наблюдаться участки нарушенной чувствительности на туловище.
Нарушения глубокой чувствительности (вибрационной, тактильной) встречаются у 20 - 50% пациентов, выражены значительно.
Выраженный болевой синдром отмечается у половины пациентов. Боль может быть корешковой (положительный симптом Ласега) либо мышечной (боли в мышцах спины, плечевого и тазового пояса, ноющего характера, усиливающиеся при движении; нередко наблюдаются мышечные спазмы, особенно в ночное время) [2, c. 94 - 96], [4, c. 99 - 100].
Встречаются более чем у 60% пациентов. Характерны нарушения со стороны сердечно-сосудистой системы: резкие колебания артериального давления, ортостатическая гипотензия, нарушения ритма (тахи- и брадиаритмии). Также характерны нарушения потоотделения (усиление или ослабление),ослабление моторики ЖКТ вплоть до развития кишечной непроходимости, нарушения мочеиспускания (задержка либо недержание мочи) [2, c. 96, 99 - 100], [4, c.100 - 101].
1) Прогрессирования (2 - 4 недели);
2) Плато (1 - 4 недели);
3) Восстановления (недели - месяцы).
Если прогрессирование симптомов продолжается в течение 4 - 8 недель, заболевание трактуется как подострая воспалительная демиелинизирующая полирадикулоневропатия. В том случае, если фаза прогрессирования длится более 8 недель, заболевание переходит в статус хронического [2, c.102 - 103], [4, c.101].
Диагностические критерии Ropper A.H.,1992 г.
Облигатные признаки : прогрессирующая слабость в верхних и нижних конечностях, арефлексия.
Признаки, свидетельствующие в пользу диагноза : нарастание симптоматики на протяжении нескольких дней или недель (до 4 недель); относительная симметричность симптомов; легкие нарушения чувствительности; вовлечение черепных нервов (особенно двустороннее поражение лицевых нервов); восстановление, начинающееся через 2 - 4 недели после прекращения прогрессирования; вегетативная дисфункция; отсутствие лихорадки в начале заболевания, высокое содержание белка в ЦСЖ при нормальном или незначительно повышенном цитозе (не более 10 клеток в мкл); типичные данные ЭМГ.
Признаки, исключающие диагноз : признаки ботулизма, миастении, полиомиелита, токсической полиневропатии; нарушение обмена порфиринов; недавно перенесенная дифтерия; изолированное нарушение чувствительности (в отсутствие мышечной слабости) [4, c.102].
- острая полиневропатия, характеризующаяся триадой симптомов: атаксия, арефлексия, офтальмоплегия. Ежегодно в мире регистрируется 1-2 новых случаев заболевания на 100 000 населения. Средний возраст пациентов - 44 года. Пики заболеваемости: 30 - 39 лет и 50 - 59 лет [8, c.312 - 313].
Особенности патогенеза : B-лимфоциты синтезируют антитела к оболочечным антигенам Campylobacter jejuni, которые перекрестно реагируют с ганглиозидом GQ1b, высокое содержание которого выявлено в нейронах мозжечка, III, IV и VI черепных нервах и спинно-мозговых ганглиях. С другой стороны, Campylobacter jejuni может провоцировать продукцию и других антител, связанных с развитием синдрома Гийена-Барре (анти-GM1, анти-GD1a). Таким образом объясняется сочетание вялого тетрапареза с поражением глазодвигательных нервов и мозжечковой атаксией. До сих пор остается открытым вопрос о локализации поражения при СМФ (является ли оно полностью периферическим или происходит с вовлечением ЦНС) [4, c.122 - 123], [8, c.317].
Первые симптомы : двоение в глазах, шаткость походки, реже - светобоязнь, птоз, дизартрия, дисфагия, слабость мимической мускулатуры. Появляются вышеперечисленные симптомы, как правило, через 1 - 3 недели после перенесенной респираторной или кишечной инфекции [4, c. 120].
- Наружная (характерно вовлечение наружных мышц глаза, при этом птоз наблюдается только у 50% пациентов);
- Внутренняя (характерно вовлечение внутренних мышц глаз, связана с парасимпатической денервацией глаза).
Начинается асимметрично, в течение нескольких дней становится симметричной и полной, может приобретать черты надъядерной или межъядерной вследствие того, что рефлекторные движения глаз восстанавливаются раньше произвольных [4, c. 120 - 121].
Характерна неустойчивость при стоянии и при ходьбе (статолокомоторная атаксия), как правило, клинически неотличима от мозжечковой атаксии.
Выпадение глубоких рефлексов с верхних и нижних конечностей.
- Парез мимической мускулатуры, связанный с поражением лицевого нерва; поражение тройничного, слухового нервов;
- Вялый парез конечностей (чаще - тетрапарез), течение легкое;
- Легкие дизестезии, чаще в дистальных отделах конечностей и на лице;
- Преходящее затруднение мочеиспускания [4, c. 120 - 122], [8, c.318].
1) Исследование цереброспинальной жидкости: выявление белково-клеточной диссоциации;
2) Выявление в крови антител к ганглиозиду GQ1b;
3) ЭМГ: снижение амплитуды потенциалов действия сенсорных нервов, отсутствие замедления проведения по моторным и сенсорным нервам [2, c.143 - 144], [4, c.126 ].
1. Полиневропатия при острой перемежающейся порфирии.
Острая перемежающаяся порфирия - заболевание с аутосомно-доминантным типом наследования, при котором активность фермента порфобилиноген-дезаминазы снижается до 50%.
Распространенность в странах Европы составляет 1 - 2 случая на 100 тыс. человек в год. Проявляется у 20% гетерозигот в виде клинических приступов, провоцирующими факторами для которых служат порфириногенные вещества (лекарственные препараты, алкоголь, инфекции, оперативные вмешательства и др.). Первый приступ обычно развивается в возрасте 15 - 35 лет. Большинство пациентов за свою жизнь переносит один или несколько приступов, которые завершаются полным выздоровлением, множественные приступы возникают менее чем у 10%. Течение острое, реже - подострое. Летальность также составляет около 10% [4, c.414 - 415].
В основе патогенеза поражения нервной системы при острой перемежающейся порфирии лежит нейротоксическое действие эпсилон - аминолевулиновой кислоты, порфобилиногена и продуктов их метаболизма на структуры как периферической (в большей степени) так и центральной нервной системы (в меньшей степени вследствие низкой проницаемости ГЭБ).
Характерна аксональная дегенерация с последующим развитием вторичной демиелинизации с вовлечением в патологический процесс толстых и тонких миелинизированных и немиелинизированных нервных волокон. Также характерно развитие дистрофии нейронов вегетативных ганглиев [4, c.415, 419 - 420].
Как правило, классический приступ острой перемежающейся порфирии начинается с вегетативных симптомов, с последующим присоединением психических нарушений и полиневропатии преимущественно моторного характера.
Вегетативные симптомы: боли в животе схваткообразного или ноющего характера разной интенсивности, рвота, запор либо диарея; задержка мочеиспускания. Характерны также синусовая тахикардия, повышение АД (иногда до очень высоких цифр, что приводит к острой гипертонической энцефалопатии), субфебрилитет.
Психические нарушения : депрессия, нарушения сна, тревожное или гипоманиакальное состояние; в тяжелых случаях - делирий, угнетение сознания, генерализованные судорожные припадки [4, c. 420 - 422].
Полиневропатия: нарастающий вялый тетрапарез, характеризующийся вовлечением первыми верхних конечностей от проксимальных отделов к дистальным (бибрахиальный парез). Парезы, как правило, симметричны, но могут быть и асимметричными (гемипарезы), в 50% случаев появляются на фоне миалгий. Возможен дебют с парестезий в конечностях, на туловище, лице и шее. Сухожильные рефлексы сохранены, по мере прогрессирования заболевания могут ослабляться и выпадать.
При тяжелом течении заболевания характерно вовлечение дыхательных мышц, поражение черепных нервов, что приводит к развитию бульбарного синдрома, пареза мимической мускулатуры, нарушений движения глаз.
Нарушение чувствительности наблюдается редко, в основном - поверхностной по типу гиперестезии [4, c.422 - 424].
Табл.1. Дифференциальная диагностика синдрома Гийена-Барре и острой перемежающейся порфирии
Синдром Гийена — Барре - симптомы и лечение
Что такое синдром Гийена — Барре? Причины возникновения, диагностику и методы лечения разберем в статье доктора Жуйкова Александра Вячеславовича, невролога со стажем в 21 год.
Над статьей доктора Жуйкова Александра Вячеславовича работали литературный редактор Елена Бережная , научный редактор Сергей Федосов

Определение болезни. Причины заболевания
Синдром Гийена — Барре (ГБС) — острое аутоиммунное заболевание, которое охватывает группу острых нарушений периферической нервной системы. Характеризуется мышечной слабостью, а также болью и ползанием мурашек в начале болезни из-за поражения чувствительных волокон. Каждый вариант нарушений характеризуется особенностями патофизиологии и клинического распределения слабости в конечностях и черепных нервах.
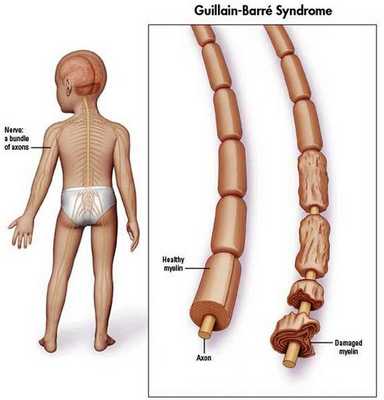
Распространённость синдрома Гийена — Барре
Синдром Гийена — Барре встречается в 1-2 случаях на 100 000 населения в год. [10]
Причины синдрома Гийена — Барре
Точная причина синдрома Гийена — Барре неизвестна. Но у 70% пациентов с ГБС наблюдались предшествующие инфекционные заболевания: респираторные, желудочно-кишечные инфекции, вирус Зика. Также синдром Гийена — Барре может развиться после заражения коронавирусом. [9]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гийена — Барре
Симптомы ОРВИ или расстройства желудочно-кишечного тракта отмечаются у 2/3 пациентов. Первыми симптомами ГБС являются парестезии пальцев конечностей, за которыми следует прогрессирующая слабость мышц нижних конечностей и нарушения походки. Болезнь прогрессирует в течение нескольких часов или дней, возникает слабость верхних конечностей и развиваются паралич черепных нервов. Параличи обычно симметричны и носят, конечно, периферический характер. У половины пациентов боль может быть первоначальной жалобой, что затрудняет диагноз. Атаксия и боль чаще встречаются у детей, чем у взрослых. Задержка мочи наблюдается у 10%-15% больных. Поражение вегетативных нервов проявляются головокружениями, гипертонией, чрезмерным потоотделением и тахикардией.
При объективном обследовании выявляется восходящая мышечная слабость, а также арефлексия. Сухожильные рефлексы нижних конечностей отсутствуют, но рефлексы верхней конечности могут вызываться. Мышечная слабость может задействовать и респираторные мышцы. Поражение черепных нервов отмечается в 35-50%, вегетативная нестабильность в 26%-50%, атаксия — в 23%, дизестезия — в 20% случаев. [1]
Наиболее распространенными признаками вегетативной дисфункции являются синусовая тахикардия или брадикардия и артериальная гипертония. У пациентов с тяжелой вегетативной дисфункцией наблюдаются изменения периферического вазомоторного тонуса с гипотензией и лабильностью артериального давления.
Нечастые варианты клинического течения болезни включают лихорадку в начале неврологических симптомов, тяжелую сенсорную недостаточность с болью (миалгии и артралгии, менингизм, корешковая боль), дисфункции сфинктеров.
Возможность ГБС должна рассматриваться у любого пациента с быстрым развитием острой нервно-мышечной слабости. На ранней стадии ГБС следует отличать от других заболеваний с прогрессирующей симметричной мышечной слабостью, включая поперечный миелит и миелопатию, острую токсическую или дифтеритическую полиневропатию, порфирию, миастению и нарушения электролитного обмена (например, гипокалиемия).
Патогенез синдрома Гийена — Барре
Нейрофизиологические процессы, лежащие в основе ГБС, подразделяются на несколько подтипов. Наиболее распространенные подтипы включают:
- острую воспалительную демиелинизирующую полирадикулопатию;
- острую двигательную аксональную невропатию;
- острую моторную и сенсорную аксональную нейропатию;
- синдром Миллера-Фишера, как вариант ГБС, характеризуется триадой признаков: офтальмоплегия, атаксия и арефлексия.
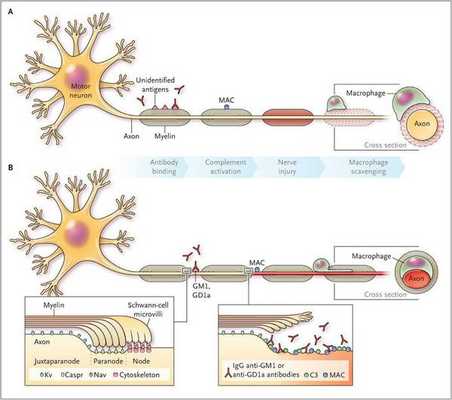
Считается, что ГБС развивается вследствие выработки антител против белка инфекционного агента, которые перекрестно реагируют с ганглиозидами нервных волокон человека. Аутоантитела связываются с миелиновыми антигенами и активируют комплемент, с формированием мембранно-атакующего комплекса на внешней поверхности клеток Шванна. Повреждение оболочек нервных стволов приводит к нарушениям проводимости и мышечной слабости (на поздней стадии может происходить и аксональная дегенерация). Демиелинизирующее поражение наблюдается по всей длине периферического нерва, включая нервные корешки.
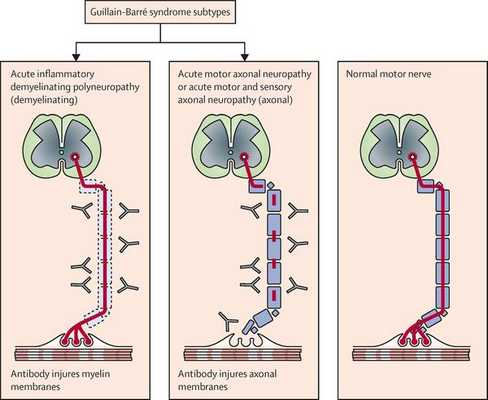
Поражаются все типы нервов, в том числе вегетативные, моторные и сенсорные волокна. Вовлечение двигательных нервов происходит значительно чаще, чем сенсорных.
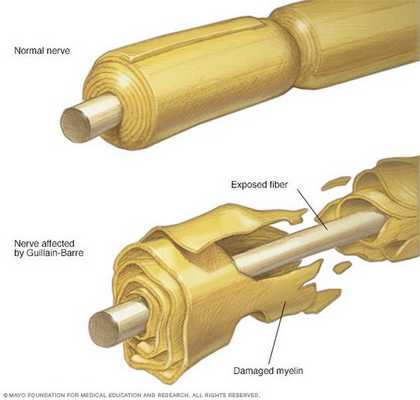
Классификация и стадии развития синдрома Гийена — Барре
В Международной классификации болезней (МКБ-10) синдром Гийена — Барре кодируется как G61.0.
Основные виды синдрома Гийена — Барре:
- Острая воспалительная демиелинизирующая полирадикулоневропатия (ОВДП) — самая распространённая форма в Северной Америке и Европе. Основным признаком ОВДП является мышечная слабость, которая сперва возникает в нижней части тела, а затем распространяется вверх.
- Синдром Миллера Фишера — проявляется параличом глаз и неустойчивостью походки. Эта форма более распространена в Азии.
- Острая моторная аксональная невропатия (ОМАН) и острая моторно-сенсорная аксональная невропатия (ОМСАН) — чаще встречаются в Китае, Японии и Мексике. [8]
Осложнения синдрома Гийена — Барре
Пациенты с ГБС подвержены риску опасных для жизни респираторных осложнений и вегетативных нарушений.
Показания для перевода в отделение интенсивной терапии включают:
- быстрое прогрессирование моторной слабости с поражением респираторных мышц;
- вентиляционную дыхательную недостаточность;
- пневмонию;
- бульбарные расстройства;
- тяжелую вегетативную недостаточность.
Осложнения проводимого лечения, требующие интенсивной терапии, включают перегрузку жидкостью, анафилаксию на введение внутривенного иммуноглобулина или гемодинамические нарушения при проведении плазмафереза.
У 15%-25% детей с ГБС развивается декомпенсированная дыхательная недостаточность, которая требует механической вентиляции легких. [2] Респираторные нарушения чаще встречается у детей с быстрым прогрессированием заболевания, слабостью верхних конечностей, вегетативной дисфункцией и поражениями черепных нервов. Интубация трахеи может потребоваться у больных для защиты дыхательных путей, проведения механической вентиляции легких. При ГБС быстрое прогрессирование, двусторонний паралич лицевого нерва и вегетативная дисфункция предопределяют повышенную вероятность интубации. Необходимо планирование ранней интубации для минимизации риска осложнений и необходимости проведения экстренной интубации.
Вегетативная дисфункция повышает риск эндотрахеальной интубации. С другой стороны, дисавтономия может увеличить риск гемодинамических реакций на препараты, используемые для индукции анестезии во время интубации.
Признаки, указывающие на необходимость механической вентиляции легких: [4]
- вентиляционная дыхательная недостаточность;
- увеличение потребности в кислороде для поддержания SpO2 выше 92%;
- признаки альвеолярной гиповентиляции (PCO2 выше 50 мм. рт. ст.);
- быстрое снижение жизненной емкости на 50% по сравнению с исходным уровнем;
- невозможность кашля
Вегетативная дисфункция является основным фактором смертности при ГБС. Фатальный сердечно-сосудистый коллапс из-за вегетативной дисфункции наблюдается у 2%-10% тяжелобольных пациентов. [3] Мониторинг частоты сердечных сокращений, артериального давления и электрокардиограммы следует продолжать до тех пор, пока пациенты нуждаются в респираторной поддержке. Чрескожная кардиостимуляция может потребоваться при выраженной брадикардии. Гипотония корректируется восполнением объема циркулирующей крови (ОЦК), и, если пациент невосприимчив к восполнению ОЦК, применяются α-агонисты, такие как норадреналин, мезатон, адреналин.
При нестабильной гемодинамике непрерывная регистрация артериального и центрального венозного давления должна проводиться для контроля объема инфузионной терапии.
Артериальная гипертензия может возникать, но это осложнение не требует специального лечения, если оно не осложняется отеком легких, энцефалопатией или субарахноидальным кровоизлиянием.
Диагностика синдрома Гийена — Барре
Сбор жалоб и анамнеза
На приёме врач в первую очередь обратит внимание на скорость распространения паралича и нарушение дыхания. Если эти признаки выражены, больному может потребоваться экстренная помощь.
Как правило, пациенты с синдромом Гийена — Барре жалуются на нарушение походки, онемение и зябкость ног, а затем и рук. Нередко пациенты рассказывают, что недавно перенесли ОРВИ.
Осмотр
Объективный неврологический осмотр — это основа диагностики при синдроме Гийена — Барре. Врач оценивает рефлексы, координацию движений, походку, чувствительность и мышечную силу.
Лабораторная диагностика
Основным видом лабораторной диагностики при синдроме Гийена — Барре является исследование спинномозговой жидкости, которую получают с помощью люмбальной пункции.
Инструментальная диагностика
ЭНМГ (Электронейромиография) — единственный инструментальный метод диагностики, позволяющий подтвердить диагноз ГБС и уточнить характер патологических изменений (демиелинизирующий или аксональный) и их распространенность. [3]
Игольчатая электромиография характеризуется наличием признаков текущего денервационно-реиннервационного процесса при полинейропатии. Исследуют дистальные мышцы верхних и нижних конечностей (например, переднюю большеберцовую мышцу, общий разгибатель пальцев), а при необходимости и проксимальные мышцы (например, четырёхглавую мышцу бедра).
ЭНМГ-исследование у больных с ГБС зависит от клинических проявлений:
- при дистальных парезах исследуются длинные нервы на руках и ногах: не менее четырех двигательных и четырех чувствительных (двигательные и чувствительные порции срединного и локтевого нервов; малоберцовый, большеберцовый, поверхностный малоберцовый и икроножный нервы с одной стороны).
Оценка основных ЭНМГ- параметров:
Первые признаки денервационного процесса появляются через две-три недели после начала заболевания, признаки реиннервационного процесса — через месяц.
Дифференциальная диагностика
Синдром Гийена — Барре следует отличать от следующих заболеваний:
- и клещевого энцефалита (чувствительность не нарушена, поражены преимущественно черепные нервы); (отягощённый эпидемиологический анамнез, например посещение эндемичных стран); (чувствительность не нарушена, рефлексы снижены незначительно);
- обменно-метаболических полиневропатий (течение хроническое).
Лечение синдрома Гийена — Барре
Показания для госпитализации
Практически во всех случаях требуется госпитализация. Экстренная госпитализация необходима пациентам с нарушениями дыхания — в таких случаях лечение проводят в условиях реанимации.
Общие принципы лечения синдрома Гийена — Барре
Лечение острой демиелинизирующей полирадикулоневропатии комплексное. Основа — плазмаферез, иммуноглобулины и кортикостероиды. В ряде случаев требуется искусственная вентиляция лёгких, коррекция нарушений кровообращения, профилактика инфекционных и тромбоэмболических осложнений. Обязательным условием успешного лечения является уход.
Общее поддерживающее лечение и уход
Пациенты, требующие интенсивной терапии, требуют тщательного общего ухода. Запор наблюдается более чем в 50% случаев пациентов с ГБС в результате динамической непроходимости кишечника. Может потребоваться искусственное питание.
Медикаментозное лечение и плазмаферез
В лечении ГБС предпринимаются различные виды иммуномодулирующей терапии. [1] [2]
Внутривенный иммуноглобулин назначают в виде ежедневной инфузии (в дозе 0,4 гр/кг/день) в течение 5 дней в первые 2 недели болезни. Второй курс иммуноглобулина может потребоваться 5%-10% пациентов, при отрицательной динамике после первоначального улучшения. Механизм действия внутривенного иммуноглобулина, вероятно, многофакторный и, как полагают, включает модуляцию активации комплемента, нейтрализацию идиотипических антител, подавление воспалительных медиаторов (цитокины, хемокины).
Побочные эффекты иммуноглобулина включают головную боль, миалгию и артралгию, гриппоподобные симптомы, лихорадку. У пациентов с дефицитом IgA может развиться анафилаксия после первого курса внутривенного иммуноглобулина.
Плазмаферез способствует удалению антител, вовлеченных в патогенез ГБС. В течение каждого сеанса 40-50 мл/кг плазмы заменяют смесью 0,9% раствора хлорида натрия и альбумина. Проведение плазмафереза приводит к сокращению времени выздоровления и снижению потребности в искусственной вентиляции. Эти преимущества очевидны, если плазмаферез проводится в течение первых двух недель после начала болезни. Осложнения, связанные с плазмаферезом, включают гематому в области венопункции, пневмоторакс после катетеризации подключичной вены и сепсис. Плазмаферез противопоказан пациентам с тяжелой гемодинамической нестабильностью, кровотечением и сепсисом. Комбинация плазмафереза и иммуноглобулина не показала клинических преимуществ.
Симптоматическое лечение синдрома Гийена — Барре:
- при боли применяют парацетамол;
- катадолон и трамадол применяют при выраженном болевом синдроме;
- при нейропатической боли эффективны карбамазепин и габапентин.
Оперативное лечение
При тяжёлом течении может потребоваться длительная респираторная поддержка и наложение трахеостомы. Если пациент находится на искусственном питании, то накладывают гастростому.
Прогноз. Профилактика
ГБС остается серьезным заболеванием, несмотря на улучшение результатов лечения. По сравнению со взрослыми, у детей чаще отмечается более благоприятное течение заболевания, с полным, а не частичным выздоровлением. Причинами неблагоприятного исхода при ГБС являются дыхательная недостаточность, осложнения искусственной вентиляции легких (пневмония, сепсис, острый респираторный дистресс-синдром и тромбоэмболические осложнения), остановка сердца, вторичная по отношению к дисавтономии.
Восстановление обычно начинается через две-четыре недели после прекращения прогрессирования симптомов. Среднее время от начала заболевания до полного выздоровления составляет 60 дней. Данные относительно долгосрочного исхода ГБС ограничены. 75% - 80% пациентов полностью выздоравливают. Около 20% пациентов не могут ходить через полгода.
Младшая возрастная группа (менее 9 лет), быстрое прогрессирование и максимальная мышечная слабость, потребность в искусственной вентиляции легких являются важными предикторами длительного двигательного дефицита. [4]
Синдром Гийена-Барре у детей
Синдром Гийена-Барре (СГБ, Guillain-Barré syndrome) - острое поражение периферической нервной системы дизиммунной природы, характеризующееся быстро прогрессирующей мышечной слабостью с формированием вялых параличей и/или парестезии конечностей (монофазная иммуноопосредованная нейропатия).

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
G61.0 - Синдром Гийена-Барре
• Острая краниальная полинейропатия
1) острая воспалительная демиелинизирующая полинейропатия (классическая форма синдрома Гийена-Барре),
5) острая панавтономная нейропатия (острый панавтономный синдром Гийена-Барре, острая пандизавтономия),
Этиология и патогенез
Этиологические факторы синдрома Гийена-Барре окончательно не изучены, что позволяет именовать болезнь идиопатической полинейропатией. Имеются основания рассматривать целый ряд патогенных микроорганизмов в качестве этиологически значимых, поскольку инфицирование ими зачастую (в 66% случаев) предшествует развитию СГБ. В их числе фигурируют следующие: цитомегаловирус, вирус Epstein-Barr, Haemophilus influenzae тип b, Mycoplasma pneumoniae, Campylobacter jejuni и многие другие возбудители инфекционных болезней и процессов.
При этом Campylobacter jejuni является причинно-значимым инфекционным агентом у трети пациентов, а молекулярная мимикрия между ганглиозидами и липосахаридами (эпитопы GM1, GM1b, GD1a, GQ1b, GalNAc-GD1a) данного микроорганизма способствует выработке антиганглиозидных антител. Высокие титры антиганглиозидных антител классов IgM, IgG и IgA, вступающие в реакции с эпитопами аксоплазматического отдела аксонов и миелиновой оболочки, обнаруживаются в сыворотке крови у 40% больных с СГБ.
Не исключается этиологическая роль некоторых видов профилактической иммунизации (противополиомиелитной, антирабической, противодифтерийной, противогриппозной и др.) в развитии СГБ. Риск развития болезни после противогриппозной вакцинации (H1N1) составляет порядка 1-2 случая на 1 миллион привитых.
В ходе генетических исследований выявлена связь между антигенами HLA-54, HLA-CW1, HLA-DQB*3 и СГБ. Обнаружена положительная корреляция между острой воспалительной демиелинизирующей полинейропатией и аллелем DQB1*0603 с уникальным эпитопом DQβED, а также отрицательная корреляции - с аллелями AQB1*0503, DQB1*0601, DQB1*0602 и DQB1*0603, характеризуемыми эпитопом RDP. Считается, что класс HLA является определяющим при различных вариантах СГБ, а сама болезнь представляет комплексное генетическое нарушение, на исход которого оказывают влияние генетические факторы и окружение.
K.H. Chang и соавт. (2012) описали 256 генов и 18 генных сетей, достоверно ассоциированных с СГБ; среди них наиболее частыми генами оказались FOS, PTGS2, HMGB2 и MMP9.
Болезнь вызывается аномальным Т-клеточным ответом, индуцированным инфекционным процессом. Возникает воспалительная нейропатия с перекрестной реактивностью между антителами к инфекционным агентам и антителами к нейроантигенам, поскольку липоолигосахариды в клеточной стенке бактерий напоминают ганглиозиды, а антиганглиозидные антитела формируются в ответ острые инфекции.
Таким образом, СГБ рассматривается, как приобретенная иммуноопосредованная нейропатия, развивающаяся вследствие аберрантной иммунной реакции на предшествующее иммуноактивирующее событие (перенесение вирусной инфекции, вакцинация и т.д.). Иммунопатологические реакции приводят к аутоиммунному повреждению тканей, ассоциированному с механизмами молекулярной мимикрии, участием суперантигенов и стимуляцией цитокинов.
Обнаружение дезоксирибонуклеиновой кислоты (ДНК) Campylobacter jejuni в миеломоноцитарных клетках позволяет предполагать представление нейритогенных антигенов Т-клеткам комплексом HLA класса II.
Начальным этапом в иммунопатогенезе болезни является представление антигена наивным Т-клеткам с их последующей активацией, циркуляцией по кровотоку и привязыванием к венулярному эндотелию периферических нервов. Затем Т-клетки пересекают гематоэнцефалический барьер, мигрируют через эндотелиальный слой в периваскулярную область и направляются в эндоневрий, задействуя механизмы молекул адгезии (селектины, лейкоцитарные интегрины и их контрарецепторы). Заключительным этапом в патогенезе СГБ является вхождение Т-клеток и аутоантител в эндоневрий вместе с макрофагами, где при помощи антительных и Т-клеточных механизмов идентифицируются аутоантигены на аксональных или шванновских клетках. Описываемый процесс приводит к выраженному повреждению тканей, чему способствует акивный фагоцитоз клеток моноцитарно-макрофагальной линии.
При классической форме СГБ (острая воспалительная демиелинизирующая полинейропатия) происходит поражение волокон двигательных и чувствительных нейронов. При этом основными структурами, подвергающимися патологическому воздействию, являются преимущественно корни мотонейронов и смежные проксимальные сплетения. Характерен феномен выраженной сегментарной воспалительной демиелинизации, сопровождающейся очаговой и диффузной инфильтрацией Т-лимфоидными и моноцитарно-макрофагальными клетками на всех уровнях периферической нервной системы. Воспалительные клетки аккумулируются вокруг мелких сосудов эндоневрия/эпиневрия. Комплемент-опосредованное связывание антител с эпитопами, расположенных на поверхностной мембране шванновских клеток, предшествует Т-клеточной инфильтрации. Клинические проявления болезни являются прямым следствием нарушения сальтаторной проводимости по миелинизированным волокнам (возникает блок проведения).
Аксональные варианты СГБ (острая моторно-аксональная и моторно-сенсорная нейропатии) характеризуются отсутствием выраженных признаков воспаления и наличием аксональной дегенерации нервных волокон. Изменения в ЦНС при этих вариантах болезни вторичны по отношению к дегенерации аксонов. При острой моторно-аксональной нейропатии «иммунной атаке» подвержены, в первую очередь, двигательные узлы Ранвье. Острая моторно-аксональная нейропатия (ОМАН) и острая моторно-сенсорная аксональная нейропатия (ОМСАН) ассоциированы с антиганглиозидными антителами (GM1/GD1a/GM1b/GalNAc-GD1a); предполагается, что оба аксональных варианта СГБ вызываются антительно-опосредованной первичной аксональной дегенерацией или антительно-опосредованной ингибицией вольтаж-зависимых натриевых каналов.
Точные механизмы патогенеза синдрома Миллера-Фишера окончательно не изучены, но известно, что болезнь ассоциирована c повышенными титрами антиганглиозидных антител (преимущественно к GQ1b), как и стволовой энцефалит Бикерстаффа [1, 2, 9, 10, 20, 21].
Эпидемиология
Синдром Гийена-Барре - редкий вид острой демиелинизирующей патологии периферической нервной системы, встречающейся с частотой 1,0-1,9 случая на 100 тысяч населения у взрослых и 0,34-1,34 на 100 000 населения у детей.
Острая воспалительная демиелинизирующая полинейропатия (ОВДП) встречается в 77-78%, на долю аксональных вариантов синдрома Гийена-Барре (острая моторно-аксональная нейропатия - ОМАН, острая моторно-сенсорная аксональная нейропатия - ОМСАН) в странах Запада приходятся 3-5%, а в Азии и Латинской Америке - 30-50%.
На долю синдрома Миллера-Фишера, острой панавтономной нейропатии, стволового энцефалита Бикерстаффа и других форм СГБ приходится не более 2% и более точных данных в доступной литературе не представлено [1, 2, 5, 10, 20].
Клиническая картина
Cимптомы, течение
Классическими проявлениями болезни считаются прогрессирующий (восходящий) паралич мышц конечностей и дыхательной мускулатуры, что сопровождается расстройствами чувствительности по полинейропатическому типу; впоследствии у пациентов возникают вегетативно-трофические нарушения. Характерно внезапное появление неврологической симптоматики: болевой синдром (до 80%) и парестезии (20%); типичны атаксия, парезы мышц конечностей и параличи черепных нервов.
Поражение симпатической нервной системы проявляется различными вегетативными нарушениями (гипертензия, постуральная гипотензия, профузное потоотделение, нарушения терморегуляции и т.д.). Паралич дыхательной мускулатуры является типичным и тяжелым осложнением острой воспалительной демиелинизирующей полинейропатии, требующим проведения ИВЛ и/или трахеостомии у взрослых, в то время как у детей наблюдается примерно в 3% случаев.
Клиническая картина практически не отличима от ОВДП, за исключением того, что не отмечается поражения сенсорных волокон периферических нервов. В большинстве случаев заболевание протекает более тяжело, чаще пациентам требуется ИВЛ, чаще формируется остаточный моторный дефицит.
Клинические проявления этого варианта СГБ практически неотличимы от симптомов острой воспалительной демиелинизирующей полинейропатии (мышечная слабость и сенсорный дефицит), но заболевание протекает более тяжело, а прогноз является гораздо более серьезным.
Отличительными чертами этого варианта болезни является наличие клинической триады в виде сочетания наружной офтальмоплегии (главный признак), атаксии и арефлексии, появляющееся в пределах первой недели после начала заболевания. Наиболее ранними симптомами синдрома Миллера-Фишера служат диплопия, иногда может наблюдаться двухсторонний парез лицевого нерва. Встречаются параличи/парезы лицевого нерва и бульбарные расстройства. По достижении максимальной выраженности, описываемые симптомы обычно сохраняются на протяжении 1-2 недель, иногда до 4-х недель, после чего отмечается постепенное восстановление неврологических функций (обычно оно бывает полным или практически полным).
Характерным также является: отсутствие мышечной слабости в конечностях; отсутствие нарушения сознания или признаков вовлечения кортико-спинального тракта; повышение белка в ЦСЖ при цитозе менее 50 мононуклеарных клеток; нормальные результаты по данным электромионейрографии (ЭНМГ) или изолированное поражение чувствительных нервов.
Частыми симптомами болезни являются нарушения потоотделения, отсутствие слезообразования, фотофобия, тошнота, дисфагия, сухость слизистых оболочек носа и ротовой полости, сухость и отслойка кожи, а также нарушения дефекации (запоры, диарея). В числе ранних неспецифических проявлений заболевания фигурируют головная боль, летаргия, усталость, сниженная мотивация (к принятию инициативных решений), а также признаки вегетативных нарушений (ортостатическое головокружение, размытость зрения, сухость глаз, нарушения мочеиспускания). Крайне редко эта форма СГБ наблюдается у детей. В дебюте заболевания наиболее часты симптомы в виде нарушений, ассоциированных с ортостатической непереносимостью, а также расстройства со стороны желудочно-кишечного тракта и нарушение функции потовых желез (судомоторная дисфункция). Могут отмечаться парасимпатические расстройства (боли в животе, рвота, запор, илеус, задержка мочи, расширение и ареактивность зрачков, потеря аккомодации). Крайне редко эта форма СГБ наблюдается у детей.
Характеризуется острым, внезапным дебютом в виде офтальмоплегии, атаксии, нарушения сознания, гиперрефлексии и наличия симптома Бабинского. Течение болезни монофазное или реже ремиттирующее-рецидивирующее. Крайне редко эта форма СГБ наблюдается у детей.
Характеризуется изолированной слабостью в лицевых, ротоглоточных, шейных мышцах, а также в мускулатуре верхних конечностей (без вовлечения нижних конечностей). Крайне редко эта форма СГБ наблюдается у детей.
Проявляется вовлечением в патологический процесс только черепных нервов. Крайне редко эта форма СГБ наблюдается у детей [1, 2, 5, 10, 20, 21]
В большинстве случаев течение болезни монофазное. В течении болезни принято выделять 3 стадии (периода): 1) период прогрессирования (не более 4-х недель), 2) период стойкой симптоматики (не более 4-х недель), 3) период восстановления (до 1 года).
В разграничении разных форм СГМ ведущим является электронейромиографическое исследование (ЭНМГ), по результатам которого выявляется демиелинизирующий или аксональных тип поражения периферических нервов. При СГБ в периоде прогрессирования невозможно прогнозировать течение заболевания, поэтому все пациенты с подозрением на СГБ в периоде прогрессирования должны быть госпитализированы, так как возможно дальнейшее нарастание тяжести парезов с развитием дыхательных и сердечно-сосудистых нарушений. В периоде прогрессирования необходимо проводить мониторинг неврологических нарушений (степени парезов, нарушения глотания, изменения тембра голоса), артериального давления, частоты сердечных сокращений, частоты дыхания, электрокардиографии (ЭКГ) и ЖЕЛ.
Синдром Гийена-Барре
Синдром Гийена-Барре (СГБ) — острая воспалительная демиелинизирующая полирадикулоневропатия аутоиммунной этиологии. Характерный признак заболевания — периферические параличи и белково-клеточная диссоциация в ликворе (в большинстве случаев). Диагноз синдрома Гийена-Барре устанавливается при наличии нарастающей слабости и арефлексии в более чем 1 конечности. При этом следует исключить другие неврологические заболевания, сопровождающиеся периферическими парезами: полиомиелит, острый период стволового инсульта, токсические поражения ЦНС и др. Лечение пациентов с синдромом Гийена-Барре проводится в стационаре, т. к. больному может потребоваться ИВЛ.
МКБ-10
Общие сведения
Синдром Гийена-Барре (СГБ) — острая демиелинизирующая воспалительная полиневропатия аутоиммунной этиологии. Характерный признак заболевания — периферические параличи и белково-клеточная диссоциация в ликворе (в большинстве случаев). В настоящее время в рамках СГБ выделяют четыре основных клинических варианта:
- классическая форма СГБ — острая воспалительная демиелинизирующая полирадикулоневропатия (до 90% случаев)
- аксональная форма СГБ — острая моторная аксональная невропатия. Характерный признак данной формы СГБ — изолированное поражение двигательных волокон. При острой моторно-сенсорной аксональной невропатии поражаются как двигательные, так и чувствительные волокна (до 15%)
- синдром Миллера-Фишера — форма СГБ, характеризующаяся офтальмоплегией, мозжечовой атаксией и арефлексией при слабовыраженных парезах (до 3%)
Помимо вышеуказанных форм синдрома Гийена-Барре, в последнее время выделяют еще несколько атипичных форм заболевания — острую сенсорную невропатию, острую пандизавтономию, острую краниальную полиневропатию, встречающиеся довольно редко.
Клиническая картина синдрома Гийена-Барре
Первыми проявлениями синдрома Гийена-Барре являются, как правило, мышечная слабость и/или сенсорные расстройства (чувство онемения, парестезии) в нижних конечностях, которые, спустя несколько часов (суток) распространяются на верхние конечности. В некоторых случаях заболевание манифестирует болями в мышцах конечностей и пояснично-крестцовой области. Очень редко первым проявлением становятся поражения ЧН (глазодвигательные расстройства, нарушение фонации и глотания). Степень двигательных нарушений при синдроме Гийена-Барре значительно варьируется — от минимальной мышечной слабости до тетраплегии. Парезы, как правило, симметричные и больше выражены в нижних конечностях.
Типична гипотония и существенное снижение (либо полное отсутствие) сухожильных рефлексов. В 30% случаев развивается дыхательная недостаточность. Расстройства поверхностной чувствительности проявляются в виде легкой или умеренной гипо- или гиперкинезии по полиневритическому типу. Приблизительно у половины пациентов наблюдаются расстройства глубокой чувствительности (иногда вплоть до полной ее утраты). Поражения ЧН, выявляемые у большинства больных, проявляются парезом мимических мышц и бульбарными нарушениями. Из вегетативных нарушений наиболее часто наблюдаются сердечные аритмии, артериальная гипертензия, расстройство потоотделения, нарушение функций ЖКТ и тазовых органов (задержка мочи).
Диагноз синдрома Гийена-Барре
При сборе анамнеза необходимо обратить внимание на наличие провоцирующих факторов, так как более чем в 80% случаев развитию СГБ предшествуют те или иные заболевания и состояния (перенесенные инфекции ЖКТ, верхних дыхательных путей, вакцинация, оперативные вмешательства, интоксикация, опухоль). Неврологическое обследование направлено на выявление и оценку выраженности основных симптомов синдрома Гийена-Барре — чувствительных, двигательных и вегетативных расстройств.
Необходимо проведение общеклинических исследований (общий анализ мочи, общий анализ крови), биохимического анализа крови (газовый состав крови, концентрация электролитов сыворотки), исследования ликвора, серологических исследований (при подозрении на инфекционную этиологию заболевания), а также электромиографию, результаты которой имеют принципиальное значение для подтверждения диагноза и определения формы СГБ. В тяжелых случаях заболевания (быстрое прогрессирование, бульбарные нарушения) следует проводить суточное мониторирование АД, ЭКГ, пульсовую оксиметрию и исследование функции внешнего дыхания (спирометрия, пикфлоуметрия).
Для подтверждения диагноза необходимо наличие прогрессирующей мышечной слабости более чем в одной конечности и отсутствие сухожильных рефлексов (арефлексия). Наличие стойких тазовых нарушений, выраженной стойкой ассиметрии парезов, полиморфноядерных лейкоцитов, а также четкого уровня расстройств чувствительности должны вызывать сомнения в диагнозе «синдром Гийена-Барре». Кроме того, существует ряд признаков, абсолютно исключающих диагноз СГБ, среди них: недавно перенесенная дифтерия, симптомы интоксикации свинцом либо доказательства интоксикации свинцом, наличие исключительно сенсорных нарушений, нарушение обмена порфиринов.
Дифференциальный диагноз
В первую очередь синдром Гийена-Барре невролог дифференцирует от иных заболеваний, которые также проявляются периферическими парезами (полиомиелит), а также других полиневропатий. Полиневропатия при острой перемежающейся порфирии может напоминать синдром Гийена-Барре, но, как правило, сопровождается разнообразной психопатологической симптоматикой (галлюцинации, бред) и выраженными абдоминальными болями. Симптоматика, схожая с признаками СГБ, возможна при обширных инсультах ствола головного мозга с развитием тетрапареза, который в острый период принимает черты периферического. Основные отличия миастении от СГБ — вариабельность симптоматики, отсутствие чувствительных расстройств, характерные изменения сухожильных рефлексов.
Лечение синдрома Гийена-Барре
Все пациенты с диагнозом «синдром Гийена-Барре» подлежат госпитализации в стационар с отделением интенсивной терапии и реанимации. Приблизительно в 30% случаев СГБ в виду развития тяжелой дыхательной недостаточности возникает необходимость в ИВЛ, продолжительность которой определяют индивидуально, ориентируясь на ЖЁЛ, восстановление глотания и кашлевого рефлекса. Отключение от аппарата ИВЛ проводят постепенно с обязательным этапом перемежающейся принудительной вентиляции.
В тяжелых случаях с выраженными парезами особое значение для предупреждения осложнений, связанных с длительной обездвиженностью пациента (инфекции, пролежни, тромбоэмболии легочной артерии), имеет правильный уход. Необходима периодическая (не менее одного раза в 2 часа) смена положения пациента, уход за кожей, контроль над функциями мочевого пузыря и кишечника, пассивная гимнастика, профилактика аспирации. При стойкой брадикардии с угрозой развития асистолии может потребоваться установка временного электрокардиостимулятора.
В качестве специфической терапии синдрома Гийена-Барре, направленной на купирование аутоиммунного процесса, в настоящее время применяют пульс-терапию иммуноглобулинами класса G и плазмаферез. Эффективность каждого из методов сравнительно одинакова, поэтому их одновременное применение считается нецелесообразным. Мембранный плазмаферез значительно уменьшает выраженность парезов и продолжительность ИВЛ. Проводят, как правило, 4-6 сеансов с интервалом в один день. В качестве замещающих сред используют 0,9% раствор натрия хлорида или декстран .
Следует помнить о противопоказаниях к проведению плазмафереза (инфекции, нарушения свертываемости крови, печеночная недостаточность), а также о возможных осложнениях (нарушение электролитного состава, гемолиз, аллергические реакции). Иммуноглобулин класса G, как и плазмаферез, уменьшает продолжительность пребывания на ИВЛ; его вводят внутривенно ежедневно в течение 5 дней в дозе 0,4 г/кг. Возможные побочные эффекты: тошнота, головные и мышечные боли, лихорадка.
Симптоматическая терапия при синдроме Гийена-Барре проводится для коррекции нарушений кислотно-основного и водно-электролитного баланса, коррекции уровня артериального давления, профилактики тромбоза глубоких вен и тромбоэмболии. Оперативное вмешательство может понадобиться для трахеостомии в случае продолжительной ИВЛ (более 10 суток), а также гастростомии при тяжелых и длительных бульбарных нарушениях.
Прогноз при синдроме Гийена-Барре
У большинства пациентов с диагнозом «синдром Гийена-Барре» наблюдается полное функциональное восстановление в течение 6-12 месяцев. Стойкая резидуальная симптоматика сохраняется приблизительно в 7-15% случаев. Частота рецидивов СГБ составляет около 4%, летальность — 5%. Возможные причины смерти — дыхательная недостаточность, пневмония или другие инфекции, тромбоэмболия легочной артерии. Вероятность летального исхода в большой степени зависит от возраста пациента: у детей в возрасте до 15 лет она не превышает 0,7%, в ВТО время как у пациентов старше 65 лет достигает 8%.
Профилактика синдрома Гийена-Барре
Специфических методов профилактики синдрома Гийена-Барре не существует. Однако следует уведомить пациента о запрете на прививки в течение первого года от дебюта заболевания, так как любая прививка способна вызвать рецидив заболевания. Дальнейшая иммунизация разрешена, при этом должна быть обоснована ее необходимость. Кроме того, развившийся в течение 6 месяцев после какой-либо вакцинации синдром Гийена-Барре — сам по себе является противопоказанием к применению данной вакцины в будущем.
Читайте также:
- Психогенные причины соматических заболеваний. Невроз органа
- Классификация переломов зубного отростка второго шейного позвонка (осевого позвонка)
- Истерический вариант развития слепого. Депривации у слепорожденных
- Ультразвуковое исследование сосудов. Этапы диагностики заболеваний артерий.
- Типы пищеварения. Собственный тип пищеварения. Аутолитический тип. Внутриклеточное пищеварение. Внеклеточное пищеварение.