Синдром Гольдштейна (Goldstein) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 21.01.2026
Синдром Гетчинсона-Гилфорда или прогерия (сенильный нанизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения. Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом. Этиология прогерии неясна. Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у больных. Однако исследования зарубежных ученых позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения. Характерен вид лица: с экзофтальмом, тонким клювовидным носом, большим мозговым и малым лицевым черепом, голос тонкий, имеются скелетные аномалии. Пубертат обычно не наступает, наружные гениталии гипоплазированы. Интеллект средний или выше среднего. Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Ключевые слова
Для цитирования:
Фофанова О.В. Синдром Гетчинсона-Гилфорда (прогерия). Проблемы Эндокринологии. 1995;41(4):24-26.
For citation:
Fofanova O.V. Syndrome Hutchinson-Gilford (progeria). Problems of Endocrinology. 1995;41(4):24-26. (In Russ.)
Синдром Гетчинсона-Гилфорда (Hutchinson-Gilford Progeria Syndrome), или прогерия (сенильный папизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения.
Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом.
Этнология прогерии неясна.
Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у бальных. Однако исследования зарубежных ученых [2-4, 7] позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Отмечается влияние возраста отца как возможной причины новых мутаций. Так, средний возраст отцов составляет 35-37 лет.
Аутосомно-рецессивный тип наследования синдрома также обсуждается в литературе [7, 8, И]. Этот тип наследования был впервые предположен Gabr и соавт. в 1960 г. при описании двух моиозиготных сестер, а впервые сообщил о семейном случае прогерии Paterson в 1922 г., хотя описание двух больных братьев было неполным и без фотографий.
Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения.
Дети рождаются нормальными, но к 1-му году жизни наблюдается выраженная задержка роста и массы тела. Конечный рост в среднем достигает 100 см. В первые годы жизни развивается тотальная алопеция. Кожа топкая, лоснящаяся, сухая, тугонатянутая (па кистях и стопах, наоборот, морщинистая). В нижней части живота и бедрах кожные изменения напоминают склеродермию. С возрастом появляются коричневые пигментные пятна. Потовые и сальные железы атрофируются.
Подкожный жировой слой полностью отсутствует, за исключением лобковой области. На черепе выражена подкожная венозная сеть. Нощи дистрофичные, влоть до аплазии. Зубы прорезываются с задержкой, аномально расположены, с ранним разрушением как молочных, так и постоянных зубов.
Характерен вид лица: с экзофтальмом, гонким клювовидным носом, большим мозговым и малым лицевым черепом. Голос тонкий.
Скелетные аномалии включают резорбцию ключицы с замещением фиброзной тканью, резорбцию конечных фаланг кистей (акроостеолиз), истончение длинных трубчатых костей и ребер, тугоподвижность сутавов пальцев, увеличение локтевых и коленных суставов. Часты асептические некрозы головки бедренной кости и вывих тазобедренного сустава.
Пубертат обычно не наступает, наружные гениталии ги- поплазированы.
Интеллект средний или выше среднего.
Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Следует отметить, что заболевания, характерные для нормального процесса старения (катаракта, опухоли, сахарный диабет), встречаются при прогерии крайне редко.
Прогноз для жизни неблагоприятный: продолжительность жизни колеблется от 7 до 28 лет, в среднем составляя 12-13,5 года [1, 4, 5, 7, 10]. Основные причины летальных исходов - острый инфаркт миокарда, застойная сердечная недостаточность, инсульты. На аутопсии выявляются распространенный атеросклероз, гипоплазия гонад, иногда гипоплазия надпочечников и значительная гиперплазия тимуса, истончение коркового слоя костей.
Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Гормональные исследования у детей [4] выявляют нормальную ночную секрецию соматотропного гормона гипофиза, но крайне низкий уровень инсулиноподобного фактора роста (ИФР-1) в плазме крови. Это позволяет предполагать наличие у данных больных бионеактивпого пула СТГ в крови, либо периферическую резистентность к эндогенному СТГ. либо выраженный дефицит питания. Трехмесячный период высококалорийного питания не увеличивает уровень ИФР-1 в крови, но ускоряет линейный рост.
Прогерия считается состоянием, связанным с ннсулиноре- зистентностыо умеренной степени. В 1983 г.[9] впервые была описана девочка, у которой в 2-легнсм возрасте уровень инсулина в крови натощак составлял 20-40 мкг/дл. В 4 года через 3 мес после удаления кисты яичника развилась гипергликемия натощак (до 250 мкг/дл) с высоким уровнем инсулина (более 2200 мкЕД/мл) и С-пептида (32,4 нг/мл) в крови. Уровень гемоглобина А1 достигал 10%. Связывание инсулина с рецепторами эритроциов было в пределах нормальных значений.
Иммунологический аспект в патогенезе прогерии впервые был выдвинут Walford в 1970 г. В 1973-1976 г. Singal и Goldstein выявили отсутствие либо резкое снижение экспрессии HLA культурой фибробластов кожи у детей с прогерией. Вместе с тем другие исследователи, анализируя экспрессию HLA у детей с прогерией и их здоровых родственников [2], нс выявили ни количественного, ни качественного дефицита в экспрессии HLA в фибробластах кожи. Различие в частоте встречаемости ряда HLA-антнгенов у больных прогерией и здоровых людей не дает право в настоящее время говорить о специфической ассоциации HLA и прогерии в связи с малым числом исследуемого материала.
Биохимическими исследованиями показано, что одним из биомаркеров старения является мочевая экскреция гиалуроновой кислоты. В норме у детей и подростков содержание ее составляет менее 1% от уровня общих гликозаминогликанов и увеличивается с возрастом до 5-6%. У детей с прогерией выявлено значительное повышение (до 10-20%) экскреции гиалуроновой кислоты с мочой по сравнению со здоровыми людьми [4]. Данное повышение не наблюдается ни при одном генетическом заболевании, кроме синдрома Вернера, или “прогерии взрослых” [4]. Считается, что гиалуроновая кислота является ключевым фактором антиангиогенеза в процессе созревания и старения.
Изучение культуры фибробластов кожи от пациентов с прогерией выявляет значительное снижение клеточного роста вследствие подавления митотической активности. С другой стороны, отмечается нормальное распределение типов коллагена в коже, характерное для детей, а именно, преобладание коллагена 3-го типа над коллагеном 1-го типа [8].
Имеются данные, что в основе прогерии лежит дефицит метаболизма витамина Е [7].
Ряд исследователей связывают прогерию с генетически обусловленной ошибкой в синтезе внутриклеточных белков. Так, показано, что эритроциты больных детей содержат повышенную термолабильную фракцию ферментов: глюкозо-6- фосфатдегидрогепазу и б-фосфоглюконатдегидрогепазу |6]. Другие работы не подтверждают эту концепцию [3].
Данные олене или детей с прогерией крайне малочисленны.
Патогенетически оправданными считаются терапия витамином Е для восполнения его дефицита (Лугез и МШап, 15974) и усиленное белковое питание.
Приводим описание собственного клинического случая.
Боль пая Н., 3 лет 9 мес., поступила в детское отделение ЭНЦ РАМН с жалобами на отставание в росте и массе, сниженный аппетит, облысение, резкую головную боль.
Раннее развитие: держит голову с 1 мес жизни, сидит с 6 .мес жизни, ходит с 1 года 2 мес, зубы появились в 1 год 1 мес, говорит с 1,5 лет. Грудное вскармливание - до 1 года 8 мес.
Перенесенные заболевания: дисплазия тазобедренный суставов (в 1 мес жизни), двусторонний врожденный вывих бедер (диагностирован в 6 мес жизни), стоматит, легкая форма (в 3 года).
Аллергологический анамнез не отягощен.
Наследственность по низкорослости не отягощена. Мать 24 лет, рост 165 см, родственники. со стороны матери: бабушка - рост 157 см, дедушка - 180 см, тетя - 168 см. Отец 26 лет, рост 176 см; родственники со стороны отца: бабушка - рост 157 см, дедушка - 170 см. Отягощена наследственность по сахарному диабету II типа, который имеется у бабушки со стороны отца и у прабабушки со стороны матери. Отягощена наследственность по бронхиальной астме, тяжелая форма которой имеется у прадедушки, со стороны матери и у двоюродной прабабушки со стороны матери.
Анамнез заболевания: с 2-месячного возраста замечены уплотнение кожных покровов и подкожной жировой клетчатки, лоснящаяся кожа на бедрах, животе, ягодицах, цианоз носогубного треугольника. В 3-месячном возрасте диагностирована легкая форма склеродермии. При исследовании биоптата кожи выявлен гиперкератоз эпителия и умеренный склероз дермы. Консультирована дерматологом: диагноз склеродермии был снят, больше данных, свидетельствующих о склеродерме. Получала лечение преднизолоновой мазыо в течение 3 мес с умеренным эффектом - блеск и плотность кожных покровов уменьшились. В возрасте 1 года появилась венозная сеть на голове. Отмечалась гипотрофия П-Ш степени. Невропатолог диагностировал перинатальную энцефалопатию, компенсированную гидроцефалию. В 1 год 7 мес девочка была впервые консультирована эндокринологом: диагностирована задержка физического развития смешанного генеза. Масса тела 7800 г, костный возраст соответствовал паспортному. В 1 год 10 мес начали выпадать полосы на голове. В 1 год 11 мес впервые консультирована генетиком, поставлен диагноз: “Синдром Гетчинсона—Гилфорда”. Девочка была обследована в стационаре по месту жительства: рост 73 см, масса 7900 г. На ЭКГ: метаболические изменения миокарда
Данные обследования в ЭНЦ РАМН. Хронологический возраст 3,9 года. Рост 81,2 см. Коэффициент стандартного отклонения (SDS роста) - 4,35. Масса тела 9,5 кг. Рост сидя 48,5 см, коэффициент “верхний ссгмент/пижпий сегмент" 1,48. Окружность головы 49 см.
Отмечаются ярко выраженные черты синдрома Гетчинсона - Гилфорда: 1) крупная голова с диспропорционально большим мозговым черепом и малым лицевым. Вдавлен) гость височных костей; 2) выраженная венозная сеть на голове; 3) тотальная алопеция; 4) узкий, деформированный нос с истончением кожи на нем, цианоз носогубного треугольника; 5) истончение кожи на туловище, конечностях, морщинистость кожи на ладонях и ступнях. Склеродермоподобные изменения кожи на животе, спине и ягодицах - очаги депигментации диаметром 0,5-0,7 см, множественные, уплотненные. Депигментация сосков; 6) подкожная жировая клетчатка развита слабо, распределена равномерно; 7) гипоплазия ногтей кистей и стоп; утолщение межфаланговых суставов и концевых фаланг кистей и стоп; варусная девиация верхней трети предплечий, короткая шея (см. рисунок).
Область сердца визуально не изменена. Тоны сердца ясные, ритмичные. Часота сердечных сокращений 104 удара в минуту, АД 80/50 мм рт.ст. Дыхание везикулярное, хрипов нет. Зев чистый. Живот мягкий, безболезненный. Печень не увеличена, селезенка не пальпируется. Стул регулярный. Дизурических расстройств нет. Симптом Пастернацкого отрицательный с обеих сторон. Эндокринный статус: щитовидная железа не увеличена, симптомов нарушения функции нет. Симптомы гипокортицизма отсутствуют. Половой статус препубертатный: Ах 0, Р 0, Ма 0, Me 0.
Гормональное исследование крови: кортизол 321,4 нмоль/л, общий трийодтиронин 2,48 нмоль/л, общий тироксин 100,0 нмоль/л, ТТГ 2,10 мкЕД/л, пролактин 248,0 мкЕД/л, ЛГ 1,60 ЕД/л, ФСГ 5,50 ЕД/о, 17-оксипрогестерон 1,5 нг/мл. Соматотропный гормон на фоне стимуляционной пробы с клофелином: 0 мин - 1,7 нг/мл, 30 мин - 1,5 нг/мл, 60 мин - 2,3 нг/мл, 120 мин - 71,0 нг/мл, 150 мин - 29,3 нг/мл.
Гормональное исследование мочи: свободный кортизол 1115,0 нмоль/л на 1 г креатинина (креатинин 0,24 г/л). Дс- гидроэпиандростерон-сульфат — следы.
Показатели гуморального аутоиммунитета: у больного ребенка не выявлено антител пи к тиреоглобулину человека, ни к микросомальному антигену тиреоцигов, ни к поверхностным антигенам клеток аденогипофиза и клеток коры надпочечников крысы.
Вместе с тем у матери ребенка обнаружены антитела к поверхностным антигенам клеток аденогипофиза крысы, а у бабушки по материнской линии - слабо положительная реакция па наличие антител к микросомальному антигену тиреоцигов при отсутствии антител к другим изучаемым антигенам.
Рентгенографическое исследование: на рентгенограмме черепа и кистей структура костей не изменена. Форма и размеры турецкого седла обычные. Сосудистый рисунок костей свода усилен. Дифференцирование скелета соответствует 12- 15 мес. Ногтевые фаланги деформированы, треугольной формы. На рентгенограмме стоп отмечается небольшое уплотнение стенок arteria dorsalis pedis.
Компьютерная томография головного мозга: на серии компьютерных томограмм изменения плотности мозговой ткани не выявлено. Желудочковая система не изменена. Хиазмальная цистерна расширена. Несколько расширена межполушарная щель. Данных, свидетельствующих об объемном процессе головного мозга, не выявлено.
ЭКГ: ЧСС 120 в минуту, ритм синусовый. Электроэнцефалограмма: на фоне умеренных диффузных изменений биоритмики с признаками диэнцефальной заинтересованности отмечаются признаки ирригации стволово-диэнцефальных структур с легким акцентом справа, в теменно-затылочной области. Эхоэнцефалограмма: эхо-пульсация неустойчиво усилена до 55%, смещения срединных структур нет, легкое расширение 111 желудочка (до 6 мм), вентрикулярный индекс умеренно выше нормы. Заключение: смещения срединных структур не выявлено. Расширение боковых желудочков.
Ультразвуковое исследование: щитовидная железа: типично расположена, контуры ровные, структура гомогенная. Правая доля 2,3x1,1хо,9 см, слева - 2,4x1,2x0,9 см, толщина перешейка 0,4 см. Объем щитовидной железы 2,55 мл. Надпочечники: нс увеличены. Органы малого таза: размеры матки и яичников соответствуют возрастной норме. Матка: 3,1x1,3x1,1 см, правый яичник: 1,6x1,2x0,8 см, левый яичник: 1,4x1,2x1,0 см. Печень: увеличена правая доля - 8,5 см, левая доля 2,6 см. Структура паренхимы гомогенная, внутри- печеночные протоки не расширены. Воротная вена не расширена. Желчный пузырь конкрементов не содержит, Почки: топография не изменена, размеры в пределах возрастной нормы. Структуры хорошо дифференцированы, без гидро- нефротических изменений и достоверных эхо-признаков дополнительных объемных включений. Паренхима гомогенна, толщина се соответствует возрастной норме.
Консультация окулиста: моторно-зрачковых нарушений не выявлено. Передний отрезок и среды без патологии. Глазное дно: диски бледно-розовые, сосуды умеренно расширены, полнокровны, извиты по всей протяженности сетчатки. Сетчатка на периферии разряжена, небольшие отеки. Заключение: повышение внутричерепного давления.
Консультация невропатолога: субкомпепсированпая гидроцефалия на фоне скудной церебральной симптоматики.
Консультация дерматолога: Alopecia totalis. Рекомендованы курсы лечения препаратами, улучшающими микроциркуляцию (трептал, троксевазип), поливитамины (Bj, В2, В(„ В15, А, Е); наружно — втирание с массажем головы геля актовегина, геля троксевазина, димексида, 01. Ricini.
Учитывая имеющиеся в литературе данные об эффективности применения гормона роста у детей с прогерией [4], для увеличения линейного роста девочке был назначен пробный 3-месячный курс лечения рекомбинантным гормоном роста человека “SAIZEN” (ARES-SERONO). Недельная доза составила 15 ЕД/м 2 , суточная доза — 1 ЕД, подкожно, ежедневно, 7 раз в неделю.
Лечение рекомендовано проводить под контролем гликемии и суточной глюкозурии.
Помимо гормона роста, девочке назначено лечение, рекомендованное дерматологом, и полноценное белковое питание.
Таким образом, представленный материал, основанный на данных анамнеза, жалоб, результатах клинического обследования и лабораторно-инструментальных исследований, подтверждает наличие у ребенка классического синдрома Гетчинсона-Гилфорда (прогерии).
ГОЛЬДШТЕЙНА СИНДРОМ
ГОЛЬДШТЕЙНА СИНДРОМ (описан немецким неврологом K. Goldstein) - мозжечковые расстройства воспалительного или травматического генеза: нарушения равновесия, адиадохокинез (невозможность быстро чередовать противоположные по направлению движения), гиперметрия (чрезмерность, несоразмерность движений), мегалография (изменение почерка в виде резкого увеличения размеров букв), брадилалия (замедленность речи вследствие затруднения артикуляции), интенционный тремор, каталепсия, хореиформные гиперкинезы и др.
K. Goldstein, F. Reichmann. Beitrage zur Kasuistik und Symptomatologie der kleinhirnerkrankungen (im besonderen zu den Storungen der Bewegungen, der Gewichts,- Raum- und Zeitschatzung). Archiv fUr Psychiatrie und Nervenkrankheiten, 1915-16, 56: 466-521.
Энциклопедический словарь по психологии и педагогике . 2013 .
Смотреть что такое "ГОЛЬДШТЕЙНА СИНДРОМ" в других словарях:
Синдром Гольдштейна-Райхманна мозжечковый — Расстройство статики и координации движений, адиадохокинез, гиперметрия, мегалография, снижение мышечного тонуса, интенционное дрожание, мозжечковая каталепсия, нарушение чувства массы тела. Симптомокомплекс мозжечковой патологии. Описал в 1916 г … Энциклопедический словарь по психологии и педагогике
Синдром мозжечковый Гольдштейна-Райхмана — Расстройства статики и координации движений, асинергии (см.), интенционное дрожание (см.), снижение мышечного тонуса, гиперметрия, мегалография (см.), нарушение чувства массы (веса) предмета, находящегося в руках, возможна мозжечковая каталепсия … Энциклопедический словарь по психологии и педагогике
Антисемитизм в СССР — Часть серии статей об антисемитизме … Википедия
описан немецким неврологом K. Goldstein) - мозжечковые расстройства воспалительного или травматического генеза: нарушения равновесия, адиадохокинез (невозможность быстро чередовать противоположные по направлению движения), гиперметрия (чрезмерность, несоразмерность движений), мегалография (изменение почерка в виде резкого увеличения размеров букв), брадилалия (замедленность речи вследствие затруднения артикуляции), интенционный тремор, каталепсия, хореиформные гиперкинезы и др.
K. Goldstein, F. Reichmann. Beitrage zur Kasuistik und Symptomatologie der kleinhirnerkrankungen (im besonderen zu den Storungen der Bewegungen, der Gewichts,- Raum- und Zeitschatzung). Archiv fUr Psychiatrie und Nervenkrankheiten, 1915-16, 56: 466-521.
Похожие термины:
Синдром мозжечковый Гольдштейна-Райхмана
Расстройства статики и координации движений, асинергии (см.), интенционное дрожание (см.), снижение мышечного тонуса, гиперметрия, мегалография (см.), нарушение чувства массы (веса) предмета, находящ
Синдром Гольдштейна-Райхманна мозжечковый
Расстройство статики и координации движений, адиадохокинез, гиперметрия, мегалография, снижение мышечного тонуса, интенционное дрожание, мозжечковая каталепсия, нарушение чувства массы тела. Си
Синдром Гетчинсона-Гилфорда (прогерия)
Синдром Гийена — Барре - симптомы и лечение
Что такое синдром Гийена — Барре? Причины возникновения, диагностику и методы лечения разберем в статье доктора Жуйкова Александра Вячеславовича, невролога со стажем в 21 год.
Над статьей доктора Жуйкова Александра Вячеславовича работали литературный редактор Елена Бережная , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Гийена — Барре (ГБС) — острое аутоиммунное заболевание, которое охватывает группу острых нарушений периферической нервной системы. Характеризуется мышечной слабостью, а также болью и ползанием мурашек в начале болезни из-за поражения чувствительных волокон. Каждый вариант нарушений характеризуется особенностями патофизиологии и клинического распределения слабости в конечностях и черепных нервах.
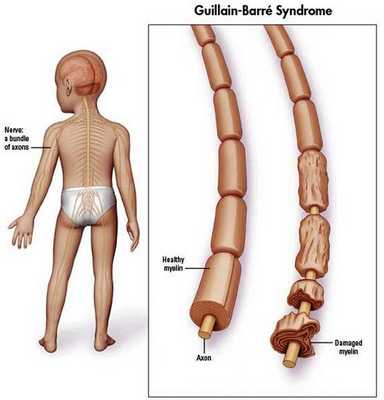
Распространённость синдрома Гийена — Барре
Синдром Гийена — Барре встречается в 1-2 случаях на 100 000 населения в год. [10]
Причины синдрома Гийена — Барре
Точная причина синдрома Гийена — Барре неизвестна. Но у 70% пациентов с ГБС наблюдались предшествующие инфекционные заболевания: респираторные, желудочно-кишечные инфекции, вирус Зика. Также синдром Гийена — Барре может развиться после заражения коронавирусом. [9]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гийена — Барре
Симптомы ОРВИ или расстройства желудочно-кишечного тракта отмечаются у 2/3 пациентов. Первыми симптомами ГБС являются парестезии пальцев конечностей, за которыми следует прогрессирующая слабость мышц нижних конечностей и нарушения походки. Болезнь прогрессирует в течение нескольких часов или дней, возникает слабость верхних конечностей и развиваются паралич черепных нервов. Параличи обычно симметричны и носят, конечно, периферический характер. У половины пациентов боль может быть первоначальной жалобой, что затрудняет диагноз. Атаксия и боль чаще встречаются у детей, чем у взрослых. Задержка мочи наблюдается у 10%-15% больных. Поражение вегетативных нервов проявляются головокружениями, гипертонией, чрезмерным потоотделением и тахикардией.
При объективном обследовании выявляется восходящая мышечная слабость, а также арефлексия. Сухожильные рефлексы нижних конечностей отсутствуют, но рефлексы верхней конечности могут вызываться. Мышечная слабость может задействовать и респираторные мышцы. Поражение черепных нервов отмечается в 35-50%, вегетативная нестабильность в 26%-50%, атаксия — в 23%, дизестезия — в 20% случаев. [1]
Наиболее распространенными признаками вегетативной дисфункции являются синусовая тахикардия или брадикардия и артериальная гипертония. У пациентов с тяжелой вегетативной дисфункцией наблюдаются изменения периферического вазомоторного тонуса с гипотензией и лабильностью артериального давления.
Нечастые варианты клинического течения болезни включают лихорадку в начале неврологических симптомов, тяжелую сенсорную недостаточность с болью (миалгии и артралгии, менингизм, корешковая боль), дисфункции сфинктеров.
Возможность ГБС должна рассматриваться у любого пациента с быстрым развитием острой нервно-мышечной слабости. На ранней стадии ГБС следует отличать от других заболеваний с прогрессирующей симметричной мышечной слабостью, включая поперечный миелит и миелопатию, острую токсическую или дифтеритическую полиневропатию, порфирию, миастению и нарушения электролитного обмена (например, гипокалиемия).
Патогенез синдрома Гийена — Барре
Нейрофизиологические процессы, лежащие в основе ГБС, подразделяются на несколько подтипов. Наиболее распространенные подтипы включают:
- острую воспалительную демиелинизирующую полирадикулопатию;
- острую двигательную аксональную невропатию;
- острую моторную и сенсорную аксональную нейропатию;
- синдром Миллера-Фишера, как вариант ГБС, характеризуется триадой признаков: офтальмоплегия, атаксия и арефлексия.
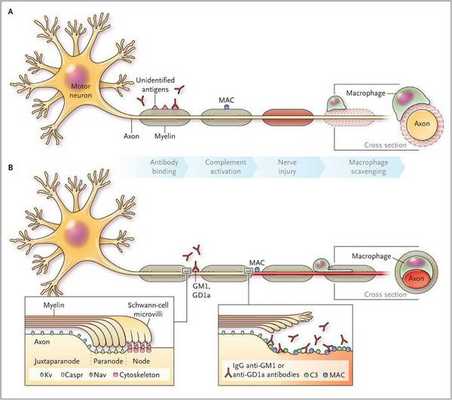
Считается, что ГБС развивается вследствие выработки антител против белка инфекционного агента, которые перекрестно реагируют с ганглиозидами нервных волокон человека. Аутоантитела связываются с миелиновыми антигенами и активируют комплемент, с формированием мембранно-атакующего комплекса на внешней поверхности клеток Шванна. Повреждение оболочек нервных стволов приводит к нарушениям проводимости и мышечной слабости (на поздней стадии может происходить и аксональная дегенерация). Демиелинизирующее поражение наблюдается по всей длине периферического нерва, включая нервные корешки.
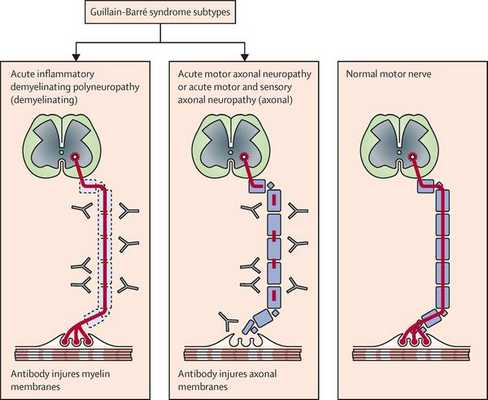
Поражаются все типы нервов, в том числе вегетативные, моторные и сенсорные волокна. Вовлечение двигательных нервов происходит значительно чаще, чем сенсорных.
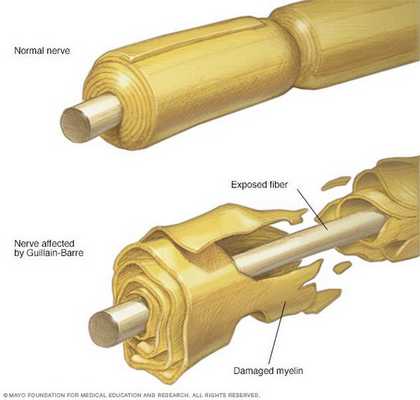
Классификация и стадии развития синдрома Гийена — Барре
В Международной классификации болезней (МКБ-10) синдром Гийена — Барре кодируется как G61.0.
Основные виды синдрома Гийена — Барре:
- Острая воспалительная демиелинизирующая полирадикулоневропатия (ОВДП) — самая распространённая форма в Северной Америке и Европе. Основным признаком ОВДП является мышечная слабость, которая сперва возникает в нижней части тела, а затем распространяется вверх.
- Синдром Миллера Фишера — проявляется параличом глаз и неустойчивостью походки. Эта форма более распространена в Азии.
- Острая моторная аксональная невропатия (ОМАН) и острая моторно-сенсорная аксональная невропатия (ОМСАН) — чаще встречаются в Китае, Японии и Мексике. [8]
Осложнения синдрома Гийена — Барре
Пациенты с ГБС подвержены риску опасных для жизни респираторных осложнений и вегетативных нарушений.
Показания для перевода в отделение интенсивной терапии включают:
- быстрое прогрессирование моторной слабости с поражением респираторных мышц;
- вентиляционную дыхательную недостаточность;
- пневмонию;
- бульбарные расстройства;
- тяжелую вегетативную недостаточность.
Осложнения проводимого лечения, требующие интенсивной терапии, включают перегрузку жидкостью, анафилаксию на введение внутривенного иммуноглобулина или гемодинамические нарушения при проведении плазмафереза.
У 15%-25% детей с ГБС развивается декомпенсированная дыхательная недостаточность, которая требует механической вентиляции легких. [2] Респираторные нарушения чаще встречается у детей с быстрым прогрессированием заболевания, слабостью верхних конечностей, вегетативной дисфункцией и поражениями черепных нервов. Интубация трахеи может потребоваться у больных для защиты дыхательных путей, проведения механической вентиляции легких. При ГБС быстрое прогрессирование, двусторонний паралич лицевого нерва и вегетативная дисфункция предопределяют повышенную вероятность интубации. Необходимо планирование ранней интубации для минимизации риска осложнений и необходимости проведения экстренной интубации.
Вегетативная дисфункция повышает риск эндотрахеальной интубации. С другой стороны, дисавтономия может увеличить риск гемодинамических реакций на препараты, используемые для индукции анестезии во время интубации.
Признаки, указывающие на необходимость механической вентиляции легких: [4]
- вентиляционная дыхательная недостаточность;
- увеличение потребности в кислороде для поддержания SpO2 выше 92%;
- признаки альвеолярной гиповентиляции (PCO2 выше 50 мм. рт. ст.);
- быстрое снижение жизненной емкости на 50% по сравнению с исходным уровнем;
- невозможность кашля
Вегетативная дисфункция является основным фактором смертности при ГБС. Фатальный сердечно-сосудистый коллапс из-за вегетативной дисфункции наблюдается у 2%-10% тяжелобольных пациентов. [3] Мониторинг частоты сердечных сокращений, артериального давления и электрокардиограммы следует продолжать до тех пор, пока пациенты нуждаются в респираторной поддержке. Чрескожная кардиостимуляция может потребоваться при выраженной брадикардии. Гипотония корректируется восполнением объема циркулирующей крови (ОЦК), и, если пациент невосприимчив к восполнению ОЦК, применяются α-агонисты, такие как норадреналин, мезатон, адреналин.
При нестабильной гемодинамике непрерывная регистрация артериального и центрального венозного давления должна проводиться для контроля объема инфузионной терапии.
Артериальная гипертензия может возникать, но это осложнение не требует специального лечения, если оно не осложняется отеком легких, энцефалопатией или субарахноидальным кровоизлиянием.
Диагностика синдрома Гийена — Барре
Сбор жалоб и анамнеза
На приёме врач в первую очередь обратит внимание на скорость распространения паралича и нарушение дыхания. Если эти признаки выражены, больному может потребоваться экстренная помощь.
Как правило, пациенты с синдромом Гийена — Барре жалуются на нарушение походки, онемение и зябкость ног, а затем и рук. Нередко пациенты рассказывают, что недавно перенесли ОРВИ.
Осмотр
Объективный неврологический осмотр — это основа диагностики при синдроме Гийена — Барре. Врач оценивает рефлексы, координацию движений, походку, чувствительность и мышечную силу.
Лабораторная диагностика
Основным видом лабораторной диагностики при синдроме Гийена — Барре является исследование спинномозговой жидкости, которую получают с помощью люмбальной пункции.
Инструментальная диагностика
ЭНМГ (Электронейромиография) — единственный инструментальный метод диагностики, позволяющий подтвердить диагноз ГБС и уточнить характер патологических изменений (демиелинизирующий или аксональный) и их распространенность. [3]
Игольчатая электромиография характеризуется наличием признаков текущего денервационно-реиннервационного процесса при полинейропатии. Исследуют дистальные мышцы верхних и нижних конечностей (например, переднюю большеберцовую мышцу, общий разгибатель пальцев), а при необходимости и проксимальные мышцы (например, четырёхглавую мышцу бедра).
ЭНМГ-исследование у больных с ГБС зависит от клинических проявлений:
- при дистальных парезах исследуются длинные нервы на руках и ногах: не менее четырех двигательных и четырех чувствительных (двигательные и чувствительные порции срединного и локтевого нервов; малоберцовый, большеберцовый, поверхностный малоберцовый и икроножный нервы с одной стороны).
Оценка основных ЭНМГ- параметров:
Первые признаки денервационного процесса появляются через две-три недели после начала заболевания, признаки реиннервационного процесса — через месяц.
Дифференциальная диагностика
Синдром Гийена — Барре следует отличать от следующих заболеваний:
- и клещевого энцефалита (чувствительность не нарушена, поражены преимущественно черепные нервы); (отягощённый эпидемиологический анамнез, например посещение эндемичных стран); (чувствительность не нарушена, рефлексы снижены незначительно);
- обменно-метаболических полиневропатий (течение хроническое).
Лечение синдрома Гийена — Барре
Показания для госпитализации
Практически во всех случаях требуется госпитализация. Экстренная госпитализация необходима пациентам с нарушениями дыхания — в таких случаях лечение проводят в условиях реанимации.
Общие принципы лечения синдрома Гийена — Барре
Лечение острой демиелинизирующей полирадикулоневропатии комплексное. Основа — плазмаферез, иммуноглобулины и кортикостероиды. В ряде случаев требуется искусственная вентиляция лёгких, коррекция нарушений кровообращения, профилактика инфекционных и тромбоэмболических осложнений. Обязательным условием успешного лечения является уход.
Общее поддерживающее лечение и уход
Пациенты, требующие интенсивной терапии, требуют тщательного общего ухода. Запор наблюдается более чем в 50% случаев пациентов с ГБС в результате динамической непроходимости кишечника. Может потребоваться искусственное питание.
Медикаментозное лечение и плазмаферез
В лечении ГБС предпринимаются различные виды иммуномодулирующей терапии. [1] [2]
Внутривенный иммуноглобулин назначают в виде ежедневной инфузии (в дозе 0,4 гр/кг/день) в течение 5 дней в первые 2 недели болезни. Второй курс иммуноглобулина может потребоваться 5%-10% пациентов, при отрицательной динамике после первоначального улучшения. Механизм действия внутривенного иммуноглобулина, вероятно, многофакторный и, как полагают, включает модуляцию активации комплемента, нейтрализацию идиотипических антител, подавление воспалительных медиаторов (цитокины, хемокины).
Побочные эффекты иммуноглобулина включают головную боль, миалгию и артралгию, гриппоподобные симптомы, лихорадку. У пациентов с дефицитом IgA может развиться анафилаксия после первого курса внутривенного иммуноглобулина.
Плазмаферез способствует удалению антител, вовлеченных в патогенез ГБС. В течение каждого сеанса 40-50 мл/кг плазмы заменяют смесью 0,9% раствора хлорида натрия и альбумина. Проведение плазмафереза приводит к сокращению времени выздоровления и снижению потребности в искусственной вентиляции. Эти преимущества очевидны, если плазмаферез проводится в течение первых двух недель после начала болезни. Осложнения, связанные с плазмаферезом, включают гематому в области венопункции, пневмоторакс после катетеризации подключичной вены и сепсис. Плазмаферез противопоказан пациентам с тяжелой гемодинамической нестабильностью, кровотечением и сепсисом. Комбинация плазмафереза и иммуноглобулина не показала клинических преимуществ.
Симптоматическое лечение синдрома Гийена — Барре:
- при боли применяют парацетамол;
- катадолон и трамадол применяют при выраженном болевом синдроме;
- при нейропатической боли эффективны карбамазепин и габапентин.
Оперативное лечение
При тяжёлом течении может потребоваться длительная респираторная поддержка и наложение трахеостомы. Если пациент находится на искусственном питании, то накладывают гастростому.
Прогноз. Профилактика
ГБС остается серьезным заболеванием, несмотря на улучшение результатов лечения. По сравнению со взрослыми, у детей чаще отмечается более благоприятное течение заболевания, с полным, а не частичным выздоровлением. Причинами неблагоприятного исхода при ГБС являются дыхательная недостаточность, осложнения искусственной вентиляции легких (пневмония, сепсис, острый респираторный дистресс-синдром и тромбоэмболические осложнения), остановка сердца, вторичная по отношению к дисавтономии.
Восстановление обычно начинается через две-четыре недели после прекращения прогрессирования симптомов. Среднее время от начала заболевания до полного выздоровления составляет 60 дней. Данные относительно долгосрочного исхода ГБС ограничены. 75% - 80% пациентов полностью выздоравливают. Около 20% пациентов не могут ходить через полгода.
Младшая возрастная группа (менее 9 лет), быстрое прогрессирование и максимальная мышечная слабость, потребность в искусственной вентиляции легких являются важными предикторами длительного двигательного дефицита. [4]
Читайте также: