Синдром Граухана (Grauhan) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Сотрудники Курского государственного медицинского университета, Научно-исследовательского клинического института педиатрии им. академика Ю.Е. Вельтищева и РНИМУ им. Н.И. Пирогова описали клинический случай синдрома Хаммана-Рича у ребенка 8 мес, основные клинические проявления и особенности диагностики синдрома.
Синдром Хаммена-Рича (идиопатический фиброзирующий альвеолит) характеризуется патологическим процессом в альвеолах и интерстициальной ткани легких неясной природы, что приводит к прогрессирующему фиброзу и сопровождается нарастающей дыхательной недостаточностью. Первые упоминания о болезни относятся к 1935 г., когда L. Hamman и А. Rich описали ряд больных с быстропрогрессирующим легочным фиброзом и дыхательной недостаточностью, приведшим в течение нескольких месяцев к летальному исходу.
В 1999, 2002 и 2013 гг. были приняты совместные соглашения Американского торакального общества и Европейского респираторного общества по интерстициальным заболеваниям легких. В этих соглашениях были выделены и дифференцированы различные формы идиопатических интерстициальных пневмоний преимущественно по морфологическим признакам. Собственно идиопатический фиброзирующий альвеолит в настоящее время рассматривается как заболевание с морфологической картиной интерстициальной пневмонии. Другие формы интерстициальных пневмоний, в их числе острая интерстициальная пневмония (синдром Хаммана-Рича), неспецифическая интерстициальная пневмония, десквамативная интерстициальная пневмония, интерстициальная болезнь с респираторным бронхиолитом и другие типы идиопатических интерстициальных пневмоний являются самостоятельными заболеваниями.
Этиология синдрома Хаммана-Рича до конца не известна. Существовавшие многие годы теории вирусного, полиэтиологического происхождения болезни не получили подтверждения с позиции доказательной медицины. Повышенная частота полиморфизмов генов, кодирующих ряд цитокинов, профибротических молекул, матриксных металлопротеиназ, которая была показана при первоначальном изучении генетического компонента идиопатического легочного фиброза, в дальнейшем также не подтвердилась.
Гены, дефекты которых обнаружены у пациентов с идиопатическим фиброзирующим альвеолитом, включают гены, связанные с теломеразами (TERT и TERC), сурфактантными белками C ( SPC) и A2 (SPA2) и белком ELMOD2. Мутации генов TERT или TERC были выявлены примерно в 18% случаев семейного легочного фиброза (среди кровных родственников) и реже - в спорадических случаях. Эти мутации приводят к укороченной длине теломеров как у пораженных индивидуумов, так и у бессимптомных носителей, а у некоторых родственников также были продемонстрированы укороченные теломеры, что может свидетельствовать об эпигенетической природе этого феномена. Кроме того, короткие теломеры в отсутствие мутаций TERT/TERC были выявлены в 25% спорадических и 37% семейных случаев идиопатического фиброзирующего альвеолита, что указывает на другие механизмы укорочения теломеров.
Общий однонуклеотидный полиморфизм в области промотора гена MUC5B (муцин 5В) был обнаружен в 38% случаев идиопатического легочного фиброза. Следует отметить, что хотя полиморфизм промотора MUC5B, по-видимому, создает повышенный риск развития идиопатического легочного фиброза, он может быть связан с улучшением выживаемости при этом заболевании. Недавно проведенные два крупных исследования по изучению генных ассоциаций подтвердили связь синдрома Хаммана-Рича с нуклеотидными вариантами TERT, TERC и MUC5B, а также другими новыми локусами (например, TOLLIP), некоторые из которых, как полагают, участвуют в клеточно-клеточной адгезии и репарации ДНК.
С учетом патогенеза предполагается, что в интерстициальной ткани легких снижается распад коллагена и повышается его синтез фибробластами и альвеолярными макрофагами. По данным A. Ferreira и соавт., заболевание относится к аутоиммунным. Комплексы антиген-антитело откладываются в стенках мелких сосудов легких. Под влиянием циркулирующих иммунных комплексов, лизосомальных ферментов альвеолярных макрофагов и нейтрофилов происходят повреждение легочной ткани, уплотнение, утолщение межальвеолярных перегородок, облитерация альвеол и капилляров фиброзной тканью. Патоморфологические изменения в легких при этом заболевании происходят в виде трех взаимосвязанных процессов: интерстициальный отек, интерстициальное воспаление (альвеолит) и интерстициальный фиброз.
В клинической картине заболевания определяющая роль принадлежит дыхательной недостаточности. Одышка - главный симптом практически у всех больных идиопатическим легочным фиброзом, наблюдается у большинства пациентов, особенно у детей младшего возраста и служит наиболее ранним признаком заболевания. Дыхательная недостаточность вначале возникает или усиливается при физической нагрузке, но неуклонно прогрессирует. У больных, как правило, отмечается кашель - непродуктивный или со скудной слизистой мокротой. Цианоз - менее постоянный и более поздний признак болезни, возникает или усиливается при физической нагрузке, у маленьких детей при кормлении.
В процессе болезни отмечается значительное похудание детей, отставание в росте. Частый и прогностически неблагоприятный признак - утолщение концевых фаланг пальцев по типу барабанных палочек, ногтей в форме часовых стекол («пальцы Гиппократа»). С большим постоянством наблюдается деформация грудной клетки.
Обнаруживаемые при физикальном обследовании изменения в легких достаточно специфичны. У больных на вдохе прослушиваются нежные крепитирующие хрипы («треск целлофана»). Они могут быть непостоянными по своей выраженности и локализации. Несоответствие выраженной одышки относительно небольшим физикальным изменениям в легких служит одним из важнейших дифференциально-диагностических признаков, позволяющих клинически отличить синдром Хаммана-Рича от других хронических заболеваний бронхолегочной системы. На поздних стадиях заболевания, как правило, отмечаются нарастание одышки, формирование легочно-сердечной недостаточности за счет гемодинамических нарушений в малом круге кровообращения.
Важнейшими в диагностике являются рентгенологические методы обследования грудной клетки, особенно компьютерная томография высокого разрешения. На ранних стадиях болезни определяются преимущественно усиление и деформация легочного рисунка, понижение прозрачности легочных полей по типу «матового стекла», мелкоочаговые тени. По мере прогрессирования процесса деформация легочного рисунка становится более выраженной, выявляются признаки интерстициального фиброза, полостные образования, формируется картина «сотового легкого». Наиболее точная диагностика возможна при помощи оценки биопсийного материала. Биопсию легких долгое время считали «золотым стандартом» в диагностике идиопатического легочного фиброза, позволяющим не только установить диагноз, но и определить прогноз заболевания. Однако в последнее время эти позиции пересматриваются.
В качестве иллюстрации приводим историю болезни Сергея Е., 8 мес, находившегося на лечении в отделении реанимации Областной детской клинической больницы г. Курска. Ребенок от второй беременности, протекавшей на фоне тяжелой преэклампсии, от первых экстренных оперативных родов на сроке 36 нед, родился с массой 1520 г, длиной 43 см, оценка по шкале Апгар 1/4/7 баллов. Находился в отделении реанимации и интенсивной терапии, а в последующем - в отделении патологии недоношенных областного перинатального цента Курска в течение 1 мес; проводились респираторная поддержка искусственной вентиляций легких, антибактериальная, симптоматическая терапия. После выписки мальчик амбулаторно наблюдался педиатром по месту жительства, неоднократно находился на стационарном лечении с диагнозом рецидивирующий бронхит.
В возрасте 7 мес состояние ребенка ухудшилось - на фоне респираторной инфекции появился сухой частый кашель, в легких выслушивались сухие и влажные хрипы, в течение нескольких дней сохранялось повышение температуры тела до фебрильной. Отмечались вялость, общая слабость, снижение аппетита. Мальчик был госпитализирован в инфекционную больницу им. Н.А. Семашко с диагнозом: внебольничная правосторонняя среднедолевая пневмония. За время пребывания в состоянии ребенка на фоне терапии отмечалась отрицательная динамика - прогрессировали проявления дыхательной недостаточности, выраженного интоксикационного синдрома, и мальчик был переведен в областную детскую клиническую больницу, в отделение реанимации и интенсивной терапии.
Общее состояние ребенка при поступлении крайне тяжелое за счет проявлений тяжелой дыхательной и сердечной недостаточности с перегрузкой малого круга кровообращения. При осмотре физическое развитие дисгармоничное за счет дефицита массы тела 2-й степени. Масса 5840 г, рост 67 см (SDS массы -3,52; SDS роста -1,22). Кожные покровы и видимые слизистые оболочки бледные с мраморным рисунком, периоральный и периорбитальный цианоз, акроцианоз. Большой родничок 4,0×4,0 см, напряжен. Зев рыхлый, умеренная гиперемия дужек. Носовое дыхание затруднено, скудное слизистое отделяемое. Одышка в покое, усиливающаяся при физическом и психоэмоциональном напряжении. При кормлении быстро устает, вскармливается через зонд, объем питания - удерживает по 150 мл смеси через 3,5 ч, не срыгивает. Грудная клетка цилиндрическая. Дыхание с выраженным участием вспомогательной мускулатуры: втяжение межреберий, западение яремной ямки.
Перкуторно над легкими легочный звук с коробочным оттенком, местами с участками притупления. При аускультации дыхание жесткое, в нижнебоковых отделах ослаблено, выслушиваются крепитирующие хрипы с обеих сторон по всем полям. Ребенок получал респираторную поддержку, увлажненный кислород через носовые канюли со скоростью 3-3,5 л/мин. При этом сатурация крови кислородом (SpO2) 80-88%. При прекращении респираторной поддержки SpO2 снижался до 68-70%. Тоны сердца приглушены, ритм правильный, частота сердечных сокращений 138-180 в минуту, артериальное давление 101/66 мм рт.ст. Язык влажный. Живот мягкий, не вздут, безболезненный. Печень +2,5 см от края реберной дуги, селезенка не пальпируется. Стул был самостоятельно 2 раза, желтого цвета, водянистый, с примесью небольшого количества слизи, Мочеиспускание не нарушено. Диурез 2,4 мл/кг/ч.
При лабораторном обследовании в крови отмечались увеличение СОЭ до 63 мм/ч, лейкоцитоз до 13,5·109/л, проявления выраженного метаболического ацидоза, повышение активности трансаминаз, лактатдегидрогеназы, С-реактивного белка. Прокальцитониновый тест ≥2. При бактериологическом исследовании мокроты выделена Candida albicans, в отделяемом верхних дыхательных путей определялись Serrata rubidea, Stenotrophomonas maltophilia.
На рентгенограмме органов грудной клетки в прямой проекции отмечались инфильтративные изменения в средней доле правого легкого, субтотальное снижение пневматизации левого легкого. В дальнейшем отмечалась отрицательная динамика в виде нарастания отечно-инфильтративных изменений в легочной ткани с обеих сторон. На фоне тотального затемнения определялась воздушная бронхограмма. Тень средостения не дифференцировалась. Куполы диафрагмы не прослеживались.
По данным электрокардиографии определялись признаки увеличения правого желудочка; нарушение реполяризации. При ультразвуковом исследовании выявлены агенезия левой почки, компенсаторная гипертрофия правой почки, умеренная гепатомегалия, относительная недостаточность трикуспидального клапана. На компьютерной томограмме грудной клетки отмечены участки интерстициальных изменений, выраженные диффузные фиброзные изменения легочной ткани.
Ребенок был заочно проконсультирован в отделе хронических воспалительных и аллергических болезней легких НИКИ педиатрии им. акад. Ю.Е. Вельтищева. Был установлен клинический диагноз: идиопатический легочный фиброз (синдром Хаммана-Рича).
В отделении ребенку проводилась комплексная антибактериальная и противогрибковая терапии; кортикостероиды - дексаметазон 3 мг/кг/сут и суспензия будесонида 1000 мкг/сут в ингаляциях через небулайзер в течение месяца, симптоматическая терапия. Однако состояние мальчика прогрессивно ухудшалось: повысилась температура тела до фебрильной, усилилась одышка до 70-80 в 1 мин, SpO2 снизилась до 62-67%. Ребенок был переведен на искусственную вентиляцию легких с содержанием кислорода во вдыхаемой смеси FiO2 90-100%, при этом SpO2 оставалась 60-70%. На фоне интенсивной терапии сохранялась выраженная гипертермия, усиливались проявления ацидоза, полиорганной недостаточности, наступила остановка сердечной деятельности и дыхания, констатирована клиническая смерть. Патологоанатомическое исследование полностью подтвердило клинический диагноз синдрома Хаммана-Рича (Областное патологоанатомическое бюро, г. Курск).
Данное редкое клиническое наблюдение подтверждает, что идиопатический легочный фиброз в раннем возрасте характеризуется быстро прогрессирующим течением, и прогноз остается неблагоприятным даже при своевременной верификации диагноза.
Миненкова Т.А., Мизерницкий Ю.Л., Разинькова Н.С., Сережкина А.В., Костюченко М.В.
Граухана синдром
Синдром Граухана (М. Grauhan, 1886-1945, нем. хирург) - сочетание аномалий развития: незаращение верхней губы и неба, полидактилия (чаще шестипалость) и незаращение органов мочеполовой системы.
Википедия
Статьи для врачей
- 12.02.2105 Восприятие марки настроено позитивно. Диктат потребителя
- 12.02.2105 Восприятие марки настроено позитивно. Диктат потребителя
- 12.02.2105 Восприятие марки настроено позитивно. Диктат потребителя
Статьи для пациентов
SPRINT: О целевом уровне артериального давления у пожилых
Антидепрессанты. Влияние на сон и когнитивные функции у пожилых
IDSA/ ATS. Рекомендации по лечению внутрибольничных пневмоний
Головная боль у взрослых. Виды головных болей.
Головная боль. Вопросы и ответы
Мигрень
Болезнь Альцгеймера и антидепрессанты
Артериальная гипертензия. Какое из имеющихся руководств лучше
Насыщенные жиры и риск сердечно-сосудистых заболеваний. Первые шаги революции?
Терпены
Терпены - группа природных ненасыщенных углеводородов, содержащихся гл. обр. в эфирных маслах (камфора, ментол и др.); применяются, напр., при изготовлении лекарственных средств и инсектицидов.
Тандлера схема
Тандлера схема (J. Tandler, 1869-1936, австрийский анатом) - схема расчета проекции долей и извилин большого мозга на кости черепа по наружным костным ориентирам.
Сустав локтевой
Сустав локтевой (a. cubiti, PNA, BNA, JNA) - сложный сустав, объединяющий плечелоктевой, плечелучевой и проксимальный лучелоктевой сустав, заключенные в одну суставную капсулу; в суставе локтевом возм.
Синдром Гийена — Барре - симптомы и лечение
Что такое синдром Гийена — Барре? Причины возникновения, диагностику и методы лечения разберем в статье доктора Жуйкова Александра Вячеславовича, невролога со стажем в 21 год.
Над статьей доктора Жуйкова Александра Вячеславовича работали литературный редактор Елена Бережная , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Гийена — Барре (ГБС) — острое аутоиммунное заболевание, которое охватывает группу острых нарушений периферической нервной системы. Характеризуется мышечной слабостью, а также болью и ползанием мурашек в начале болезни из-за поражения чувствительных волокон. Каждый вариант нарушений характеризуется особенностями патофизиологии и клинического распределения слабости в конечностях и черепных нервах.
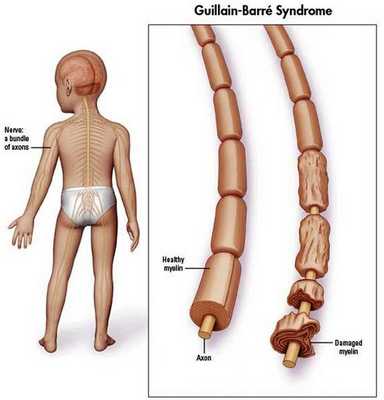
Распространённость синдрома Гийена — Барре
Синдром Гийена — Барре встречается в 1-2 случаях на 100 000 населения в год. [10]
Причины синдрома Гийена — Барре
Точная причина синдрома Гийена — Барре неизвестна. Но у 70% пациентов с ГБС наблюдались предшествующие инфекционные заболевания: респираторные, желудочно-кишечные инфекции, вирус Зика. Также синдром Гийена — Барре может развиться после заражения коронавирусом. [9]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гийена — Барре
Симптомы ОРВИ или расстройства желудочно-кишечного тракта отмечаются у 2/3 пациентов. Первыми симптомами ГБС являются парестезии пальцев конечностей, за которыми следует прогрессирующая слабость мышц нижних конечностей и нарушения походки. Болезнь прогрессирует в течение нескольких часов или дней, возникает слабость верхних конечностей и развиваются паралич черепных нервов. Параличи обычно симметричны и носят, конечно, периферический характер. У половины пациентов боль может быть первоначальной жалобой, что затрудняет диагноз. Атаксия и боль чаще встречаются у детей, чем у взрослых. Задержка мочи наблюдается у 10%-15% больных. Поражение вегетативных нервов проявляются головокружениями, гипертонией, чрезмерным потоотделением и тахикардией.
При объективном обследовании выявляется восходящая мышечная слабость, а также арефлексия. Сухожильные рефлексы нижних конечностей отсутствуют, но рефлексы верхней конечности могут вызываться. Мышечная слабость может задействовать и респираторные мышцы. Поражение черепных нервов отмечается в 35-50%, вегетативная нестабильность в 26%-50%, атаксия — в 23%, дизестезия — в 20% случаев. [1]
Наиболее распространенными признаками вегетативной дисфункции являются синусовая тахикардия или брадикардия и артериальная гипертония. У пациентов с тяжелой вегетативной дисфункцией наблюдаются изменения периферического вазомоторного тонуса с гипотензией и лабильностью артериального давления.
Нечастые варианты клинического течения болезни включают лихорадку в начале неврологических симптомов, тяжелую сенсорную недостаточность с болью (миалгии и артралгии, менингизм, корешковая боль), дисфункции сфинктеров.
Возможность ГБС должна рассматриваться у любого пациента с быстрым развитием острой нервно-мышечной слабости. На ранней стадии ГБС следует отличать от других заболеваний с прогрессирующей симметричной мышечной слабостью, включая поперечный миелит и миелопатию, острую токсическую или дифтеритическую полиневропатию, порфирию, миастению и нарушения электролитного обмена (например, гипокалиемия).
Патогенез синдрома Гийена — Барре
Нейрофизиологические процессы, лежащие в основе ГБС, подразделяются на несколько подтипов. Наиболее распространенные подтипы включают:
- острую воспалительную демиелинизирующую полирадикулопатию;
- острую двигательную аксональную невропатию;
- острую моторную и сенсорную аксональную нейропатию;
- синдром Миллера-Фишера, как вариант ГБС, характеризуется триадой признаков: офтальмоплегия, атаксия и арефлексия.
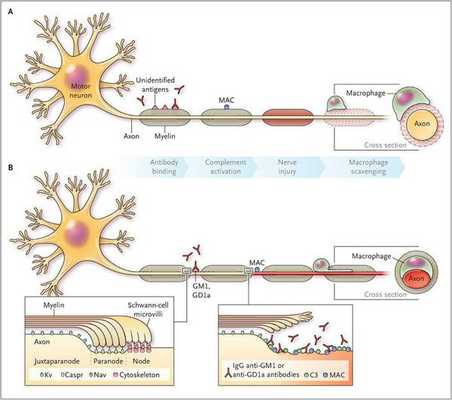
Считается, что ГБС развивается вследствие выработки антител против белка инфекционного агента, которые перекрестно реагируют с ганглиозидами нервных волокон человека. Аутоантитела связываются с миелиновыми антигенами и активируют комплемент, с формированием мембранно-атакующего комплекса на внешней поверхности клеток Шванна. Повреждение оболочек нервных стволов приводит к нарушениям проводимости и мышечной слабости (на поздней стадии может происходить и аксональная дегенерация). Демиелинизирующее поражение наблюдается по всей длине периферического нерва, включая нервные корешки.
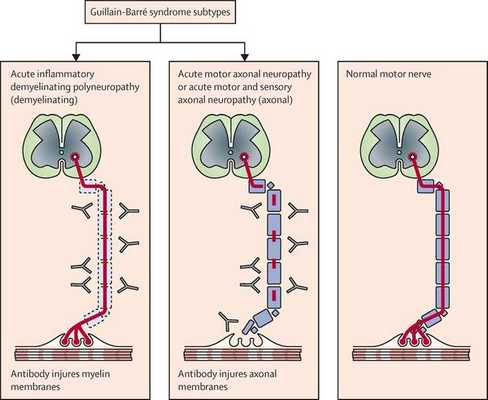
Поражаются все типы нервов, в том числе вегетативные, моторные и сенсорные волокна. Вовлечение двигательных нервов происходит значительно чаще, чем сенсорных.
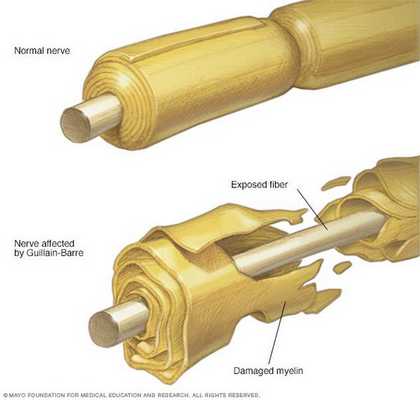
Классификация и стадии развития синдрома Гийена — Барре
В Международной классификации болезней (МКБ-10) синдром Гийена — Барре кодируется как G61.0.
Основные виды синдрома Гийена — Барре:
- Острая воспалительная демиелинизирующая полирадикулоневропатия (ОВДП) — самая распространённая форма в Северной Америке и Европе. Основным признаком ОВДП является мышечная слабость, которая сперва возникает в нижней части тела, а затем распространяется вверх.
- Синдром Миллера Фишера — проявляется параличом глаз и неустойчивостью походки. Эта форма более распространена в Азии.
- Острая моторная аксональная невропатия (ОМАН) и острая моторно-сенсорная аксональная невропатия (ОМСАН) — чаще встречаются в Китае, Японии и Мексике. [8]
Осложнения синдрома Гийена — Барре
Пациенты с ГБС подвержены риску опасных для жизни респираторных осложнений и вегетативных нарушений.
Показания для перевода в отделение интенсивной терапии включают:
- быстрое прогрессирование моторной слабости с поражением респираторных мышц;
- вентиляционную дыхательную недостаточность;
- пневмонию;
- бульбарные расстройства;
- тяжелую вегетативную недостаточность.
Осложнения проводимого лечения, требующие интенсивной терапии, включают перегрузку жидкостью, анафилаксию на введение внутривенного иммуноглобулина или гемодинамические нарушения при проведении плазмафереза.
У 15%-25% детей с ГБС развивается декомпенсированная дыхательная недостаточность, которая требует механической вентиляции легких. [2] Респираторные нарушения чаще встречается у детей с быстрым прогрессированием заболевания, слабостью верхних конечностей, вегетативной дисфункцией и поражениями черепных нервов. Интубация трахеи может потребоваться у больных для защиты дыхательных путей, проведения механической вентиляции легких. При ГБС быстрое прогрессирование, двусторонний паралич лицевого нерва и вегетативная дисфункция предопределяют повышенную вероятность интубации. Необходимо планирование ранней интубации для минимизации риска осложнений и необходимости проведения экстренной интубации.
Вегетативная дисфункция повышает риск эндотрахеальной интубации. С другой стороны, дисавтономия может увеличить риск гемодинамических реакций на препараты, используемые для индукции анестезии во время интубации.
Признаки, указывающие на необходимость механической вентиляции легких: [4]
- вентиляционная дыхательная недостаточность;
- увеличение потребности в кислороде для поддержания SpO2 выше 92%;
- признаки альвеолярной гиповентиляции (PCO2 выше 50 мм. рт. ст.);
- быстрое снижение жизненной емкости на 50% по сравнению с исходным уровнем;
- невозможность кашля
Вегетативная дисфункция является основным фактором смертности при ГБС. Фатальный сердечно-сосудистый коллапс из-за вегетативной дисфункции наблюдается у 2%-10% тяжелобольных пациентов. [3] Мониторинг частоты сердечных сокращений, артериального давления и электрокардиограммы следует продолжать до тех пор, пока пациенты нуждаются в респираторной поддержке. Чрескожная кардиостимуляция может потребоваться при выраженной брадикардии. Гипотония корректируется восполнением объема циркулирующей крови (ОЦК), и, если пациент невосприимчив к восполнению ОЦК, применяются α-агонисты, такие как норадреналин, мезатон, адреналин.
При нестабильной гемодинамике непрерывная регистрация артериального и центрального венозного давления должна проводиться для контроля объема инфузионной терапии.
Артериальная гипертензия может возникать, но это осложнение не требует специального лечения, если оно не осложняется отеком легких, энцефалопатией или субарахноидальным кровоизлиянием.
Диагностика синдрома Гийена — Барре
Сбор жалоб и анамнеза
На приёме врач в первую очередь обратит внимание на скорость распространения паралича и нарушение дыхания. Если эти признаки выражены, больному может потребоваться экстренная помощь.
Как правило, пациенты с синдромом Гийена — Барре жалуются на нарушение походки, онемение и зябкость ног, а затем и рук. Нередко пациенты рассказывают, что недавно перенесли ОРВИ.
Осмотр
Объективный неврологический осмотр — это основа диагностики при синдроме Гийена — Барре. Врач оценивает рефлексы, координацию движений, походку, чувствительность и мышечную силу.
Лабораторная диагностика
Основным видом лабораторной диагностики при синдроме Гийена — Барре является исследование спинномозговой жидкости, которую получают с помощью люмбальной пункции.
Инструментальная диагностика
ЭНМГ (Электронейромиография) — единственный инструментальный метод диагностики, позволяющий подтвердить диагноз ГБС и уточнить характер патологических изменений (демиелинизирующий или аксональный) и их распространенность. [3]
Игольчатая электромиография характеризуется наличием признаков текущего денервационно-реиннервационного процесса при полинейропатии. Исследуют дистальные мышцы верхних и нижних конечностей (например, переднюю большеберцовую мышцу, общий разгибатель пальцев), а при необходимости и проксимальные мышцы (например, четырёхглавую мышцу бедра).
ЭНМГ-исследование у больных с ГБС зависит от клинических проявлений:
- при дистальных парезах исследуются длинные нервы на руках и ногах: не менее четырех двигательных и четырех чувствительных (двигательные и чувствительные порции срединного и локтевого нервов; малоберцовый, большеберцовый, поверхностный малоберцовый и икроножный нервы с одной стороны).
Оценка основных ЭНМГ- параметров:
Первые признаки денервационного процесса появляются через две-три недели после начала заболевания, признаки реиннервационного процесса — через месяц.
Дифференциальная диагностика
Синдром Гийена — Барре следует отличать от следующих заболеваний:
- и клещевого энцефалита (чувствительность не нарушена, поражены преимущественно черепные нервы); (отягощённый эпидемиологический анамнез, например посещение эндемичных стран); (чувствительность не нарушена, рефлексы снижены незначительно);
- обменно-метаболических полиневропатий (течение хроническое).
Лечение синдрома Гийена — Барре
Показания для госпитализации
Практически во всех случаях требуется госпитализация. Экстренная госпитализация необходима пациентам с нарушениями дыхания — в таких случаях лечение проводят в условиях реанимации.
Общие принципы лечения синдрома Гийена — Барре
Лечение острой демиелинизирующей полирадикулоневропатии комплексное. Основа — плазмаферез, иммуноглобулины и кортикостероиды. В ряде случаев требуется искусственная вентиляция лёгких, коррекция нарушений кровообращения, профилактика инфекционных и тромбоэмболических осложнений. Обязательным условием успешного лечения является уход.
Общее поддерживающее лечение и уход
Пациенты, требующие интенсивной терапии, требуют тщательного общего ухода. Запор наблюдается более чем в 50% случаев пациентов с ГБС в результате динамической непроходимости кишечника. Может потребоваться искусственное питание.
Медикаментозное лечение и плазмаферез
В лечении ГБС предпринимаются различные виды иммуномодулирующей терапии. [1] [2]
Внутривенный иммуноглобулин назначают в виде ежедневной инфузии (в дозе 0,4 гр/кг/день) в течение 5 дней в первые 2 недели болезни. Второй курс иммуноглобулина может потребоваться 5%-10% пациентов, при отрицательной динамике после первоначального улучшения. Механизм действия внутривенного иммуноглобулина, вероятно, многофакторный и, как полагают, включает модуляцию активации комплемента, нейтрализацию идиотипических антител, подавление воспалительных медиаторов (цитокины, хемокины).
Побочные эффекты иммуноглобулина включают головную боль, миалгию и артралгию, гриппоподобные симптомы, лихорадку. У пациентов с дефицитом IgA может развиться анафилаксия после первого курса внутривенного иммуноглобулина.
Плазмаферез способствует удалению антител, вовлеченных в патогенез ГБС. В течение каждого сеанса 40-50 мл/кг плазмы заменяют смесью 0,9% раствора хлорида натрия и альбумина. Проведение плазмафереза приводит к сокращению времени выздоровления и снижению потребности в искусственной вентиляции. Эти преимущества очевидны, если плазмаферез проводится в течение первых двух недель после начала болезни. Осложнения, связанные с плазмаферезом, включают гематому в области венопункции, пневмоторакс после катетеризации подключичной вены и сепсис. Плазмаферез противопоказан пациентам с тяжелой гемодинамической нестабильностью, кровотечением и сепсисом. Комбинация плазмафереза и иммуноглобулина не показала клинических преимуществ.
Симптоматическое лечение синдрома Гийена — Барре:
- при боли применяют парацетамол;
- катадолон и трамадол применяют при выраженном болевом синдроме;
- при нейропатической боли эффективны карбамазепин и габапентин.
Оперативное лечение
При тяжёлом течении может потребоваться длительная респираторная поддержка и наложение трахеостомы. Если пациент находится на искусственном питании, то накладывают гастростому.
Прогноз. Профилактика
ГБС остается серьезным заболеванием, несмотря на улучшение результатов лечения. По сравнению со взрослыми, у детей чаще отмечается более благоприятное течение заболевания, с полным, а не частичным выздоровлением. Причинами неблагоприятного исхода при ГБС являются дыхательная недостаточность, осложнения искусственной вентиляции легких (пневмония, сепсис, острый респираторный дистресс-синдром и тромбоэмболические осложнения), остановка сердца, вторичная по отношению к дисавтономии.
Восстановление обычно начинается через две-четыре недели после прекращения прогрессирования симптомов. Среднее время от начала заболевания до полного выздоровления составляет 60 дней. Данные относительно долгосрочного исхода ГБС ограничены. 75% - 80% пациентов полностью выздоравливают. Около 20% пациентов не могут ходить через полгода.
Младшая возрастная группа (менее 9 лет), быстрое прогрессирование и максимальная мышечная слабость, потребность в искусственной вентиляции легких являются важными предикторами длительного двигательного дефицита. [4]
Синдром Гудпасчера
Синдром Гудпасчера - аутоиммунная патология, характеризующаяся образованием аутоантител к базальным мембранам почечных клубочков и легочных альвеол. Клинически синдром Гудпасчера проявляется рецидивирующими легочными кровотечениями, прогрессирующим гломерулонефритом и почечной недостаточностью. Диагноз синдрома Гудпасчера подтверждается выявлением антител к базальной мембране клубочков (Anti GBM), данными биопсии почек и легких, рентгенологического исследования легких. Лечение синдрома Гудпасчера включает иммуносупрессивную терапию (глюкокортикостероиды, цитостатики), плазмаферез, по показаниям - гемодиализ, трансплантацию почек.
Общие сведения
Синдром Гудпасчера - иммуно-воспалительное поражение капилляров почек и легких, протекающее с развитием гломерулонефрита и геморрагического пневмонита. Впервые признаки данной патологии были описаны в 1919 г. американским патофизиологом Э.У. Гудпасчером, за что болезнь и была названа его именем. В ревматологии синдром Гудпасчера относится к системным васкулитам и нередко обозначается как «геморрагический легочно-почечный синдром», «геморрагическая пневмония с гломерулонефритом», «идиопатический гемосидероз легких с нефритом». Частота развития синдрома Гудпасчера составляет 1 случай на 1 млн. населения. Отмечается два возрастных пика заболеваемости - в 20-30 лет и 50-60 лет; болеют преимущественно мужчины. При отсутствии лечения синдрома Гудпасчера летальность среди пациентов достигает 75-90%.
Причины синдрома Гудпасчера
Этиологические механизмы заболевания достоверно не определены. Клинические наблюдения указывают на связь развития синдрома Гудпасчера с перенесенной вирусной инфекцией (гриппом, вирусным гепатитом А и др.), приемом лекарственных препаратов (карбимазола, пеницилламина), производственными вредностями (вдыханием паров органических растворителей, лаков, бензина), переохлаждением, курением. Отмечена генетическая предрасположенность к данному синдрому у лиц-носителей HLA-DRwl5, HLA-DR4 и HLA-DRB1 аллелей. Описаны семейные случаи синдрома Гудпасчера.
Под воздействием того или иного этиологического фактора, в результате изменения толерантности иммунной системы, в организме начинается выработка аутоантител к базальным мембранам легочных альвеол и почечных клубочков. Предполагается, что в роли аутоантигена выступает структурный компонент а-3 цепи коллагена IV типа, присутствующий в базальных мембранах легочных и почечных капилляров. Сформировавшиеся антитела (GВМ-антитела) в присутствии С3-комплемента связываются с антигенами; образовавшиеся иммунные комплексы откладываются вдоль базальных мембран, индуцируя иммуно-воспалительное поражение почечных клубочков (гломерулонефрит) и альвеол (альвеолит). В развитии аутоиммунного воспаления большая роль принадлежит активации клеточных элементов (Т-лимфоцитов, эндотелиоцитов, моноцитов, альвеолярных макрофагов, полиморфноядерных лейкоцитов), цитокинов (инсулиноподобного, тромбоцитарного факторов роста, факторов некроза опухоли, интерлейкина-1), свободных радикалов, протеолитических ферментов и других факторов, повреждающих почечную и легочную ткань.
Патоморфологическими субстратами синдрома Гудпасчера служат геморрагический некротизирующий альвеолит и нефрозонефрит. При гистологическом исследовании почечной ткани обнаруживается пролиферативно-мембранозный, пролиферативный или некротизирующий гломерулонефрит, склероз клубочков и фиброз почечной паренхимы. Морфологическое исследование легочной ткани выявляет капиллярит межальвеолярных перегородок, легочные инфильтраты, гемосидероз, пневмосклероз.
Симптомы синдрома Гудпасчера
Выделяют три варианта клинического течения синдрома Гудпасчера: злокачественный, умеренный и медленный. Для злокачественного течения характерны рецидивирующая геморрагическая пневмония и стремительно прогрессирующий гломерулонефрит. При втором типе легочно-почечный синдром развивается медленнее и выражен умеренно. При третьем варианте синдрома Гудпасчера преобладают явления гломерулонефрита и ХПН; легочные проявления развиваются поздно.
Злокачественный вариант синдрома Гудпасчера дебютирует легочным кровотечением и острой почечной недостаточностью, требующими проведения интенсивной терапии (устранения водно-электролитных нарушений, возмещения кровопотери, ингаляций кислорода, ИВЛ, гемодиализа или перитонеального диализа). В других случаях заболевание может начинаться с общих симптомов: субфебрилитета, недомогания, похудания. Иногда появлению жалоб предшествует перенесенная ОРВИ. Из специфических симптомов обычно первыми развиваются признаки поражения легких - кашель, прогрессирующая одышка, цианоз, боль в грудной клетке, рецидивирующее кровохарканье или легочное кровотечение. Поражение легких при синдроме Гудпасчера нередко осложняется сердечной астмой и отеком легких.
Вскоре к легочным проявлениям добавляется почечная симптоматика: гематурия, олигурия, периферические отеки, артериальная гипертензия. У 10-15% пациентов синдром Гудпасчера манифестирует с клинических признаков гломерулонефрита. Во многих случаях течение заболевания сопровождается миалгиями, артралгиями, геморрагиями кожи и слизистых оболочек, интраретинальными кровоизлияниями, перикардитами.
Диагностика синдрома Гудпасчера
При осмотре пациентов с синдромом Гудпасчера обращает на себя внимание бледность кожных покровов, пастозность или отеки лица. В легких выслушиваются сухие и влажные хрипы, количество которых увеличивается в момент кровохарканья и после него. В общем анализе крови обнаруживается гипохромная анемия, анизоцитоз, пойкилоцитоз, лейкоцитоз, резкое увеличение СОЭ. Для общего анализа мочи характерна протеинурия, цилиндрурия, эритроцитурия; проба по Зимницкому выявляет изогипостенурию. В биохимическом анализе крови определяется нарастание уровня креатинина, мочевины, серомукоида; снижение концентрации железа. Для синдрома Гудпасчера типично обнаружение большого количества эритроцитов, сидерофагов и гемосидерина в общем анализе мокроты.
Наиболее чувствительным и специфичным методом диагностики синдрома Гудпасчера служит определение антител к базальной мембране клубочков (Anti-GBM) с помощью ИФА или РИА. На рентгенограммах легких выявляются множественные очаговые тени. Морфологическое подтверждение синдрома Гудпасчера основывается на данных биопсии легких и почек. Вспомогательное значение имеют результаты инструментальной диагностики: спирометрии, УЗИ почек, ЭКГ, ЭхоКГ.
Лечение и прогноз синдрома Гудпасчера
В остром периоде синдрома Гудпасчера показано назначение пульс-терапии метилпреднизолоном или комбинированной пульс-терапии (метилпреднизолон+циклофосфан) с последующим переводом на поддерживающую терапию после достижения клинико-лабораторной и рентгенологической ремиссии. С целью элиминации циркулирующих иммунных комплексов проводится плазмаферез. Симптоматическая терапия синдрома Гудпасчера включает переливания эритроцитарной массы и плазмы крови, назначение препаратов железа. При развитии терминальной почечной недостаточности применяются сеансы гемодиализа. Возможно выполнение нефрэктомии с последующей трансплантацией почки, однако это не исключает рецидива некротизирующего гломерулонефрита в трансплантате.
Течение синдрома Гудпасчера неуклонно прогрессирующее; прогноз - мало обнадеживающий. Гибель пациентов происходит, как правило, вследствие профузных легочных кровотечений, тяжелой почечной или дыхательной недостаточности. При злокачественном варианте летальный исход наступает в считанные недели; в остальных случаях средняя продолжительность жизни колеблется от нескольких месяцев до 1-3 лет. В литературе описаны единичные спонтанные ремиссии синдрома Гудпасчера.
Болезнь Рейно
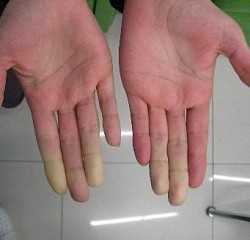
Некоторые виды сосудистых заболеваний могут быть локальными, вплоть до проявления лишь на кончиках пальцев рук или ног. Именно так ведет себя болезнь периферических сосудов Рейно, которая характеризуется приступообразной ишемией пальцев кистей и стоп. Данное состояние возникает на фоне систематического поражения мелких концевых артерий и нарушения сосудистого тонуса. Исследования показали, что женщины страдают от заболевания в пять раз чаще, чем мужчины. При этом недуг обычно сочетается у них с мигренью. Средний возраст болезни составляет 20-40 лет. Хоть она и не вызывает развитие других заболеваний, все же может доставлять массу неприятных симптомов и значительно снизить качество жизни больного.
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обратиться к профильному специалисту.

Симптомы
Выявить заболевание на ранних стадиях развития очень сложно, поскольку люди не оказывают должного внимания неприятным ощущениям в конечностях, легким приступам онемения или холода. А ведь именно так проявляются первые признаки болезни. При воздействии низких температур может наблюдаться бледность пальцев. Стоит отметить, что клиническая картина болезни Рейно всегда проявляется внезапно и приступообразно. Возникновение каких-либо симптомов может длиться от нескольких минут до нескольких часов. Иными признаками заболевания является:
- симметричная потеря чувствительности пальцев;
- похолодание в руках;
- ощущение покалывания;
- синюшный или розовый цвет кончиков пальцев;
- болезненные ощущения.
После приступа кончики пальцев становятся горячими, а любое прикосновение к ним вызывает более сильную боль. Симптомы болезни Рейно проявляются в зависимости от прогрессирования недуга. Выделяют несколько его стадий:
- ангиоспастическая;
- ангиопаралитическая;
- трофопаралитическая.
Как отмечают специалисты, болезни больше всего подвержены пианисты и люди, которые много печатают за компьютером. При продолжительном расстройстве питания кожи проявление недуга становится все более заметным: подушечки кончиков пальцев вытягиваются и уплощаются, кожа в этом месте становится неэластичная, сухая, возле ногтей могут появляться гнойнички, которые плохо заживают.
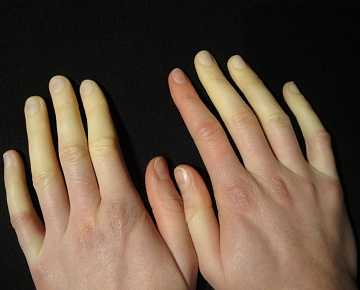
Причины заболевания
Принято считать, что болезнь Рейно чаще всего вызывают частые стрессовые ситуации и переохлаждения. Это действительно так, однако есть немало причин, которые также являются своего рода предрасположенностью к появлению недуга. Итак, факторами развития данного заболевания являются:
- гипотермия;
- частое травмирование пальцев;
- сдавливание артерий и нервов;
- эндокринные заболевания;
- тромбоцитоз;
- макроглобулинемия, криогробулинемия;
- вибрационная болезнь.
Фактор наследственности как причина возникновения болезни встречается лишь у 4% больных, при этом первые симптомы можно наблюдать в юношеском возрасте. Нередко специфические условия труда, оказывая пагубное влияние на организм, способствуют появлению недуга. Это может быть воздействие:
- помещений с низкой температурой;
- солей тяжелых металлов;
- кремниевой кислоты и др.
Не зависимо от причины, болезнь может иметь неприятные последствия и лишить человека возможности заниматься некоторыми видами трудовой деятельности. В частности, для музыкантов такой диагноз ставит крест на карьере, поскольку потеря чувствительности в конечностях приводит к неспособности играть на музыкальных инструментах на профессиональном уровне.
Методы диагностики
На первичном приеме у врача практически невозможно выявить причину, которая привела к развитию недуга. Поэтому он назначает ряд исследований, которые помогут разобраться с этиологией заболевания. Для этих целей проводится:
- Проба Аллена. Позволяет оценить проходимость локтевой и лучевой артерии. Выполняется во время осмотра. Проба обычно включена в стоимость приема, поэтому колеблется от 500 до 1500 рублей. Информативность метода 90 %.
- Капилляроскопия ногтевого ложа. Анализ определяет расширенные капиллярные петли, если диагностируется болезнь Рейно. Стоимость исследования в Москве колеблется в районе 1000 рублей, а результат свыше 90 % точности.
- Ревматологические пробы. Позволяют обнаружить заболевания по результатам исследования крови. Сделать пробы в Москве можно от 700 до 1500 рублей. Информативность метода около 90 %.
- Анализ на гемостаз. Определяет наличие и локализацию тромбоцитарных пробок. Результат определяется с точностью выше 90%.
- Общий анализ крови. Назначается для подтверждения диагноза по вышеуказанным методам. Стоимость в Москве от 400 до 900 рублей, информативность исследования 80-90 %.
Какой врач поможет?
Чтобы выяснить, являются ли неприятные ощущения в пальцах связаны с данным заболеванием, нужно обратиться к:
Читайте также:
- Трудоспособность после перенесенного отогенного менингита
- Дефицит гормона роста. Человеческий гормон роста (hGH)
- Техника пальпации кишечника. Особенности пальпации кишок
- Синдром тарзального канала: причины, симптомы и лечение
- Топография средостения. Верхнее средостение. Нижнее средостение. Переднее средостение. Среднее средостение. Заднее средостение.