Синдром Камурати-Энгельманна (Camurati-Engelmann) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 21.01.2026
ЭНГЕЛЬМАННА СИНДРОМ (синдром Энгельмана - Камурати описан итальянским ортопедом M. Camurati, затем немецким хирургом и ортопедом G. Engelmann; синонимы - врожденные системные диафизарные гиперостозы, диафизарная дисплазия) - наследственное системное заболевание скелета, характеризующееся утолщением кортикального слоя длинных трубчатых костей с соответствующим сужением костномозговой полости (возможно также вовлечение в процесс ребер, позвонков, лопаток, ключиц, костей основания черепа, костей таза, кистей и стоп). Манифестирует чаще до 10 лет мышечной слабостью, неустойчивой походкой, быстрой утомляемостью, ноющими болями в спине и конечностях, сколиозом, поясничным лордозом, контрактурами суставов, плоскостопием. Утолщение костей скелета при осмотре обнаруживается не всегда; диагноз устанавливается на основании типичной рентгенологической картины: симметричное веретенообразное утолщение диафизов длинных трубчатых костей, резкое утолщение трабекул губчатого вещества. Возможное осложнение - формирование контрактур. Тип наследования - аутосомно-доминантный. При выраженном болевом синдроме возможно применение глюкокортикоидов.
M. Camurati. Di uno raro caso di osteite simmetrica ereditaria degli arti inferiori. La Chirurgia degli organi di movimento, 1922; 6: 662-665.
G. Engelmann. Ein Fall von Osteopathia hyperostotica (sclerotisans) multiplex infantilis. Fortschritte auf dem Gebiete der Rontgenstrahlen und der Nuklearmedizin, 1929; 39: 1101-1106.
Энциклопедический словарь по психологии и педагогике . 2013 .
Полезное
Смотреть что такое "ЭНГЕЛЬМАННА СИНДРОМ" в других словарях:
КАМУРАТИ - ЭНГЕЛЬМАННА СИНДРОМ — - см. Энгельманна синдром … Энциклопедический словарь по психологии и педагогике
Камурати — Энгельманна болезнь — I Камурати Энгельманна болезнь (М. Camurati, итальянский врач 20 в., G. Engelmann, австрийский хирург, родился в 1876 г.; синоним: врожденный системный гиперостоз, прогрессирующая диафизарная дисплазия) наследственное системное заболевание костей … Медицинская энциклопедия
Остеохондродисплази́я — (греч. osteon кость + chondros хрящ + Дисплазия группа врожденных наследственных заболеваний скелета, характеризующихся нарушением эмбрионального развития костно хрящевой системы и сопровождающихся системным поражением скелета. В ряде случаев О.… … Медицинская энциклопедия
Гиперостоз — I Гиперостоз (hyperostosis; греч. hyper + osteon кость + ōsis) патологическое увеличение содержания костного вещества в неизмененной костной ткани. Является ответной реакцией костной ткани на большую нагрузку или симптомом, например остеомиелита … Медицинская энциклопедия
Кость — I Кость (os) орган опорно двигательного аппарата, построенный преимущественно из костной ткани. Совокупность К., связанных (прерывно или непрерывно) соединительной тканью, хрящом или костной тканью, образует Скелет. Общее количество К. скелета… … Медицинская энциклопедия
Гиперостоз — (др. греч. ὑπερ «сверх » + ὀστέον «кость» + ōsis) патологическое увеличение содержания костного вещества в неизмененной костной ткани. Является ответной реакцией костной ткани на большую нагрузку или симптомом, например остеомиелита,… … Википедия
КАМУРАТИ-ЭНГЕЛЬМАННА БОЛЕЗНЬ
Камурати-Энгельманна болезнь (М. Camurati, современный итальянский врач; G. Engelmann, австрийский хирург и ортопед, род. в 1876 г.; син.: врожденные системные диафизарные гиперостозы, генерализованные системные гиперостозы с инволютивной миопатией, гиперпластический периостит, прогрессирующая диафизарная дисплазия, множественный диафизарный склероз, болезнь Энгельманна) — наследственный системный диафизарный гиперостоз у детей с инволютивной миопатией, относящийся к синдромам генерализованного остеосклероза. Чаще всего поражаются бедренные, плечевые и большеберцовые кости, в остальных костях изменения умеренные. Реже других поражаются передние и средние отделы основания черепа, лобные кости, тела позвонков, ногтевые фаланги.
Камурати-Энгельманна болезнь относят к редким семейным наследственным нарушениям развития костей. Предполагается доминантная передача мутантного гена. Описаны также спорадические случаи: Синглтон (Е. Singleton, 1956) наблюдал системное утолщение стенок сосудов, питающих кости, с сужением просвета артерий разного калибра, что дает основания приписать роль патогенетического фактора редуцированному кровоснабжению костной ткани.
Костная ткань в типичных случаях с выраженным остеосклерозом (см.), с сохранением общей структуры коркового и губчатого вещества, но резким сужением каналов остеона (гаверсовых каналов) и просветов сосудов. Костные балки, как правило, значительно утолщены.
Рис. 1. Больной системным гиперостозом: характерный вид длинных утолщенных бедер цилиндрической формы, саблевидных голеней, сгибательных контрактур в крупных суставах.
Ранний период жизни больного ребенка характеризуется замедленным развитием, небольшой прибавкой в весе, гипотрофией мышц конечностей. Ходить такие дети начинают на 3— 4-м году жизни. В дальнейшем наблюдается прогрессирующая мышечная слабость и развитие так наз. утиной походки. При тщательном осмотре можно обнаружить утолщение конечностей, приобретающих цилиндрическую форму без выпуклостей и углублений; суставы почти не выделяются (рисунок 1). Кожа над утолщенными костями натянута, отмечается уменьшение объема мышц (инволютивная Миопатия). Постоянным симптомом являются локализующиеся в диафизах больших трубчатых костей интенсивные, ноющие, «грызущие» боли, обычно связанные с периодом активации болезни и усиливающиеся при физ. нагрузке, тутоподвижность суставов и скованность движений. Описаны случаи непропорционального роста больных с выраженным удлинением конечностей, нарушением сухожильных рефлексов. Редко может развиться глухота и парез лицевого нерва по периферическому типу. Изменений крови и эндокринных органов обычно нет, однако М. В. Волков в 1974 г. сообщил о больной 19 лет, у к-рой были обнаружены инфантилизм, резкая гипоплазия гениталий и отсутствие менструаций, гипопластический субнанизм с отставанием умственного развития.
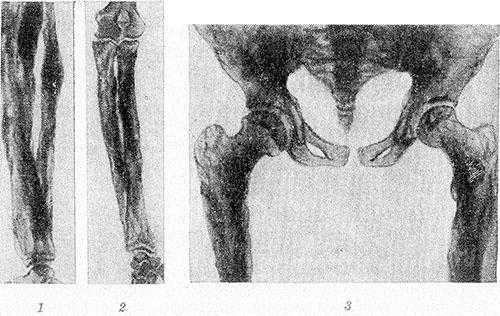
Рис. 2. Рентгенограммы костей голени (1), предплечья (2), таза и верхней трети бедер (3) больного системным гиперостозом: длинные трубчатые кости цилиндрически и булавовидно утолщены, корковый слой неплотный, резко утолщенный, имеет неравномерную пятнистую структуру с неровными контурами; кости таза утолщены.
Рентгенологически определяется симметричный гиперостоз трубчатых костей скелета. Пораженные кости громоздки, их диафизы утолщены в 2—3 раза, костно-мозговой канал равномерно сужен, но никогда не исчезает полностью. Эпифизы не изменены. Корковый слой значительно утолщен кнаружи и кнутри, с явлениями остеосклероза, но структура его не нарушена. Рисунок губчатого вещества изменен — костные балки грубы, толсты, плотны и обрывисты (рис. 2). В детском возрасте структурный рисунок может быть неравномерным, иметь рассеянный крупнопятнистый очаговый характер.
Камурати-Энгельманна болезнь следует дифференцировать с деформирующей остеодистрофией Педжета (см. Остоз деформирующий), акромегалией (см.), множественными кортикальными гиперостозами раннего возраста (см. Гиперостоз), болезнью Реклингхаузена (см. Нейрофиброматоз) и др. Важным дифференциально-диагностическим признаком, кроме типичной рентгенол, картины, отсутствия обезображивающих изменений, искривления костей и др., является отсутствие склонности к спонтанным переломам, несмотря на прогрессирование процесса.
Лечение симптоматическое. Назначают общеукрепляющие медикаменты и процедуры, при болях — анальгетики.
Течение заболевания медленное и доброкачественное. Прогноз для жизни благоприятный.
Библиография: Волков М. В. Болезни костей у детей, М., 1974; Каменева Т. И. и Трофимова 3. А. О врожденных системных, диафизарных гиперостозах у детей, Вопр. охр. мат. и дет., т. И, № 11, с. 89, 1966; Рейнберг С. А. Рентгенодиагностика заболеваний костей и суставов, кн. 1, с. 422, М., 1964; Ярандина М. П. Врожденный системный диафизарный гиперостоз — болезнь Энгельманна, Вестн, рентгенол, и радиол., № 3, с. 82, 1969; Camurati М. Symmetrical hereditary osteitis, Chir. Organi Mov., v. 6, p. 662, 1922; Engelmann G. Ein Fall von Osteopathia hyperostotica (Sclerotisans) multiplex infantilis, Fortschr. Rontgenstr., Bd 39, S. 1101, 1929.
Камурати — Энгельманна болезнь
Камурати — Энгельманна болезнь I Камура́ти — Э́нгельманна бо́лезнь (М. Camurati, итальянский врач 20 в., G. Engelmann, австрийский хирург, родился в 1876 г.; синоним: врожденный системный гиперостоз, прогрессирующая диафизарная дисплазия)
наследственное системное заболевание костей, характеризующееся утолщением кортикального слоя длинных трубчатых костей. Относится к Остеохондродисплазиям. Передается по аутосомно-доминантному типу. Описаны спорадические случаи. Частота заболевания не установлена, т.к. нередко оно протекает бессимптомно и выявляется случайно при рентгенологичеком исследовании.
Предполагают, что в основе патогенеза К. — Э. б. лежит первичный порок развития внутрикостных сосудов. Существует мнение, что при К. — Э. б. нарушается обмен кальция и фосфора, изменяется активность фосфатазы. Однако биохимические показатели, характеризующие минеральный обмен костной ткани, при этом заболевании практически не отличаются от нормы.
Первое проявление К. — Э. б. возможно в любом возрасте. Клиническое течение отличается полиморфизмом — от бессимптомных до ярко выраженных форм с болевым синдромом и контрактурой суставов. Заболевание характеризуется медленным прогрессированием симптомов. В одних случаях на первый план выступает болевой синдром. Боли постоянные, чаще ноющие, локализуются в проекции пораженных длинных трубчатых костей. Другой вариант клинического течения встречается чаще и характеризуется появлением болевого синдрома в более поздние сроки. Наиболее выражено утолщение бедренной кости и костей голени с увеличением объема всего пораженного сегмента. Вследствие этого кожа выглядит истонченной, бледной и малоподвижной, заметен ее венозный рисунок. Обращает на себя внимание цилиндрическая форма пораженных сегментов. Движения в суставах обычно не нарушены. В ряде случаев отмечаются сгибательные контрактуры в тазобедренных и коленных суставах. Другие суставы в процесс вовлекаются очень редко. Утолщение сегментов конечностей сочетается с их удлинением. На этом фоне дистальные сегменты, которые поражаются значительно реже, выглядят уменьшенными в размере. Пальпация костей безболезненна. Наряду с изменениями опорно-двигательного аппарата нередко нарушается общее состояние, отмечаются отставание в физическом развитии, слабость мышечной системы. При К. — Э. б. не бывает избытка массы тела.
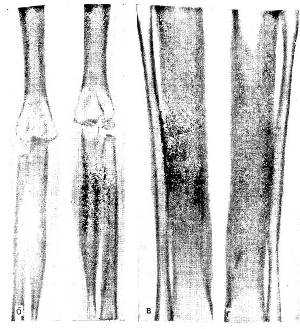
У детей младшего возраста гиперостоз в основном носит несимметричный, очаговый характер. Участки с утолщением кортикального слоя чередуются с участками, где эти изменения отсутствуют. С возрастом гиперостоз становится симметричным и приобретает генерализованный характер. Наиболее часто поражаются длинные трубчатые кости, хотя вовлечение коротких костей не является исключением. Утолщение кортикального слоя приводит к сужению костномозгового канала (рис.), но в отличие от остеопетроза (мраморной болезни) он никогда полностью не закрывается. В губчатой кости при К. — Э. б. наблюдается утолщение трабекул и грубая их структура.
Дифференциальный диагноз проводят с другими гиперостозами — младенческим кортикальным гиперостозом, мелореостозом, остеопетрозом. У детей младшего возраста из-за очагового и несимметричного характера поражения возникает необходимость дифференциальной диагностики с сирингомиелией.
Лечение в основном симптоматическое, направлено на уменьшение болевого синдрома. Прогноз для жизни благоприятный, в отношении функций конечностей — зависит от тяжести течения заболевания. См. также Гиперостоз.
II Камура́ти — Э́нгельманна боле́знь (М. Camurati, итал. врач XX в.; G. Engelmann, р. 1876 г., австрийский хирург; син.: гиперостоз генерализованный, гиперостоз системный диафизарный врожденный, дисплазия диафизарная прогрессирующая, остеосклероз системный наследственный с миопатией, Энгельманна болезнь)
наследственная болезнь, характеризующаяся гиперостозом диафизов бедренных, плечевых, большеберцовых, реже других костей, сопровождающимся уменьшением объема мышц и тугоподвижностью суставов; наследуется по аутосомно-доминантному типу.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Энгельмана синдром
Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки.
При синдроме Ангельмана отсутствуют некоторые гены из 15-й хромосомы (в большинстве случаев — частичная делеция либо мутация 15 хромосомы). При синдроме Ангельмана страдает материнская хромосома; в случае повреждения отцовской хромосомы возникает синдром Прадера-Вилли.
Кариотип 46 ХХ или ХУ, 15р-. Каждая хромосома состоит из короткого и длинного плеча. Короткое плечо называется «p», а длинное плечо — «q». Область «q» разделена на пронумерованные сегменты; сегмент q11-q13 относится к области, расположенной в середине хромосомы 15. Эта область состоит из 5-10 миллионов молекул ДНК, так что она включает много генов. Именно в этой области кроется причина возникновения синдрома Ангельмана. Обычно синдром вызывается спонтанным хромосомным дефектом, когда большая смежная область из 3-4 миллионов молекул ДНК отсутствует в области q11-q13 15-й хромосомы.
Согласно исследованиям доктора Арта Бодэ (Бэйлорский медицинский колледж), причиной возникновения синдрома Ангельмана может являться мутации в гене UBE3A. Продукт этого гена, ферментный — компонент сложной системы деградации белков. Синдром назван по имени британского педиатра Гарри Ангельмана, впервые описавшего его в 1965 г.
Частота встречаемости, по разным данным, — 1 : 10 000-20 000 живорожденных младенцев. (Однако, согласно данным Центра развития человека и отклонений в развитии (университет Вашингтона, США), можно предполагать, что доля людей с синдромом Ангельмана в действительности намного больше статистической.)
Содержание
Особенности
Для синдрома Ангельмана характерны:
- В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес;
- задержка в развитии навыков общей моторики (умение сидеть, ходить);
- задержка речевого развития, неразвитая речь (у всех детей);
- дети больше понимают, чем могут сказать или выразить;
- дефицит внимания и гиперактивность;
- сложности с обучением; (80 % случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана происходит вторичная (симптоматическая) общая эпилепсия;
- необычные движения (мелкий тремор, хаотические движения конечностей);
- частый смех без повода;
- ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками;
- размер головы меньше среднего, нередко с уплощением затылка;
- иногда особые черты лица — широкий рот, зубы с промежутками между ними, выдающийся вперед подбородок, высунутый наружу язык);
- нарушения сна; (косоглазие) в 40 % случаев; (искривление позвоночника) в 10 % случаев;
- повышенная чувствительность к высокой температуре;
- бывают сильно увлечены играми с водой.
Диагностика
Синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Родители и врачи должны обратить внимание на случаи мелкого тремора, хаотические, порывистые движения конечностей, походку с негнущимися ногами; в ряде случаев специфическое выражение лица, слишком частый смех.
Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15q11-q13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE3A.
Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана. Наука пока не дает ответ на вопрос, как это возможно.
Лечение
Синдром Ангельмана является врожденной генетической аномалией и, следовательно, не может быть излечен.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом.
В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0.3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана.
Нарушения стула регулируются назначением легких слабительных.
Приступы лечатся так же, как эпилепсия. Дети с синдромом Ангельмана часто испытывают больше одного типа приступов. Показана электроэнцефалография.
Нежелательное поведение. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложность в общении с другими людьми. Врачи США показывают, что для аутичных детей внутривенные инъекции гормона секретин (найденного в поджелудочной железе) успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана.
Риски
Оценка риска повторного рождения ребенка с синдромом Ангельмана у тех же родителей очень сложна, необходима консультация профессионального генетика. Считается, что обычная делеция является спонтанной, риск повтора меньше 1 %. В случае молекулярной микроделеции в 15q11-q13, если она наблюдается и у матери, риск теоретически до 50 %. Мутации внутри гена UBE3A могут быть случайными и неунаследованными, в этой ситуации риск повтора
Перспективы развития
Дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарем 5-10 слов. При этом дети/люди с синдромом Ангельмана любят общаться с людьми, играть, как правило, они дружелюбны и милы.
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам [1] , направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты.
Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы).
C возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки.
Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью.
Некоторые люди с синдромом Ангельмана способны есть ножом и вилкой, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний».
Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом.
Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом.
Диагностика и лечение болезни Камурати-Энгельманна
Болезнь (синдром) Камурати-Энгельманна - редко встречающееся генетическое нарушение, которому свойственны пороки и прогрессирующее увеличение ширины диафиза длинных костей. Главные проявления этого патологического состояния состоят из: болевого синдрома в костях, особенно в нижних конечностях, скелетное аномальное развитие либо слабость и гипопластические явления в разных группах мышц. Болевое ощущение и слабость мышечных волокон нижних конечностей иногда приводит к сильно шаткой походке.
Эпидемиологические данные
Синдром Камурати-Энгельманна относится к редчайшим нарушениям, он формируется у людей разных полов. Обычно, подобное нарушение становится визуально заметным у людей в тридцатилетнем возраст. Зачастую начальные проявления этого патологического состояния начинают возникать десять лет, которых в медицинской статистике насчитывается около ста случаев.
Этиологические факторы
Болезнь имеет аутосомно-доминантным тип наследования. Некоторые источники утверждают, что большая часть заболевших подобным синдромом обусловлена расстройствами либо мутационными явлениями в гене измененного фактора роста бета-1, который располагается на длинноплечевом участке девятнадцатой пары хромосомы.
Дифференциальная диагностика
Синдром необходимо дифференцировать со следующими нарушениями:
- Оребрение - особо редкое нарушение с наследственной предрасположенностью, которое характеризуется чрезмерным повышением прочности (склеротическое изменение) диафизарного участка длинных костей. В основном начинается подобное патологическое состояние по истечении пубертатного периода. Взаимосвязь между этими расстройствами до конца не изучена. Некоторая часть авторов предполагают, что это - похожие, но совершенно разные нозологические единицы. Другая же часть считает, что эти болезни являются различными фенотипами одного и того же нарушения. - патология скелетной системы, которая имеет медленно-прогрессирующее течение, характеризуется чрезмерно быстрым распадом и образованием костных структур, что ведет к возникновению плотной и хрупкой костной ткани. Главным признаком выступает болевой синдром в костях. Эта патология обычно формируется у людей среднего возрастного периода и у пожилых. Зачастую патологический процесс развивается в позвоночном столбе, черепе, тазу и в костях нижних конечностей. Часто патология имеет бессимптомное и мягкое течение. Клинические признаки характеризуются расплывчатостью. Описанную болезнь зачастую путают с многочисленными иными патологиями костной ткани.
- Краниодиафизарная дисплазия в тяжелой степени. Этому нарушению свойственный значительный рост (гиперостоз) костей головы и лица в сочетании с интенсивным склерозом. Сдавление черепно-мозговых нервов приводит к серьезным последствиям. Прогрессирующая олигофрения характерна этой патологии. Тяжесть заболевания определяется уже при рождении и по всей вероятности относится к группе патологий с аутосомно-рецессивным типом наследования.
Симптоматическая характеристика
Клинические характеристики синдрома Камурати-Энгельманна значительно различаются в каждом отдельном случае, даже в семейном очаге между заболевшими членами семьи. Возрастная показатель начала болезни также имеет высокое разнообразие. В большей части проявляется эта патология в раннем детском возрасте.
Описанному синдрому свойственно болевое ощущение в костных структурах, особенно в бедренных костях. В основной части слабость в мышечных волокнах и боль в нижних конечностях становятся причиной шаткости походки. А в более тяжелых случаях при сильной выраженности эти проявления могут совершенно обездвижить заболевшего. Примерно в 50% случаев наблюдается склеротические процессы в костях на уровне основания черепа. Такое поражение ведет к расстройствам в зрительной, слуховой системах и к параличу лица.
В особых вариантах утомляемость, боли в голове, сниженный аппетит, чрезмерное выпячивание глазного яблока, увеличение печени, уменьшение подкожно-жировой клетчатки либо отечность голеней может стать дополнительными проявлениями патологического состояния.
Лечебные мероприятия
В лечебном комплексе предусматривается применение небольших дозы кортикостероидных средств — кортизон, преднизолон либо дефлазакорт. Они показаны для облегчения симптомов у большей части заболевших с этой патологией. Коррекция хирургическим способом (остеотомия) нижних конечностей показана в определенных случаях, подбирается индивидуально. В основном такой способ применяется в глазной хирургии с целью ослабления давления на зрительные нервы. Однако, этот способ не всегда эффективен, и поэтому не всегда рекомендуется.
Читайте также: