Синдром Коутса (Coats) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 22.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Синдром Кушинга: причины появления, симптомы, диагностика и способы лечения.
Синдром Иценко-Кушинга - это сочетание клинических симптомов, вызванных хроническим повышением уровня кортизола или родственных ему кортикостероидов в крови. Болезнь Иценко-Кушинга - это синдром Кушинга, причиной которого служит избыточная гипофизарная продукция адренокортикотропного гормона (АКТГ), как правило, обусловленная небольшой доброкачественной опухолью гипофиза - аденомой. Иногда АКТГ производится опухолью, которая не связана с гипофизом, она может находиться где угодно, чаще - в легких и грудной клетке. Порой злокачественные опухоли хорошо маскируются под железы и начинают вырабатывать гормоны альдостерон и кортизол, что, в свою очередь, приводит к повышению их концентраций в человеческом организме. При этом собственные железы понемногу начинают атрофироваться - таким образом организм старается бороться с избытком гормонов.
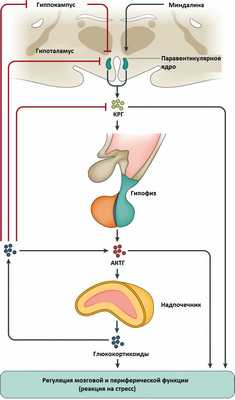
Синдром Иценко-Кушинга также возникает из-за перепроизводства кортизола надпочечниками или употребления больших доз глюкокортикоидных препаратов таких как преднизолон или дексаметазон при лечении ряда болезней (астмы, ревматоидного артрита и некоторых других аутоиммунных патологических состояний). Заболевание может возникнуть в любом возрасте, но чаще всего в 20-40 лет, оно может быть врожденным или приобретенным. Женщины поражаются в 10 раз чаще, чем мужчины.
У пациентов, страдающих алкоголизмом или тяжелыми депрессивными расстройствами, а также во время беременности, иногда наблюдается небольшое повышение уровня гормонов надпочечников и развивается псевдо-синдром Иценко-Кушинга.
Классификация заболевания. Кодирование по МКБ-10
Синдром Иценко-Кушинга (E24):
E24.0. Болезнь Иценко-Кушинга гипофизарного происхождения (гиперсекреция АКТГ гипофизом, гиперадренокортицизм гипофизарного происхождения);
E24.1. Синдром Нельсона;
E24.3. Эктопический АКТГ-синдром;
E24.4. Кушингоидный синдром, вызванный алкоголем;
E24.8. Другие состояния, характеризующиеся кушингоидным синдромом;
E24.9. Синдром Иценко-Кушинга неуточненный.
Симптомы синдрома Иценко-Кушинга
У большинства больных с различными формами гиперкортицизма: АКТГ-зависимыми (болезнь Иценко-Кушинга, аденома гипофиза, АКТГ эктопический синдром) и АКТГ-независимыми формами (аденома коры надпочечника и/или двусторонняя микро-, макроузелковая гиперплазия) клинические проявления заболевания постоянны и зависят от скорости секреции кортизола надпочечниками.
К классическим признакам синдрома Иценко-Кушинга у взрослых относятся «лунообразное» лицо багрово-красного цвета, часто возникают многочисленные угревидные высыпания, центральное ожирение с одновременной потерей жировой ткани на бедрах, ягодицах и руках, истончение кожи и ломкость капилляров, приводящие к легкому и часто спонтанному образованию синяков. За счет неправильного и неравномерного жироотложения происходит необратимая деформация позвоночника, больные сутулятся, происходит нарушение осанки (кифоз, сколиоз). На бедрах, предплечьях, животе можно увидеть растяжки ярко-красного или даже фиолетового цвета, надключичные жировые подушечки и периферические отеки. Часто происходит разрушение костной ткани, отмечается склонность к переломам. У женщин по причине избытка половых гормонов возникают признаки излишнего оволосения по мужскому типу, появляются существенные перебои менструального цикла. У детей самым ранним признаком служит избыточная масса тела при задержке роста.
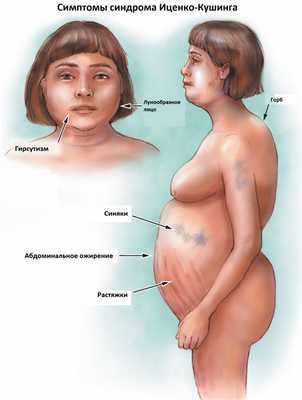
За счет повышенного уровня кортизола могут возникать гипертония, аритмия, поражение сердца и сосудов, высокий уровень глюкозы в крови, снижение зрения, приступы агрессивности, депрессия, нарушения терморегуляции (именно такие больные очень часто потеют, а также могут мерзнуть в жаркую погоду).
Диагностика синдрома Иценко-Кушинга
Ярко выраженный синдром Иценко-Кушинга не представляет особых диагностических трудностей. Достаточно лишь оценить внешний облик человека и провести с ним беседу. Но заболевание с умеренными проявлениями может вызвать у врача ряд проблем. Всегда следует исключать предварительный прием глюкокортикостероидов пациентом (экзогенный синдром Кушинга). Диагноз ставится клинически, а подтверждается данными лабораторных и визуализирующих методов исследований для установления стадии болезни и выяснения первопричины патологии.
Подтверждение избытка кортизола выполняется строго по показаниям врача одним из четырех методов:
- оценка количества кортизола - определение свободного кортизола мочи в двукратных суточных пробах;
Синонимы: Анализ мочи на кортизол; Анализ суточной мочи на свободный кортизол; Кортизол мочи. Hydrocortisone; Urine cortisol; Free Cortisol Urine Test; Urine Cortisaol Test. Краткое описание теста .
Болезнь Коатса (ретинит)
Причины
Развитие болезни Коатса связано с наследственными мутациями в генах, которые вызывают детерминированные аномалии. Для последних стадий ретинита Коатса характерно развитие отслойки сетчатки. Нередко болезнь Коатса не имеет тенденции к прогрессии, но в 20% случаев возникают серьезные осложнения.
Симптомы и стадии ретинита
В основе классификации болезни Коатса лежит возрастной критерий (зависит от возраста, в котором возникли начальные проявления ретинита). Согласно этому выделяют ювенальную форму (5-10 лет), сенильную форму, которая является довольно редкой. Последняя форма ретинита характеризуется поздним появлением первых симптомов, отложением холестерина помимо твердого экссудата.
Согласно стадиям заболевания выделяют следующие разновидности болезни Коатса:
- Начальная стадия часто протекает бессимптомно или же сопровождается неспецифическими признаками (искажение предметов, одностороннее появление пятен, краснота глаза, косоглазие). Чаще в этой стадии ретинит выявляют при плановом обследовании у офтальмолога. Врач обнаруживает в области глазного дна много небольших расширенных сосудов (микроаневризмы, телеангиэкстазии). При офтальмоскопии начальную стадию подразделяют на два подтипа: 1а, при котором выявляют большое количество небольших очагов твердого экссудата в периферической области глазного дна, 1б стадия сопровождается появлением твердого экссудата в центральной области, в том числе в макулярной зоне.
- Развитая стадия сопровождается значительным снижением зрительной функции. В области глазного дна доктор отмечает увеличение количества очагов твердого экссудата. Они склонны к слиянию, что сопровождается формированием туморообразного депозита (опухолеподобных образований). Мелкие сосуды расширяются и изменяются, они становятся похожи на красные бусы. В локальных областях сетчатки могут возникать хоны отслойки.
- При далеко зашедшей стадии зоны отслойки увеличиваются в размере, становятся пузыревидными. Также возникает воспалительная реакция в области сосудистой оболочки (увеит), катаракта и новообразование сосудов в радужной оболочке (рубеоз).
- Терминальная стадия болезни Коатса приводит к неоваскулярной глаукоме (4а стадия) и субатрофии глаза (4б стадия).
В раннем возрасте часто наблюдается тяжелое течение ретинита Коатса. При этом симптомы, начавшиеся в младенчестве, быстро нарастают. При таком течении ретинита к возрасту 5-6 лет формируется полная отслойка сетчатки и субатрофия глазного яблока.
Диагностика и лечение
Пациентам с подозрением на болезнь Коатса требуется выполнить ряд диагностических процедур:
- Биомикроскопия и нептямая офтальмоскопия. При этом врач тщательно изучает область глазного дна, оценивает наличие, количество и степень распространения очагов экссудации.
- При флуоресцентной ангиографии можно оценить наличие сосудистых аномалий, а также области сетчатки с нарушенным кровотоком.
- Периметрия необходима при оценки динамики ретинита.
- Оптическую когерентную томографию выполняют с целью детальной оценки очагов экссудации в макулярной зоне.
- УЗИ с цветной допплерографией, картированием, МРТ и КТ назначают в особых случаях, когда имеются туморообразные депозитивы. Это необходимо для дифференциальной диагностики ретинита Коатса и злокачественного новообразования в области сетчатки (ретинобластома, медуллоэпителиома), так как заболевания эти имеют сходную клиническую картину.
Все пациенты с диагностированным ретинитом Коатса должны наблюдаться офтальмологом на всех стадиях патологии (начиная от бессимптомного периода и заканчивая полным отслоением сетчатой оболочки). С целью лечения проводят спайку измененных сосудов, что предотвращает образование новых очагов экссудации.
В зависимости от степени выраженности симптомов ретинита для этого применяют:
1. Лазерную коагуляцию на ранних стадиях заболевания.
2. Криокоагуляция показана при незначительных областях отслойки.
3. Витреоретинальные вмешательства выполняют в случаях полной отслойки.
Если своевременно не провести лечение ретинита Коатса, то прогноз неблагоприятный, так как со временем происходит полная отслойка сетчатки и потеря зрительной функции. В литературе можно встретить клинические описания самопроизвольного излечения и рассасывания очагов экссудации. Однако даже в этом случае зрение к пациенту не возвращается.
В медицинском центре «Московская Глазная Клиника» все желающие могут пройти обследование на самой современной диагностической аппаратуре, а по результатам - получить консультацию высококлассного специалиста. Клиника консультирует детей от 4 лет. Мы открыты семь дней в неделю и работаем ежедневно с 9 ч до 21 ч. Наши специалисты помогут выявить причину снижения зрения, и проведут грамотное лечение выявленных патологий.
Что такое синдром Лойса - Дитца у ребенка?

Синдром Лойса-Дитца — это новая болезнь, которую описали менее 20 лет назад. Эксперты утверждают, что даже не все врачи знакомы с таким заболеванием и нередко неверно диагностируют его как синдром Марфана. Что же это за новый синдром и как он проявляется у детей?
Что такое синдром Лойса-Дитца?
Это генетическое заболевание, которое разрушает соединительные ткани. Те самые, что поддерживают и придают гибкость мышцам, кровеносным сосудам и костям.
Изменения в соединительных тканях влияют на формирование костей, а также на развитие артерий. Обычно симптомы проявляются в подростковом возрасте, но постановка точного диагноза может быть затруднена до взрослого возраста.
Каковы генетические причины болезни?

Существует пять различных версий синдрома Лойса-Дитца в зависимости от того, какой из генов мутировал. Хотя каждая версия патологии вызывается мутацией разных генов, но все пять из этих генов вовлечены в один и тот же клеточный сигнальный путь — путь трансформирующего бета фактора роста.
Этот путь контролирует, как клетки функционируют во время развития ребёнка. Он также помогает в развитии внеклеточного матрикса — сети белков и других молекул, соединяющих клетки.
Мутировавшие гены производят «сломанные» белки, которые не работают должным образом. Симптомы синдрома Лойса-Дитца являются результатом этих нарушений.
Все мутации при Лойса-Дитца являются доминантными, а это означает, что ребёнку достаточно унаследовать только одну мутировавшую копию от одного родителя, чтобы иметь синдром.
Однако по новым данным исследования самих первооткрывателей этого синдрома, врачей Dietz H и Loeys B, в 75% всех случаев данные мутации возникают спонтанно, поэтому семейного анамнеза болезни может и не быть.
Каковы симптомы синдрома Лойса-Дитца?
Четыре основные характеристики предполагают, что у ребёнка это заболевание. Эти особенности обычно не возникают все вместе при других расстройствах соединительной ткани как основные характеристики. Признаки включают:
- Аневризмы (расширение артерий), которые можно наблюдать с помощью методов визуализации. Они чаще всего наблюдаются в корне аорты (основание артерии, ведущее от сердца), но их можно увидеть в других артериях по всему телу.
- Артериальная извилистость (скрученные или спиральные артерии), чаще всего встречающиеся в сосудах шеи.
- Гипертелоризм — необычно широко расположенные глаза на лице.
- Разделенный или широкий небный язычок, или увула (маленький кусочек плоти, который свисает в задней части рта).
Важно: эти симптомы не всегда наблюдаются у всех пациентов, однако присутствуют в большинстве случаев.
Кроме этого набора у детей с диагнозом синдрома Лойса-Дитца могут быть отличительные признаки во внешности: плоские щеки (маларская гипоплазия), краниосиностоз (раннее зарастание родничков), расщелина неба, синеватые склеры глаз, маленький или отсутствующий подбородок, очень длинные пальцы и/или их срастание, деформации, сколиоз, полупрозрачная, очень мягкая кожа с быстро возникающими синяками и широкими шрамами
Синдром Лойса-Дитца также вызывает врожденные пороки сердца, грыжи, близорукость, частые пищевые аллергии и болезни желудка и кишечника, астму, мигрень и так далее.
Как ставят диагноз «синдром Лойса-Дитца» у ребёнка?

Если есть подозрение на синдром Лойса-Дитца, в первую очередь рекомендуется консультация генетика, который знаком с расстройствами соединительной ткани. Во время первоначального визита будет проведен сбор семейного анамнеза и истории болезни, комплексное физическое обследование для оценки скелетных, черепно-лицевых и связанных с кожей признаков.
Если подозрение на болезнь продолжается, должна быть выполнена эхокардиография (ультразвуковая визуализация сердца), чтобы оценить, есть ли увеличение аорты и/или другие структурные дефекты сердца, которые согласуются с диагнозом. Консультация с кардиологом потребуется, чтобы помочь интерпретировать результаты обследования.
Врач также может предложить дальнейшую визуализацию артерий по всему телу. Ее проводят с помощью КTA ( КТ-ангиография, или обследование сосудов при компьютерной томографии) или МРА (магнитно-резонансная томография в ангиорежиме) всего артериального дерева (голова, шея, грудь, таз и живот). Эти исследования помогут обнаружить аневризмы в других артериях.
Генетическое тестирование на мутации в генах TGFBR1, TGFBR2, SMAD3, TGFB2 и TGFB3 проводятся, если есть высокое подозрение наличия синдрома. Если у ребёнка обнаружена мутация гена, обычно рекомендуется проверить родителей на ту же мутацию, чтобы дать точную информацию о риске рецидива.
Ожидаемая продолжительность жизни людей с синдромом Лойса-Дитца по данным экспертов (Genetics in Medicine) оценивается примерно в 37 лет, но некоторые люди с этим расстройством могут жить намного дольше — иногда до семидесяти лет.
Доступное лечение синдрома Лойса-Дитца: что возможно?
Терапия при синдроме Лойса-Дитца зависит от симптомов ребёнка: если что-то беспокоит или грозит развитием патологии, специалисты концентрируют усилия в этих направлениях. Российские ученые из Санкт-Петербурга уточняют, что основной командой специалистов для ребёнка с такой болезнью должны стать педиатр, кардиолог, невролог и ортопед, а обследование сосудов сердца надо проходить как минимум ежегодно, даже если ничего не беспокоит.
К общим целям лечения относятся:
- Снижение нагрузки на артерии.
- Управление различными скелетными и мышечными проблемами, которые развиваются, и болью, которую они могут вызвать.
- Управление любыми проблемами иммунной системы — с помощью образа жизни, диеты, лекарств, вакцинации.
В случаях, когда у ребёнка развивается аневризма, врач будет в зависимости от размера и локализации советовать либо оперативное лечение, либо наблюдение.
Если у ребёнка синдром Лойса-Дитца, ему следует избегать напряженных, повторяющихся действий, таких как приседания и отжимания. Однако сама физическая нагрузка полезна, с учетом основной рекомендации: на пике усилий ребёнок должен дышать достаточно эффективно, чтобы спокойно поддерживать разговор. Полезна также целенаправленная закалка организма. Как ее проводить, читайте в статье «Закаливание детей: виды, принципы, рекомендации».
Синдром Маршалла (PFAPA синдром)
Что такое синдром Маршалла, клиническая картина, симптомы, лечение.

3.90 (Проголосовало: 20)
- Что это такое?
- Клиническая картина
- Диагностика
- Важными диагностическими критериями являются:
- Может ли ребенок посещать детский сад или школу?
- Можно ли водить ребенка в спортивную секцию?
- Можно ли ребенку делать прививки?
Что это такое?
Синдром Маршалла - синдром периодической лихорадки, афтозного стоматита, фарингита и шейного лимфаденита.
Заболевание проявляется в детском возрасте, чаще у мальчиков.
Характеризуется периодическими эпизодами лихорадки (38 - 41ºC) с регулярными интервалами от 2 до 8 недель и клиническими проявлениями афтозного стоматита, фарингита, шейного лимфаденита. Между эпизодами клинические проявления отсутствуют.
На сегодняшний день причина заболевания не известна, возможно, имеет генетический характер. Распространённость также не известна.
Клиническая картина
Периодическая лихорадка - главный признак, по которому можно заподозрить синдром Маршалла. На фоне полного здоровья у ребенка повышается температура до 38 - 41 ºC. И держится от 2 до 7 дней, иногда до 10 дней и исчезает самостоятельно. Как правило, ребенку назначают антибиотики и жаропонижающие препараты, на фоне которых нет эффекта.
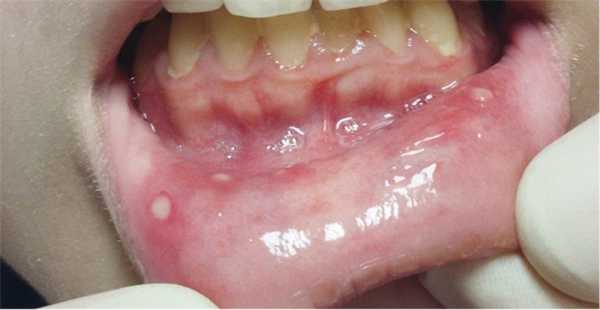
Афтозный стоматит - язвы располагаются на слизистой оболочки губ и щёк. Этот признак проявляется примерно у 40-80% пациентов.
![]()
Фарингит с экссудативным компонентом на миндалинах встречается у 65-100% пациентов.
Шейная лимфаденопатия (увеличение лимфоузлов) встречается 60-100% пациентов. Шейные лимфоузлы могут быть уплотнены, болезненны при пальпации.
Диагностика
К сожалению, на данный момент специфической диагностики нет. Диагноз ставится на основании жалоб и эпизодов лихорадки.
В общем анализе крови могут быть незначительные изменения - увеличение СОЭ, нейтропения (уменьшение количества нейтрофилов). Посевы на микрофлору из горла часто не дают никаких результатов при данном заболевании.
PFAPA синдром - это диагноз исключения. В первую очередь исключаются другие причины рецидивирующей лихорадки: инфекция, воспалительные заболевания кишечника, лихорадка при лимфоме Ходжкина, циклическая нейтропения и другие.
Важными диагностическими критериями являются:
- Более 3 эпизодов лихорадки продолжительностью до 5 дней и возникающих регулярно на фоне полного здоровья;
- Фарингит и лимфаденопатия или афтозные язвы;
- Хорошее здоровье между эпизодами и нормальный рост;
- Разрешение симптомов в течение нескольких часов после лечения преднизолоном в виде однократной дозы или двух доз с интервалом от 12 до 48 часов.
Лечение синдрома Маршалла
Для лечения в период обострения используют глюкокортикостероиды (преднизолон) с интервалом введения. Но фоне приема которых симптомы заболевания проходят после 1-2 инъекции.
Назначение жаропонижающих препаратов иногда приводит к снижению температуры.
Хирургическое лечение - тонзиллэктомия (удаление миндалин), назначается пациентам, которые не реагируют на терапию гормональными препаратами или у которых заболевание вызывает серьёзное ухудшение качества жизни. После тонзиллэктомии симптомы заболевания купируются, в том числе и лихорадка, кроме афтозного стоматита.
Прогноз благоприятный. С возрастом клиническая картина улучшается, периоды ремиссии становятся длиннее, а симптомы менее выраженными. У большинства пациентов проявления заболевания прекращаются к 10 годам.
Наиболее частые вопросы от родителей
Может ли ребенок посещать детский сад или школу?
- Да, может и должен, но не в момент обострения.
Можно ли водить ребенка в спортивную секцию?
- Единственное противопоказание - это период обострения.
Можно ли ребенку делать прививки?
- Да, ребенка можно и нужно вакцинировать. Врач-педиатр должен быть проинформирован о вашем заболевании. Доктор также может составить индивидуальный план прививок. Особое внимание нужно уделить, когда ребенку вводится живая или ослабленная вакцина.
Своевременное обращение к врачу поможет сохранить Ваше здоровье.
Не откладывайте лечение, звоните прямо сейчас. Мы работаем круглосуточно в Москве.Синдром Дресслера
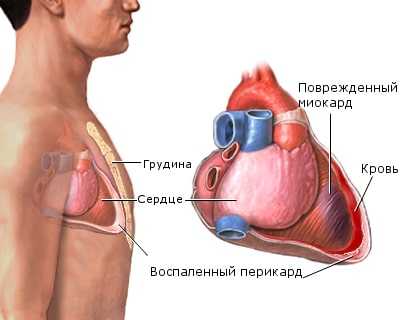
![Синдром Дресслера - аутоиммунный процесс в перикарде и суставах]()
Постинфарктный синдром (или синдром Дресслера) — реактивное аутоиммунное осложнение инфаркта миокарда, развивающееся через 2—6 недель после его начала.
Частота развития
Первоначально считалось, что синдром Дресслера возникает примерно у 4% больных, перенесших инфаркт миокарда. C учетом атипичных и малосимптомных форм частота его развития значительно выше - 15-23 %, а по некоторым источникам достигает 30 %. Однако в последние годы частота синдрома Дресслера уменьшилась. Причинами могут быть широкое использование нестероидных противовоспалительных средств (ацетилсалициловой кислоты) и распространение реперфузионных методов лечения ИМ, уменьшающих объем повреждения мышцы сердца. Другой причиной снижения частоты развития синдрома Дресслера может быть включение в комплекс терапии инфаркт миокарда ингибиторов ангиотензинпревращающего фермента, антагонистов альдостерона и статинов, вследствие их иммуномодулирующего и противовоспалительного действия. Постинфарктый синдром развивается в подостром периоде (не ранее 10-го дня от момента заболевания) у 3—4 % пациентов, перенёсших инфаркт миокарда.
Причины развития
Основная причина синдрома Дресслера - инфаркт миокарда. Считается, что синдром Дресслера чаще развивается после крупноочаговых и осложненных инфарктов, а также после кровотечений в полость перикарда. Синдром Дресслера, точнее синдром постповреждения сердца, может развиваться после кардиохирургических вмешательств (постперикардиотомический синдром, посткомиссуротомический синдром). Помимо этого, типичные признаки синдрома Дресслера могут появляться после других повреждений сердца (ранение, контузия, непроникающий удар в область грудной клетки, катетерная абляция). В настоящее время синдром Дресслера рассматривается как аутоиммунный процесс, обусловленный аутосенсибилизацией к миокардиальным и перикардиальным антигенам. Определенное значение придается также антигенным свойствам крови, попавшей в полость перикарда.
При постинфарктном синдроме наблюдаются изменения и в клеточном иммунитете. Так, имеются данные, что при синдроме Дресслера значительно повышен уровень цитотоксических T-клеток. Этиологическим фактором синдрома Дресслера может быть инфекция, в частности вирусная, поскольку у больных, у которых этот синдром развился после кардиохирургических вмешательств, часто регистрируют повышение титра противовирусных антител.
Симптомы и течение
Развивается на 2-4-й неделе инфаркта миокарда, однако эти сроки могут уменьшаться - «ранний синдром Дресслера» и увеличиваться до нескольких месяцев, «поздний синдром Дресслера». Иногда течение синдрома Дресслера принимает агрессивный и затяжной характер, он может длиться месяцы и годы, протекать с ремиссиями и обострениями. Основные клинические проявления синдрома: лихорадка, перикардит, плеврит, пневмонит и поражение суставов. Лихорадка при синдроме Дресслера не имеет какой-либо строгой закономерности. Как правило, она бывает субфебрильной, хотя в отдельных случаях может быть фебрильной или вообще отсутствовать.
Перикардит является обязательным элементом синдрома Дресслера. Клинически он проявляется болью в перикардиальной зоне, которая может иррадиировать в шею, плечо, спину, брюшную полость. Боль может быть острой приступообразной (плевритическая) или давящей, сжимающей (ишемической). Она может усиливаться при дыхании, кашле, глотании и ослабевать в вертикальном положении или лежа на животе. Она длительная и исчезает или ослабевает после появления в полости перикарда воспалительного экссудата. Главный аускультативный признак перикардита - шум трения перикарда: в первый день болезни при внимательной аускультации он определяется у абсолютного большинства (до 85 %) больных. Шум лучше всего выслушивается у левого края грудины, при задержке дыхания и наклоне туловища пациента вперед. В классическом варианте он состоит из трех компонентов - предсердного (определяется в систолу) и желудочкового (систолического и диастолического). Как и боль, шум трения перикарда уменьшается или исчезает вовсе после появления в полости перикарда выпота, раздвигающего трущиеся листки перикарда. Обычно перикардит протекает нетяжело: уже через несколько дней боли стихают, а экссудат в полости перикарда почти никогда не накапливается в таком количестве, чтобы ухудшить кровообращение, хотя иногда могут появиться признаки тяжелой тампонады сердца. Иногда воспалительный процесс в перикарде при синдроме Дресслера принимает затяжной рецидивирующий характер и заканчивается развитием констриктивного перикардита. При применении антикоагулянтов на фоне синдрома Дресслера возможно также развитие геморрагического перикардита, хотя подобное осложнение может быть и при отсутствии антикоагулянтной терапии.
Плеврит. Проявляется болью в боковых отделах грудной клетки, усиливающейся при дыхании, затруднением дыхания, шумом трения плевры, притуплением перкуторного звука. Он может быть сухим и экссудативным, односторонним и двусторонним. Нередко плеврит носит междолевой характер и не сопровождается типичными физикальными симптомами.
Пневмонит. Пневмонит при синдроме Дресслера выявляется реже, чем перикардит и плеврит. Если очаг воспаления достаточно велик, также отмечается притупление перкуторного звука, ослабленное или жесткое дыхание, появление фокуса мелкопузырчатых хрипов. Возможен кашель и выделение мокроты, иногда с примесью крови, что всегда вызывает определенные диагностические трудности.
Поражение суставов. Для синдрома Дресслера характерно появление так называемого «синдрома плеча»: болезненных ощущений в области плечелопаточных суставов, чаще слева, ограничение подвижности этих суставов. Вовлечение в процесс синовиальных оболочек нередко приводит к возникновению болей и в крупных суставах конечностей.
Другие проявления. Проявлением постинфарктного синдрома может быть сердечная недостаточность вследствие диастолической дисфункции, геморрагический васкулит и острый гломерулонефрит.
Методы исследования
Лабораторные данные. Часто отмечается повышение СОЭ и лейкоцитоз, а также эозинофилия. Весьма характерно резкое повышение уровня С-реактивного белка. У больных с синдромом Дресслера регистрируются нормальные уровни маркеров повреждения миокарда (МВ-фракция креатинфосфокиназы (МВ-КФК), миоглобин, тропонины), хотя иногда отмечается их незначительное повышение, что требует проведения дифференциальной диагностики с рецидивом инфаркта миокарда.
Электрокардиография (ЭКГ). При наличии перикардита на ЭКГ определяются диффузный подъем сегмента ST и, периодически, депрессия сегмента PR, за исключением отведения aVR, в котором наблюдаются депрессия ST и подъем PR. По мере накопления экссудата в полости перикарда может снизиться амплитуда комплекса QRS.
Эхокардиография. При накоплении жидкости в полости перикарда выявляется сепарация его листков и могут появиться признаки тампонады сердца. Для синдрома Дресслера не характерен большой объем жидкости в полости перикарда - как правило, сепарация листков перикарда не достигает 10 мм в диастолу.
Рентгенография. Обнаруживают скопление жидкости в плевральной полости, междолевой плеврит, расширение границ сердечной тени, очаговые тени в легких.
Компьютерная или магнитнорезонансная томография также выявляют жидкость в полости плевры или перикарда и легочную инфильтрацию.
Плевральная и перикардиальная пункция. Извлеченный из полости плевры или перикарда экссудат может быть серозным или серозно-геморрагическим. При лабораторном исследовании в нем определяется эозинофилия, лейкоцитоз и высокий уровень С-реактивного белка.
Лечение
Нестероидные противовоспалительные препараты (НПВС). Препаратом выбора при синдроме Дресслера традиционно считается ибупрофен (400- 800 мг/сут). Реже используют аспирин. Хирургическое лечение применяется при констриктивном перикардите.
Осложнения
Тампонада сердца, геморрагический или констриктивный перикардит, окклюзия (сдавление) коронарного шунта и редко - анемия.
Прогноз
Прогноз при синдроме Дресслера, как правило, благоприятный. Вместе с тем его течение иногда принимает затяжной рецидивирующий характер. Кроме того, имеются данные о том, что выживаемость в течение 5 лет среди перенесших этот синдром, хотя и незначительно, но снижается.
Аутоиммунный процесс развивающийся после инфаркта миокарда и проявляющийся перикардитом, плевритом, воспалительными заболеваниями суставов и сосудов. Лечение проводится с помощью противовоспалительных препаратов.
Читайте также:
- Общие сведения об аллергических реакциях
- Укорочение конечностей
- Рак бронхов: симптомы онкологии, диагностика опухоли и лечение бронхогенного рака легких
- Закрытие просвета при нарушении целостности кишки. Наложение шва по Серебрянникову и Снежковой. Наложение анастомоза.
- Дифференциальный диагноз мигрени и головной боли напряжения. Доброкачественная рецидивирующая головная боль.