Синдром Гренова (Groenouw) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Синдром Марфана: причины появления, симптомы, диагностика и способы лечения.
Определение
Синдромом Марфана, или Марфана-Ашара, называют наследственное заболевание соединительной ткани с преимущественным поражением сердечнососудистой системы, скелета и органа зрения. Частота синдрома в популяции составляет от 1:3000 до 1:15000. Впервые его описали французские врачи Антонин Бернард Марфан (1896) и Эмиль Шарль Ашар (1902).
Причины появления синдрома Марфана
Синдром Марфана имеет аутосомно-доминантный тип наследования (то есть дети получают патологический ген от родителей, которые страдают данным заболеванием). Молекулярной основой синдрома Марфана является нарушение синтеза одного из белков соединительной ткани - фибриллина, который в норме придает ей эластичность и сократительную способность.
При СМ наблюдается дефицит фибриллина или его аномальное строение, поэтому соединительная ткань обладает повышенной растяжимостью и теряет способность выдерживать физиологические нагрузки.
Классификация заболевания
В зависимости от количества пораженных систем организма выделяют несколько форм синдрома Марфана:
- стертую - со слабо выраженными изменениями в 1-2 системах;
- выраженную - со слабо выраженными изменениями в 3 системах; значительно выраженными изменениями хотя бы в 1 системе; выраженными изменениями в 2-3 и более системах.
Принципиальную роль в определении прогноза болезни играет характер ее течения.
Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, с каждым годом жизни пациента возрастают риски фатальных осложнений.
Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Симптомы синдрома Марфана
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ весьма разнообразна.
Симптомы патологии сердечно-сосудистой системы
- Изменения сердечно-сосудистой системы отмечаются у большинства больных. Их основная причина - потеря способности стенок артерий и клапанных структур сердца выдерживать естественные гемодинамические нагрузки. Наиболее частая сердечная патология при синдроме Марфана -недостаточность митрального клапана, когда наблюдается поражение эластических структур створок и сухожильных нитей клапана с развитием его дисфункции. Со временем у многих больных эта дисфункция переходит в умеренную или тяжелую митральную недостаточность, требующую хирургической коррекции. Реже можно наблюдать аортальную и трикуспидальную недостаточность. Стенозы клапанов для СМ не характерны. При небольшой или умеренной хронической митральной недостаточности жалобы обычно отсутствуют (особенно при медленном нарастании недостаточности). Со временем появляются одышка, ощущение быстрой утомляемости и учащенное сердцебиение.
Клапанные пороки нередко осложняются инфекционным эндокардитом (воспалением внутренней оболочки сердца) - внезапно повышается температура, появляются озноб, боль в суставах, бледность кожных покровов и слизистых).
Кроме того, формируется геморрагическая сыпь, деформируются фаланги пальцев и ногтевые пластины (симптом «барабанных палочек» и «часовых стекол»).
Самую большую опасность представляют, пожалуй, патологические изменения аорты. Поскольку внутренний слой стенки сосудов также состоит из волокон соединительной ткани, сосуды постепенно изнашиваются. Принимая во внимание тот факт, что давление крови в аорте выше, чем на других участках сосудистого русла, это приводит к постепенному ее расширению, патологическому скоплению крови между сосудистыми стенками и формированию аневризмы или спонтанному разрыву. Основные симптомы аневризмы аорты следующие: осиплость голоса; нехватка воздуха; боль в плече; кровохарканье, боль в спине.
Симптомы поражения опорно-двигательного аппарата
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести заболевания и особенностей организма пациента. Люди с синдромом Марфана обычно отличаются высоким ростом. У детей с СМ очень длинные, непропорциональные росту руки и патологически удлиненные, тонкие пальцы, так называемые пальцы паука (арахнодактилия).
Лицо у людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Для пациентов характерны деформации грудной клетки, которые могут быть двух вариантов: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь).
Часто определяются различной степени выраженности сколиоз (отклонения позвоночного столба в сторону) или кифоз (формирование горба).
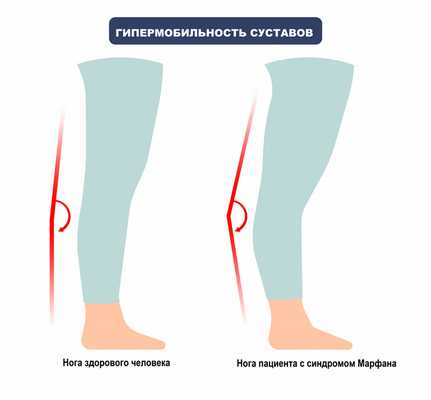
Кроме того, у пациентов с СМ нередко диагностируют плоскостопие, повышенную подвижность суставов, слабость связочного аппарата, неразвитость мышечных структур и подкожно-жирового слоя.
Симптомы поражения кожи
Высокий темп роста и нарушение выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется повышенной растяжимостью кожного покрова с образованием растяжек (стрий).
Симптомы поражения органа зрения
Чаще всего повреждения глаз у пациентов с синдромом Марфана включают:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Симптомы поражения органов дыхания
В легких пациентов с СМ может разрастаться может патологически разрастаться соединительная ткань, приводя к сужению бронхов и легочному фиброзу. Нередко на фоне генетической мутации манифестирует бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет риск развития спонтанного пневмоторакса — неотложной ситуации, при которой в плевральную полость попадает воздух, и легкое спадается: у пациента внезапно появляется одышка, резкая или ноющая боль в груди, сухой кашель.
Симптомы поражения желудочно-кишечного тракта
У людей с FBN1 мутацией нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз. Пациент испытывает привкус горечи во рту, тяжесть в области правого подреберья, в надчревной области. Часто беспокоит изжога, отрыжка, вздутие живота.
Симптомы поражения мочевыделительной системы
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Признаком нефроптоза легкой степени являются периодические одно- или двусторонние боли в проекции почек. Как правило, они проходят после непродолжительного пребывания в горизонтальном положении. Боль обычно тупая, ноющая, слабая или умеренная. Усугубление опущения ведет к нарушению оттока мочи и создает условия для развития инфекционных процессов и гидронефроза (скопления жидкости в почке). При присоединении инфекции может повышаться температура тела, появляться слизь и гной в моче.
По мере снижения функции почек у больного развиваются отеки, гипертензия и другие системные нарушения.
Симптомы поражения нервной системы
Расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. У таких пациентов развивается синдром хронической усталости, проявляющийся астенией и депрессивными расстройствами.
Интеллектуальная деятельность в большинстве случаев не нарушена, наоборот - среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Диагностика синдрома Марфана
Диагностика генетической аномалии включает комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7-18 лет — измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку 10 см.
- Лабораторное обследование.
Для подтверждения нарушений развития соединительной ткани и оценки степени выраженности мутации гена FBN1 пациентам с подозрением на синдром Марфана назначают:
Электрокардиография (ЭКГ) - повсеместно распространенный метод изучения работы сердца, в основе которого лежит графическое изображение электрических импульсов с�.
Детская прогерия (синдром Гетчинсона-Гилфорда)
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Детская прогерия (син. синдром Гетчинсона-Гилфорда) - редкое, вероятно, генетически гетерогенное заболевание, с преимущественно аутосомно-рецессивным типом наследования, не исключается возможность новой доминантной мутации. Характеризуется старческими изменениями организма в детском возрасте с летальным исходом, часто наступающим до 15-летнего возраста от осложнений, обусловленных атеросклерозом.
Код по МКБ-10
Эпидемиология
Причины детской прогерии
Причины детской прогерии неизвестны.
Патогенез
Выявляют атрофические изменения эпидермиса и дермы, истончение подкожной жировой клетчатки. В зоне уплотнения кожи эпидермис обычной толщины и строения, дерма резко утолщена, в ее нижней части отмечается гиалинизация коллагеновых волокон, распространяющаяся в подкожную клетчатку в виде прослоек. В верхней части дермы - умеренные периваскулярные воспалительные инфильтраты. Концевые отделы потовых желез расположены более высоко по сравнению с нормой.
В культуре фибробластов, полученных от больных и их гетерозиготных родителей, обнаружены замедление клеточного роста, снижение митотической активности, синтеза ДНК и клонирующей способности. В гибриде фибробластов больных с клетками асцита Эрлиха мышей поглощение тимидина резко снижено.
Симптомы детской прогерии
Кожа истонченная, сухая, морщинистая, с просвечивающими венами, выражена атрофия мышц и подкожной клетчатки, дистрофия зубов и ногтей, изменения костно-суставного аппарата, миокарда, помутнение хрусталика, нарушение липидного обмена. Заболевание проявляется обычно в 6-12-месячном возрасте замедлением роста, выпадением волос на волосистой части головы, в области бровей, ресниц. Отмечают несоответствие между объемом черепа и маленьким лицом, недоразвитие нижней челюсти, клювообразную форму носа, цианоз вокруг рта. Кожа туловища тонкая, пигментированная, со склероподобными бляшками. Наблюдается гипоплазия половых органов, вторичные половые признаки отсутствуют.
Синдром Гетчинсона-Гилфорда (прогерия)
Синдром Гетчинсона-Гилфорда или прогерия (сенильный нанизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения. Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом. Этиология прогерии неясна. Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у больных. Однако исследования зарубежных ученых позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения. Характерен вид лица: с экзофтальмом, тонким клювовидным носом, большим мозговым и малым лицевым черепом, голос тонкий, имеются скелетные аномалии. Пубертат обычно не наступает, наружные гениталии гипоплазированы. Интеллект средний или выше среднего. Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Ключевые слова
Для цитирования:
For citation:
Синдром Гетчинсона-Гилфорда (Hutchinson-Gilford Progeria Syndrome), или прогерия (сенильный папизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения.
Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом.
Этнология прогерии неясна.
Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у бальных. Однако исследования зарубежных ученых [2-4, 7] позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Отмечается влияние возраста отца как возможной причины новых мутаций. Так, средний возраст отцов составляет 35-37 лет.
Аутосомно-рецессивный тип наследования синдрома также обсуждается в литературе [7, 8, И]. Этот тип наследования был впервые предположен Gabr и соавт. в 1960 г. при описании двух моиозиготных сестер, а впервые сообщил о семейном случае прогерии Paterson в 1922 г., хотя описание двух больных братьев было неполным и без фотографий.
Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения.
Дети рождаются нормальными, но к 1-му году жизни наблюдается выраженная задержка роста и массы тела. Конечный рост в среднем достигает 100 см. В первые годы жизни развивается тотальная алопеция. Кожа топкая, лоснящаяся, сухая, тугонатянутая (па кистях и стопах, наоборот, морщинистая). В нижней части живота и бедрах кожные изменения напоминают склеродермию. С возрастом появляются коричневые пигментные пятна. Потовые и сальные железы атрофируются.
Подкожный жировой слой полностью отсутствует, за исключением лобковой области. На черепе выражена подкожная венозная сеть. Нощи дистрофичные, влоть до аплазии. Зубы прорезываются с задержкой, аномально расположены, с ранним разрушением как молочных, так и постоянных зубов.
Характерен вид лица: с экзофтальмом, гонким клювовидным носом, большим мозговым и малым лицевым черепом. Голос тонкий.
Скелетные аномалии включают резорбцию ключицы с замещением фиброзной тканью, резорбцию конечных фаланг кистей (акроостеолиз), истончение длинных трубчатых костей и ребер, тугоподвижность сутавов пальцев, увеличение локтевых и коленных суставов. Часты асептические некрозы головки бедренной кости и вывих тазобедренного сустава.
Пубертат обычно не наступает, наружные гениталии ги- поплазированы.
Интеллект средний или выше среднего.
Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Следует отметить, что заболевания, характерные для нормального процесса старения (катаракта, опухоли, сахарный диабет), встречаются при прогерии крайне редко.
Прогноз для жизни неблагоприятный: продолжительность жизни колеблется от 7 до 28 лет, в среднем составляя 12-13,5 года [1, 4, 5, 7, 10]. Основные причины летальных исходов - острый инфаркт миокарда, застойная сердечная недостаточность, инсульты. На аутопсии выявляются распространенный атеросклероз, гипоплазия гонад, иногда гипоплазия надпочечников и значительная гиперплазия тимуса, истончение коркового слоя костей.
Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Гормональные исследования у детей [4] выявляют нормальную ночную секрецию соматотропного гормона гипофиза, но крайне низкий уровень инсулиноподобного фактора роста (ИФР-1) в плазме крови. Это позволяет предполагать наличие у данных больных бионеактивпого пула СТГ в крови, либо периферическую резистентность к эндогенному СТГ. либо выраженный дефицит питания. Трехмесячный период высококалорийного питания не увеличивает уровень ИФР-1 в крови, но ускоряет линейный рост.
Прогерия считается состоянием, связанным с ннсулиноре- зистентностыо умеренной степени. В 1983 г.[9] впервые была описана девочка, у которой в 2-легнсм возрасте уровень инсулина в крови натощак составлял 20-40 мкг/дл. В 4 года через 3 мес после удаления кисты яичника развилась гипергликемия натощак (до 250 мкг/дл) с высоким уровнем инсулина (более 2200 мкЕД/мл) и С-пептида (32,4 нг/мл) в крови. Уровень гемоглобина А1 достигал 10%. Связывание инсулина с рецепторами эритроциов было в пределах нормальных значений.
Иммунологический аспект в патогенезе прогерии впервые был выдвинут Walford в 1970 г. В 1973-1976 г. Singal и Goldstein выявили отсутствие либо резкое снижение экспрессии HLA культурой фибробластов кожи у детей с прогерией. Вместе с тем другие исследователи, анализируя экспрессию HLA у детей с прогерией и их здоровых родственников [2], нс выявили ни количественного, ни качественного дефицита в экспрессии HLA в фибробластах кожи. Различие в частоте встречаемости ряда HLA-антнгенов у больных прогерией и здоровых людей не дает право в настоящее время говорить о специфической ассоциации HLA и прогерии в связи с малым числом исследуемого материала.
Биохимическими исследованиями показано, что одним из биомаркеров старения является мочевая экскреция гиалуроновой кислоты. В норме у детей и подростков содержание ее составляет менее 1% от уровня общих гликозаминогликанов и увеличивается с возрастом до 5-6%. У детей с прогерией выявлено значительное повышение (до 10-20%) экскреции гиалуроновой кислоты с мочой по сравнению со здоровыми людьми [4]. Данное повышение не наблюдается ни при одном генетическом заболевании, кроме синдрома Вернера, или “прогерии взрослых” [4]. Считается, что гиалуроновая кислота является ключевым фактором антиангиогенеза в процессе созревания и старения.
Изучение культуры фибробластов кожи от пациентов с прогерией выявляет значительное снижение клеточного роста вследствие подавления митотической активности. С другой стороны, отмечается нормальное распределение типов коллагена в коже, характерное для детей, а именно, преобладание коллагена 3-го типа над коллагеном 1-го типа [8].
Имеются данные, что в основе прогерии лежит дефицит метаболизма витамина Е [7].
Ряд исследователей связывают прогерию с генетически обусловленной ошибкой в синтезе внутриклеточных белков. Так, показано, что эритроциты больных детей содержат повышенную термолабильную фракцию ферментов: глюкозо-6- фосфатдегидрогепазу и б-фосфоглюконатдегидрогепазу |6]. Другие работы не подтверждают эту концепцию [3].
Данные олене или детей с прогерией крайне малочисленны.
Патогенетически оправданными считаются терапия витамином Е для восполнения его дефицита (Лугез и МШап, 15974) и усиленное белковое питание.
Приводим описание собственного клинического случая.
Боль пая Н., 3 лет 9 мес., поступила в детское отделение ЭНЦ РАМН с жалобами на отставание в росте и массе, сниженный аппетит, облысение, резкую головную боль.
Раннее развитие: держит голову с 1 мес жизни, сидит с 6 .мес жизни, ходит с 1 года 2 мес, зубы появились в 1 год 1 мес, говорит с 1,5 лет. Грудное вскармливание - до 1 года 8 мес.
Перенесенные заболевания: дисплазия тазобедренный суставов (в 1 мес жизни), двусторонний врожденный вывих бедер (диагностирован в 6 мес жизни), стоматит, легкая форма (в 3 года).
Аллергологический анамнез не отягощен.
Наследственность по низкорослости не отягощена. Мать 24 лет, рост 165 см, родственники. со стороны матери: бабушка - рост 157 см, дедушка - 180 см, тетя - 168 см. Отец 26 лет, рост 176 см; родственники со стороны отца: бабушка - рост 157 см, дедушка - 170 см. Отягощена наследственность по сахарному диабету II типа, который имеется у бабушки со стороны отца и у прабабушки со стороны матери. Отягощена наследственность по бронхиальной астме, тяжелая форма которой имеется у прадедушки, со стороны матери и у двоюродной прабабушки со стороны матери.
Анамнез заболевания: с 2-месячного возраста замечены уплотнение кожных покровов и подкожной жировой клетчатки, лоснящаяся кожа на бедрах, животе, ягодицах, цианоз носогубного треугольника. В 3-месячном возрасте диагностирована легкая форма склеродермии. При исследовании биоптата кожи выявлен гиперкератоз эпителия и умеренный склероз дермы. Консультирована дерматологом: диагноз склеродермии был снят, больше данных, свидетельствующих о склеродерме. Получала лечение преднизолоновой мазыо в течение 3 мес с умеренным эффектом - блеск и плотность кожных покровов уменьшились. В возрасте 1 года появилась венозная сеть на голове. Отмечалась гипотрофия П-Ш степени. Невропатолог диагностировал перинатальную энцефалопатию, компенсированную гидроцефалию. В 1 год 7 мес девочка была впервые консультирована эндокринологом: диагностирована задержка физического развития смешанного генеза. Масса тела 7800 г, костный возраст соответствовал паспортному. В 1 год 10 мес начали выпадать полосы на голове. В 1 год 11 мес впервые консультирована генетиком, поставлен диагноз: “Синдром Гетчинсона—Гилфорда”. Девочка была обследована в стационаре по месту жительства: рост 73 см, масса 7900 г. На ЭКГ: метаболические изменения миокарда
Данные обследования в ЭНЦ РАМН. Хронологический возраст 3,9 года. Рост 81,2 см. Коэффициент стандартного отклонения (SDS роста) - 4,35. Масса тела 9,5 кг. Рост сидя 48,5 см, коэффициент “верхний ссгмент/пижпий сегмент" 1,48. Окружность головы 49 см.
Отмечаются ярко выраженные черты синдрома Гетчинсона - Гилфорда: 1) крупная голова с диспропорционально большим мозговым черепом и малым лицевым. Вдавлен) гость височных костей; 2) выраженная венозная сеть на голове; 3) тотальная алопеция; 4) узкий, деформированный нос с истончением кожи на нем, цианоз носогубного треугольника; 5) истончение кожи на туловище, конечностях, морщинистость кожи на ладонях и ступнях. Склеродермоподобные изменения кожи на животе, спине и ягодицах - очаги депигментации диаметром 0,5-0,7 см, множественные, уплотненные. Депигментация сосков; 6) подкожная жировая клетчатка развита слабо, распределена равномерно; 7) гипоплазия ногтей кистей и стоп; утолщение межфаланговых суставов и концевых фаланг кистей и стоп; варусная девиация верхней трети предплечий, короткая шея (см. рисунок).
Область сердца визуально не изменена. Тоны сердца ясные, ритмичные. Часота сердечных сокращений 104 удара в минуту, АД 80/50 мм рт.ст. Дыхание везикулярное, хрипов нет. Зев чистый. Живот мягкий, безболезненный. Печень не увеличена, селезенка не пальпируется. Стул регулярный. Дизурических расстройств нет. Симптом Пастернацкого отрицательный с обеих сторон. Эндокринный статус: щитовидная железа не увеличена, симптомов нарушения функции нет. Симптомы гипокортицизма отсутствуют. Половой статус препубертатный: Ах 0, Р 0, Ма 0, Me 0.
Гормональное исследование крови: кортизол 321,4 нмоль/л, общий трийодтиронин 2,48 нмоль/л, общий тироксин 100,0 нмоль/л, ТТГ 2,10 мкЕД/л, пролактин 248,0 мкЕД/л, ЛГ 1,60 ЕД/л, ФСГ 5,50 ЕД/о, 17-оксипрогестерон 1,5 нг/мл. Соматотропный гормон на фоне стимуляционной пробы с клофелином: 0 мин - 1,7 нг/мл, 30 мин - 1,5 нг/мл, 60 мин - 2,3 нг/мл, 120 мин - 71,0 нг/мл, 150 мин - 29,3 нг/мл.
Гормональное исследование мочи: свободный кортизол 1115,0 нмоль/л на 1 г креатинина (креатинин 0,24 г/л). Дс- гидроэпиандростерон-сульфат — следы.
Показатели гуморального аутоиммунитета: у больного ребенка не выявлено антител пи к тиреоглобулину человека, ни к микросомальному антигену тиреоцигов, ни к поверхностным антигенам клеток аденогипофиза и клеток коры надпочечников крысы.
Вместе с тем у матери ребенка обнаружены антитела к поверхностным антигенам клеток аденогипофиза крысы, а у бабушки по материнской линии - слабо положительная реакция па наличие антител к микросомальному антигену тиреоцигов при отсутствии антител к другим изучаемым антигенам.
Рентгенографическое исследование: на рентгенограмме черепа и кистей структура костей не изменена. Форма и размеры турецкого седла обычные. Сосудистый рисунок костей свода усилен. Дифференцирование скелета соответствует 12- 15 мес. Ногтевые фаланги деформированы, треугольной формы. На рентгенограмме стоп отмечается небольшое уплотнение стенок arteria dorsalis pedis.
Компьютерная томография головного мозга: на серии компьютерных томограмм изменения плотности мозговой ткани не выявлено. Желудочковая система не изменена. Хиазмальная цистерна расширена. Несколько расширена межполушарная щель. Данных, свидетельствующих об объемном процессе головного мозга, не выявлено.
ЭКГ: ЧСС 120 в минуту, ритм синусовый. Электроэнцефалограмма: на фоне умеренных диффузных изменений биоритмики с признаками диэнцефальной заинтересованности отмечаются признаки ирригации стволово-диэнцефальных структур с легким акцентом справа, в теменно-затылочной области. Эхоэнцефалограмма: эхо-пульсация неустойчиво усилена до 55%, смещения срединных структур нет, легкое расширение 111 желудочка (до 6 мм), вентрикулярный индекс умеренно выше нормы. Заключение: смещения срединных структур не выявлено. Расширение боковых желудочков.
Ультразвуковое исследование: щитовидная железа: типично расположена, контуры ровные, структура гомогенная. Правая доля 2,3x1,1хо,9 см, слева - 2,4x1,2x0,9 см, толщина перешейка 0,4 см. Объем щитовидной железы 2,55 мл. Надпочечники: нс увеличены. Органы малого таза: размеры матки и яичников соответствуют возрастной норме. Матка: 3,1x1,3x1,1 см, правый яичник: 1,6x1,2x0,8 см, левый яичник: 1,4x1,2x1,0 см. Печень: увеличена правая доля - 8,5 см, левая доля 2,6 см. Структура паренхимы гомогенная, внутри- печеночные протоки не расширены. Воротная вена не расширена. Желчный пузырь конкрементов не содержит, Почки: топография не изменена, размеры в пределах возрастной нормы. Структуры хорошо дифференцированы, без гидро- нефротических изменений и достоверных эхо-признаков дополнительных объемных включений. Паренхима гомогенна, толщина се соответствует возрастной норме.
Консультация окулиста: моторно-зрачковых нарушений не выявлено. Передний отрезок и среды без патологии. Глазное дно: диски бледно-розовые, сосуды умеренно расширены, полнокровны, извиты по всей протяженности сетчатки. Сетчатка на периферии разряжена, небольшие отеки. Заключение: повышение внутричерепного давления.
Консультация невропатолога: субкомпепсированпая гидроцефалия на фоне скудной церебральной симптоматики.
Консультация дерматолога: Alopecia totalis. Рекомендованы курсы лечения препаратами, улучшающими микроциркуляцию (трептал, троксевазип), поливитамины (Bj, В2, В(„ В15, А, Е); наружно — втирание с массажем головы геля актовегина, геля троксевазина, димексида, 01. Ricini.
Учитывая имеющиеся в литературе данные об эффективности применения гормона роста у детей с прогерией [4], для увеличения линейного роста девочке был назначен пробный 3-месячный курс лечения рекомбинантным гормоном роста человека “SAIZEN” (ARES-SERONO). Недельная доза составила 15 ЕД/м 2 , суточная доза — 1 ЕД, подкожно, ежедневно, 7 раз в неделю.
Лечение рекомендовано проводить под контролем гликемии и суточной глюкозурии.
Помимо гормона роста, девочке назначено лечение, рекомендованное дерматологом, и полноценное белковое питание.
Таким образом, представленный материал, основанный на данных анамнеза, жалоб, результатах клинического обследования и лабораторно-инструментальных исследований, подтверждает наличие у ребенка классического синдрома Гетчинсона-Гилфорда (прогерии).
Болезнь прогерия, или Синдром Хатчинсона-Гилфорда

Прогерия, также известная как синдром Хатчинсона-Гилфорда, является чрезвычайно редким, прогрессирующим генетическим заболеванием, которое заставляет детей быстро стареть, начиная с первых двух лет их жизни.
Дети с этим расстройством при рождении ничем не отличаются внешне от других младенцев, однако в течение первого года начинаются проявляться первые признаки - замедленный рост и выпадение волос.
Средняя продолжительность жизни ребенка с прогерией составляет около 13 лет, однако в некоторых случаях она достигает 20 лет.
К сожалению, на данном этапе лекарства от синдрома Хатчинсона-Гилфорда нет, однако исследования продолжаются.
Симптомы синдрома Хатчинсона-Гилфорда
Обычно в течение первого года жизни рост ребенка с прогерией заметно замедляется, но двигательное развитие и интеллект остаются в норме.
Признаки этого прогрессирующего расстройства включают в себя специфический внешний вид:
- замедленный рост и вес ниже среднего;
- узкое лицо, маленькая нижняя челюсть, тонкие губы и клювастый нос;
- голова непропорционально большая для лица;
- выпуклые глаза и неполное смыкание век;
- выпадение волос, включая ресницы и брови;
- истонченная, пятнистая, морщинистая кожа;
- видимые вены;
- высокий голос.
Признаки и симптомы также включают проблемы со здоровьем:
- Тяжелое прогрессирующее заболевание сердца и кровеносных сосудов (сердечно-сосудистые заболевания);
- Отвердение и стягивание кожи на туловище и конечностях (по аналогии со склеродермией);
- Замедленное и неправильное формирование зубов;
- Частичная потеря слуха;
- Потеря жира под кожей и мышечной массы;
- Скелетные аномалии и хрупкие кости;
- Жесткие суставы;
- Вывих бедра;
- Резистентность к инсулину.
Когда следует обращаться к врачу?
Прогерия обычно обнаруживается в младенчестве или раннем детстве, часто при регулярных осмотрах, когда у ребенка впервые проявляются характерные признаки преждевременного старения.
Причины прогерии
Причина синдрома Хатчинсона-Гилфорда - мутация гена.
Ген, известный как ламин А/C (LMNA), создает белок, необходимый для удержания ядра клетки. Когда этот ген имеет дефект (мутацию), образуется аномальная форма белка ламина, называемого прогерином, и делает клетки нестабильными. Это, по-видимому, приводит к процессу старения.
В отличие от многих генетических мутаций, cиндром Хатчинсона-Гилфорда редко передается по наследству.
Другие подобные синдромы
Есть и другие прогероидные синдромы, которые передаются по наследству и вызывают быстрое старение, сокращая продолжительность жизни:
- Синдром Видемана-Раутенштрауха, также известный как неонатальный прогероидный синдром, начинается в утробе матери, причем признаки и симптомы старения проявляются при рождении.
- Синдром Вернера, также известный как взрослая прогерия, начинается в подростковом возрасте или в раннем взрослом возрасте, вызывая преждевременное старение и состояния, типичные для старости, такие как катаракта и диабет.
Фактор риска
Нет никаких известных факторов, таких как образ жизни или экологические проблемы, которые увеличивают риск возникновения прогерии или рождения ребенка с синдромом Хатчинсона-Гилфорда.
Для родителей, у которых был один ребенок с прогерией, вероятность рождения второго ребенка с синдромом Хатчинсона-Гилфорда составляет 2-3%.
Осложнения, вызванные прогерией
У детей с прогерией обычно развивается тяжелое затвердение артерий (атеросклероз). Это состояние, при котором стенки артерий - кровеносных сосудов, несущих питательные вещества и кислород от сердца к остальной части тела, - застывают и уплотняются, часто ограничивая кровоток.
Большинство детей с прогерией умирают от осложнений, связанных с атеросклерозом, в том числе:
- проблем с кровеносными сосудами, которые снабжают сердце (сердечно-сосудистые проблемы), приводящих к сердечному приступу и застойной сердечной недостаточности;
- проблем с кровеносными сосудами, которые снабжают мозг (цереброваскулярные проблемы), приводящих к инсульту;
- других проблем со здоровьем, часто связанных со старением - таких как артрит, катаракта.
Постановка диагноза синдрома Хатчинсона-Гилфорда
Врачи могут заподозрить прогерию на основании признаков и симптомов, характерных для этого синдрома. Для подтверждения диагноза используется генетический тест на мутации LMNA.
Тщательное физическое обследование ребенка включает в себя:
- измерение роста и веса;
- построение измерений на графике кривой нормального роста;
- проверку слуха и зрения;
- измерение жизненно важных показателей, включая кровяное давление;
- поиск видимых признаков и симптомов, характерных для прогерии.
Лечение прогерии
Нет лекарства от прогерии, но регулярное наблюдение за состоянием сердца и кровеносных сосудов (сердечно-сосудистых заболеваний) помогает в облегчении симптомов. Во время медицинских визитов измеряются вес и рост ребенка с последующим нанесением на график нормальных значений.
Дополнительные регулярные обследования, включая электрокардиограммы и осмотры зубов, зрения и слуха, могут быть рекомендованы врачом для проверки наличия изменений. Некоторые методы могут облегчить или отсрочить некоторые признаки и симптомы.
Лечение зависит от состояния и симптомовребенка. Они могут включать в себя:
- Аспирин в малых дозах. Суточная доза может помочь предотвратить сердечные приступы и инсульт.
- Другие лекарственные препараты. В зависимости от состояния врач может назначить другие лекарства, такие как статины для снижения уровня холестерина, лекарства для снижения артериального давления, антикоагулянты для предотвращения образования тромбов и лекарства для лечения головных болей и судорог.
- Физиотерапия и трудотерапия. Эти методы лечения помогают при тугоподвижности суставов и проблемах с бедрами.
- Питание. Питательные, высококалорийные продукты и добавки необходимы для поддержания полноценного питания.
- Стоматологическая помощь. Проблемы с зубами часто встречаются при прогерии.
Потенциал дальнейшего лечения прогерии
Современные исследования направлены на понимание прогерии и выявление новых вариантов лечения.
Некоторые области исследований включают в себя:
- Изучение генов и течения болезни, чтобы понять, как она прогрессирует. Это может помочь выявить новые методы лечения.
- Изучение способов профилактики заболеваний сердца и сосудов. Проведение клинических испытаний с использованием препаратов, известных как ингибиторы фарнезилтрансферазы (ФТИ), таких как лонафарниб, которые были разработаны для лечения рака, но могут быть эффективны для лечения прогерии, помогая с увеличением веса и повышением гибкости кровеносных сосудов.
Образ жизни при прогерии
Вот несколько шагов, которые можно предпринять, чтобы помочь ребенку с прогерией:
Синдром Шегрена
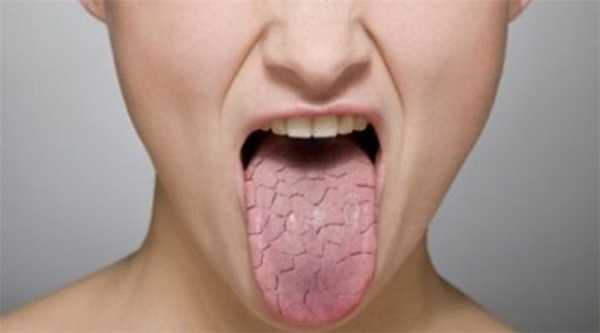
Синдром Шегрена («сухой синдром») проявляется снижением функции желез внешней секреции, вследствие такой патологии появляется выраженная сухость кожных покровов и слизистой оболочки влагалища, трахеи, носоглотки, глаз, полости рта, также наблюдается уменьшение секреции пищеварительных ферментов, которые вырабатывает поджелудочная железа.
Чаще всего данный синдром сопровождает целый ряд аутоиммунных патологий соединительной ткани - дерматомиозит, склеродермию, и в таких случаях носит название - вторичный синдром Шегрена. Если же патология развивается самостоятельно, то название звучит, как первичный синдром Шегрена, или болезнь Шегрена.
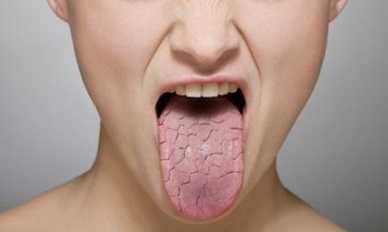
Что это такое?
Синдром Шегрена — аутоиммунное системное поражение соединительной ткани, проявляющееся вовлечением в патологический процесс желез внешней секреции, главным образом слюнных и слёзных, и хроническим прогрессирующим течением.
Патоморфология
Основным морфологическим признаком является инфильтрация желез внешней секреции лимфо- и плазмоцитами. В первую очередь страдают слезные и слюнные железы, чуть реже - железы бронхов, пищеварительного тракта и влагалища.
- Поражаются и крупные, и мелкие железы. Сначала, на раннем этапе болезни, в процесс вовлекаются лишь мелкие протоки; по мере прогрессирования ее инфильтрат распространяется далее, на собственно ткань железы, в результате чего железистая ткань атрофируется и замещается соединительной. В ряде случаев описанные выше инфильтраты возникают не только в экзокринных (внешней секреции) железах, но и в других органах и системах организма, в частности, в мышцах, легких и почках. Это в конечном итоге приводит к нарушению функции пораженного органа.
- У 30-40% больных в материале, взятом путем биопсии слюнных желез, определяется метаплазия (видоизменение) выстилающих протоки клеток: появляются миоэпителиальные островки.
Дольки пораженных желез у ряда больных разрушены, а у других дольковая структура сохранена. Железы либо увеличены в размере, либо находятся в пределах нормы.
Что интересно, даже при отсутствии яркой клинической симптоматики синдрома Шегрена, у больного каким-либо заболеванием соединительной ткани вероятнее всего обнаружатся гистологические признаки воспаления слюнных желез.
Причины развития
Причины возникновения синдрома Шегрена пока до конца не установлены. Среди наиболее вероятных является теория о патологической реакции иммунной системы организма. Такая реакция развивается в ответ на повреждение клеток внешних желез ретровирусом, в частности это - вирус Эпштейн-Барр, VI вирус герпеса, цитомегаловирус, вирус иммунодефицита человека. Несмотря на значительное сходство иммунологических нарушений с изменениями в организме, пораженном вирусом, прямых доказательств роли вируса, как причины развития патологии, не получено.
Сами вирусы и измененные от их воздействия эпителиальные клетки желез воспринимаются иммунной системой как антигены (чужеродные агенты). Иммунная система продуцирует антитела против таких клеток и постепенно вызывает разрушение тканей железы. Заболевание часто встречается как наследственное или семейное, особенно часто среди близнецов, что позволяет предположить о том, что существует генетическая предрасположенность.
Таким образом, предполагают, что в механизме развития и возникновения патологии важное значение имеет сочетание множества факторов:
- стрессовой реакции организма, которая возникает вследствие иммунного ответа;
- иммунная регуляция с участием половых гормонов, о чем говорит редкая заболеваемость среди лиц до 20 лет, при этом среди детей чаще всего болеют девочки;
- иммунный контроль с помощью Т-лимфоцитов;
- вирусный;
- генетический.
Есть два типа синдрома Шегрена: первичный — симптомы болезни являются первыми проявлениями ее, и вторичный, когда симптоматика проявляется у пациентов, страдающих иными ревматическими заболеваниями, такими как склеродермия, ревматоидный артрит или системная красная волчанка. Первичный и вторичный варианты синдрома встречаются примерно с одинаковой частотой. Это иногда затрудняет точную постановку диагноза. Синдром Шегрена достаточно распространен: в Великобритании, например, насчитывается около полумиллиона больных. Наиболее часто поражаются женщины в возрасте от 40 до 60 лет, тогда как только один пациент из 13 - мужчина.
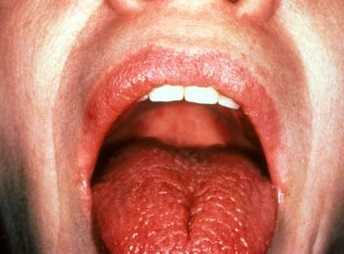
Симптомы синдрома Шегрена
Симптоматику заболевания делят на две группы:
- Железистые проявления, при которых поражаются эпителиальные железы и нарушаются их функции.
- Внежелизистые проявления. Симптоматика данной группы очень различается, вследствие поражения различных органов человека.
Железистые симптомы, можно выделить следующие:
- Патология слезных желез. В данном случае возникают неприятные и болезненные ощущения в глазах (жжение, глаза как будто начинает резать, ощущения песка в них), при этом появляется характерный зуд возле глаз, покраснение. В результате снижается зрение, возникновение точечных кровоизлияний, отеков, чувствительность к свету, глазные боли.
- Патология слюнных желез. Проявление воспаления главным образом околоушных желез, которые увеличены в размере, иногда с проявлением болезненных ощущений. Также возникает сухость слизистой оболочки рта, глотание пищи затруднено, нередко больным приходится запивать еду водой, вследствие с проблемой проглатывания. При этом слизистая оболочка рта принимает ярко-розоватый оттенок.
- Сухость дыхательных путей, которая приводит воспалению бронхов, трахеи, легких.
- Кожные нарушения, в результате чего кожа становится сухой.
- Поражение слизистой оболочки носа. Возникновение сухости в носу, покрытие внутриносовой коркой, вследствие чего развивается воспаление.
Внежелизистым проявлениям характерны следующие признаки:
- Возникновение трахеобронхита, сопровождающейся кашлем, отдышкой. Не редко при обследовании у больного обнаруживается пневмония или фиброз легких.
- Увеличение лимфатических узлов.
- Поражение периферической нервной системы. При этом начинают проявляться болевые ощущения (покалывание, жжение).
- Повышенная температура;
- Боли в суставах и мышцах.
- Поражение щитовидной железы. Является редким нарушением, при котором начинают проявляться аллергические реакции на различные продукты питания, лекарства и другие средства.
- Увеличенные лимфоузлы (подчелюстные, шейные, затылочные). Не редко наблюдается увеличение печени и селезенки.
- Воспаление сосудов, которое протекает на форе атеросклероза нижних конечностей, вследствие нарушения кровообращения. При нарушении функционирования сосудов, проявляются кожные заболевания (сыпь), которые сопровождаются зудом, жжением и повышенной температурой.
Нередко на фоне описываемой нами патологии у больных развивается повышенная индивидуальная чувствительность к некоторым лекарственным препаратам, в частности, к нестероидным противовоспалительным лекарственным средствам, некоторым антибиотикам (пенициллину), препаратам так называемой базисной терапии, цитостатикам.
Диагностика
Жжение глаз и сухость полости рта не всегда может означать, что речь идет о наличии у человека данного синдрома. Диагностировать синдром Шегрена можно лишь при наличии воспалительного поражения желез. Однако встречаются случаи, когда к подобному результату могут привести различные заболевания обмена веществ, при которых значительно снижается секреция слюны (чаще всего это сахарный диабет).
Вследствие этого у людей старческого возраста преждевременно снижаются функции слюнных и слезных желез. Данные формы сухости глаз и рта не имеют никакого отношения к синдрому Шегрена. Также для диагностики данного синдрома проводится исследование тканей. Для данного вида исследования берут небольшие фрагменты слизистой оболочки полости рта, которые исследуют при помощи микроскопа. Таким образом устанавливается поражение слизистых желез.
Осложнения
Распространенные последствия синдрома Шегрена:
- лимфомы (новообразования, поражающие кровь, лимфоузлы);
- васкулит (воспалительный процесс в сосудах, который может возникать повсеместно);
- присоединение вторичной инфекции;
- развитие онкологических заболеваний;
- угнетение кровообразования, сокращение в крови лейкоцитов, эритроцитов и/или тромбоцитов.
Если больной долго не предпринимает никаких мер по лечению заболевания Шегрена или ему была назначена неверная терапия, патология прогрессирует, приводя к серьезным нарушениям работы органов и систем.
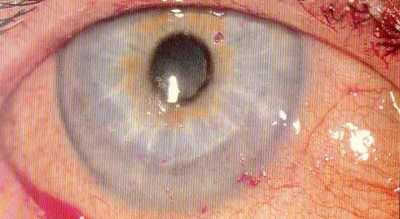
Лечение синдрома Шегрена
При наличии синдрома Шегрена лечение проводится в зависимости от стадии заболевания и наличия системных проявлений.
В целях стимуляции функции желез проводится:
- капельное введение контрикала.
- подкожное введение галантамина.
- с общеукрепляющей целью проводятся курсы витаминотерапии.
- в качестве симптоматического лечения назначаются «искусственные слезы» (капли в глаза) — с низкой вязкостью — Лакрисифи (200-250руб), Слеза натуральная (250 руб), средней вязкости Лакрисин, высокой вязкости Офтагель 180 руб, Видисик 200 руб, Лакропос 150 руб.
На начальных этапах при отсутствии поражения других систем и невыраженных лабораторных изменениях назначаются длительные курсы глюкокортикостероидов (преднизолон, дексаметазон) в небольших дозах.
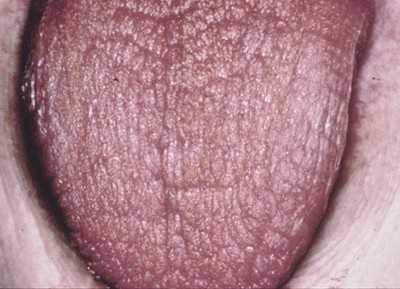
Если симптоматика и лабораторные показатели значительно выражены, но нет системных проявлений, к кортикостероидам добавляются цитостатические иммунодепрессивные препараты — циклофосфамид, хлорбутин, азатиоприн. Поддерживающая терапия проводится этими же средствами в течение нескольких лет.
- При наличии симптомов системного поражения независимо от стадии заболевания сразу назначаются в высоких дозах кортикостероиды и иммунодепрессанты в течение нескольких дней с постепенным переводом на поддерживающие дозы.
- При генерализованном полиневрите, васкулите, поражении почек и других тяжелых проявлениях заболевания к вышеперечисленному лечению добавляются такие методы, экстракорпоральное лечение — плазмаферез, гемосорбция, плазменная ультрафильтрация.
- Остальные препараты назначаются в зависимости от осложнений и сопутствующих заболеваний — холецистит, гастрит, пневмония, эндоцервицит и др.
В определенных случаях необходимо соблюдение диетического питания и ограничение физических нагрузок.
Профилактика
Для профилактики нарушения необходимо соблюдать все указания лечащего врача для предотвращения обострений. Также необходимо соблюдать несколько простых правил:
- ограничить нагрузки на органы зрения;
- если синдром возник вследствие другого заболевания, нужно лечить причину возникновения.
- избегать стрессовых ситуаций.
Прогноз
Синдром Шегрена может повреждать жизненно важные органы с переходом в стабильное состояние, постепенным прогрессированием или, наоборот, длительной ремиссией. Такое поведение характерно и для других аутоиммунных заболеваний.
- Некоторые больные могут иметь слабо выраженные симптомы сухости глаз и ротовой полости, тогда как у других развиваются серьёзные осложнения. Одним пациентам полностью помогает симптоматическое лечение, другим приходится постоянно бороться с ухудшением зрения, постоянным дискомфортом в глазах, часто рецидивирующими инфекциями ротовой полости, отеком околоушной слюнной железы, затруднением жевания и глотания. Постоянный упадок сил и суставная боль серьёзно снижают качество жизни. У части пациентов в патологический процесс вовлекаются почки — гломерулонефрит, ведущий к протеинурии, нарушению концентранционной способности почек и дистальному почечному тубулярному ацидозу.
- Больные синдромом Шегрена имеют более высокий риск возникновения неходжкинской лимфомы по сравнению со здоровыми людьми и людьми, больными другими аутоиммунными заболеваниями. У около 5 % пациентов развивается та или иная форма лимфомы.
Кроме того, установлено, что у детей женщин, больных синдромом Шегрена во время беременности, более высокий риск развития неонатальной красной волчанки с врожденной блокадой сердца.
Читайте также:
- ЭКГ при хронической обструктивной болезни легких (ХОБЛ). ЭКГ при легочной эмболии
- Синдром Дюпюитрена (Dupuytren)
- Лингвальная дуга Мершона. Характеристика дуговых аппаратов.
- Причины дегенеративных заболеваний межпозвонкового диска (остеохондроза)
- КТ, МРТ при ограниченной трансляции мыщелка височно-нижнечелюстного сустава (ВНЧС)