Синдром Леннокса-Гасто (СЛГ): причины, клиника, диагностика, лечение
Добавил пользователь Alex Обновлено: 21.01.2026
ГБОУ ВПО »Кировская государственная медицинская академия», Киров, России
Кировская государственная медицинская академия, Киров
ГОУ ВПО «Ульяновский государственный университет», ГУЗ «Центральная клиническая медико-санитарная часть», Ульяновск ,Кировская государственная медицинская академия, Кировская городская клиническая больница №1, Киров
Эффективность и безопасность лечения эпилепсии руфинамидом (по данным метаанализа)
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2016;116(8): 40‑43
Цель исследования. Сопоставление результатов эффективности и безопасности руфинамида по совокупности данных клинических исследований диагностически гетерогенных групп пациентов с тяжелыми и рефрактерными к фармакотерапии формами эпилепсии с помощью метаанализа. Материал и методы. Из 164 публикаций, доступных в медико-клинических базах данных, были отобраны 15 с численностью пациентов, включенных в исследования, 1847. В массив данных вошли сведения о применении руфинамида при синдроме Леннокса-Гасто (СЛГ), сходных энцефалопатических формах эпилепсий, а так же при парциальных формах эпилепсии, устойчивых к лечению стандартными противоэпилептическими препаратами (ПЭП). Руфинамид получали 1169 участников в дополнение к другим ПЭП (экспериментальная группа). Проходили лечение без назначения руфинамида 686 пациентов (группа контроля). Результаты и заключение. По числу пациентов с редукцией приступов на 50% и более группа, принимавшая руфинамид, статистически достоверно превзошла контрольную. Из осложнений приема ПЭП среди получавших руфинамид достоверно чаще наблюдались такие, как головная боль или головокружение и тошнота и рвота. Таким образом, руфинамид в качестве дополнительной терапии эффективен в отношении разных типов эпилептических приступов, имеет благоприятный профиль безопасности и переносимости и может широко применяться в неврологической практике.
Эпилепсия, резистентная к противоэпилептическим препаратам (ПЭП), представляет собой серьезную медико-социальную проблему. Во-первых, в данную группу попадают редкие формы тяжелой эпилепсии с неврологическими осложнениями и неблагоприятным прогнозом. Установление диагноза при таких формах может означать ограничения социального функционирования, неизбежность значительных затрат на медицинскую и социальную помощь [1, 2]. Во-вторых, крайне редко удается в полном объеме провести дифференциальную диагностику и подробное выяснение этиологии резистентной к лечению эпилепсии [3]. В особенности это касается медицинского обслуживания малообеспеченных слоев населения. Остаются актуальными и такие проблемы, как недостаток времени, отведенного на осмотр одного больного, высокая стоимость инструментальных, биохимических и других лабораторных методов диагностики, отсутствие регулярного поступления реагентов для исследований и других расходных материалов, возможность мониторинга типа и продолжительности приступов. Поэтому информацию о больных приходится собирать со слов родственников, опекунов и самих пациентов. Эти факторы снижают качество диагностики, особенно при таких типах эпилепсии, как синдром Драве (обусловлен мутацией SCN1A; манифестирует полиморфными приступами продолжительностью более 15 мин, судорожной активностью на фоне лихорадки и развивающимися впоследствии когнитивными расстройствами) [4]. В-третьих, новые ПЭП часто регистрируются не для всех типов эпилептических синдромов, а только для некоторых из них. Например, такой препарат, как руфинамид, в 2008 г. получил разрешение в Европе и США на применение только для лечения синдрома Леннокса-Гасто (СЛГ) [3, 5], хотя в литературе есть данные о его применении и при других формах эпилепсии [6, 7]. Поэтому практически важным становится вопрос о правомерности назначения, подтверждения его клинической эффективности и безопасности при более широких показаниях, нежели предусмотренных регламентом полученной производителем регистрации.
Одним из таких показаний может быть резистентная к терапии эпилепсия.
Материал и методы
Цель данной работы - оценка эффективности и безопасности лечения руфинамидом пациентов с рефрактерной эпилепсией по данным метаанализа.
В доступных базах данных были найдены 164 публикации, из которых для систематизированного обзора и метаанализа были отобраны 15 [7-11, 12-18, 19-21]. При их включении в исследование авторы ориентировались на сходство изучавшихся нозологических форм; в обновленные массивы данных включались результаты как контролируемых, так и обсервационных исследований.
Возраст больных, вошедших в эту диагностически гетерогенную выборку, составил от 4 мес до 80 лет. По данным оригинальных исследований клинический статус пациентов оценивался не менее чем за последний месяц болезни (в некоторых работах дольше). Затем следовали периоды титрации дозы руфинамида (последующее клиническое наблюдение с фиксацией суррогатных/конечных точек) длительностью от 3 мес (строгие временные рамки превалировали в слепых контролируемых исследованиях [12, 19]) до года и дольше (что более соответствует обстановке реальной практики с неоднородной комплаентностью больных с заболеваниями ЦНС.
Окончательные группы больных, отобранных в 15 упоминавшихся исследованиях [7-11, 12-18, 19-21], составили 1169 пациентов, получавших руфинамид, назначенный в дополнение к препаратам базовых схем терапии (экспериментальная группа), и 686 пациентов контрольной выборки с комбинациями стандартных ПЭП. В частности, в контрольную группу вошли больные из более ранних сравнительных исследований, посвященных оценке эффективности и безопасности ламотриджина и топирамата при СЛГ [18, 20].
В качестве критериев оценки действия ПЭП, в том числе у руфинамида, рассматривали долю пациентов (от их общей численности в группе сравнения), у которых: удалось снизить частоту приступов всех типов на 50% и более; наблюдались нежелательные побочные явления в виде сонливости, тошноты и рвоты, сыпи, анорексии и снижения аппетита, раздражительности, утомляемости, головной боли и головокружения.
По каждому из соответствующих показателей оценивали статистическую значимость различий в группах сравнения по непараметрическому критерию χ 2 и вычисляли следующие унифицированные показатели клинической эффективности [22]: 1) отношение шансов (ОШ) позитивного исхода или наступления нежелательного эффекта (ОШ) с 95% доверительным интервалом (95% ДИ) [23]; 2) частоту результатов лечения руфинамидом позитивных или нежелательных исходов (ЧИЛ, %); 3) частоту результатов лечения позитивных или нежелательных исходов в контрольной группе (ЧИК, %); 4) повышение абсолютной пользы (АП) или абсолютного риска (ПАР) нежелательных эффектов при лечении руфинамидом (ПАП=ЧИЛ-ЧИК, %); 5) повышение относительной пользы (ПОП) или относительного риска (ПОР) нежелательных эффектов от лечения руфинамидом (ПОП= ПАП·100%/ЧИК, %); 6) число больных, которым необходимо лечение руфинамидом для достижения с позитивным или наступления неблагоприятного эффекта (ЧБНЛ=1·100%/ПАП, усл.ед.).
Почти двукратное преобладание по численности экспериментальной группы объясняется включением в нее нескольких крупных простых обсервационных исследований, в которых группа сравнения не формировалась.
Результаты и обсуждение
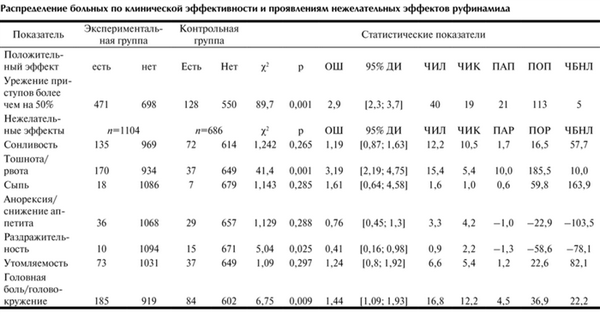
Распределение больных по клинической эффективности и проявлениям нежелательных эффектов руфинамида
Доля пациентов с позитивным клиническим эффектом в группе руфинамида оказалась в два раза больше, чем в группе сравнения.
При сравнении числа респондеров в группе руфинамида и контрольной группах по методу χ 2 достигнута высокая мощность критерия согласия (89,7) при р=0,0001 (см. таблицу). Таким образом, превосходящая эффективность лечения с включением в схему руфинамида при сопоставлении с группой сравнения показана с высокой степенью статистической достоверности. Об этом же свидетельствует значение ОШ 2,9 (95% ДИ от 2,3 до 3,7), которое означает почти трехкратное повышение вероятности наступления позитивного клинического эффекта при дополнительном применении руфинамида по сравнению с традиционным лечением.
Динамика неблагоприятных побочных явлений в больших гетерогенных группах больных рефрактерной эпилепсией по нашим данным оказалась иной, чем в доступных зарубежных обзорах [5]. Так, в итоговых сравнениях настоящего метаанализа частота сонливости и утомляемости в группе больных леченных с применением руфинамида практически не отличались от аналогичных показателей в группе сравнения. Об этом свидетельствует исчезающе малая мощность критерия χ 2 при значениях статистической достоверности р=0,265 и 0,297 (статистически недостоверная межгрупповая разница). Значения О.Ш. 1,19 и 1,24 при нижних границах доверительного интервала менее 1,0 для обоих показателей, также не позволяют говорить о них как о специфичных для руфинамида побочных эффектах (см. таблицу). Напротив, у лечившихся руфинамидом пациентов достоверно чаще (р=0,009) встречались головная боль и головокружение, хотя мощность χ 2 для них была сравнительно невелика, да и ОШ не сильно отличается от 1,0. Побочные эффекты в виде раздражительности и ажитации, в группе руфинамида встречались реже, чем в группе сравнения (р=0,025). Симптомы значительно снижающие качество жизни тошнота/рвота предсказуемо преобладали в основной группе (χ 2 =41,4 при р=0,0001; ОШ=3,19 с 95% ДИ от 2,19 до 4,75). Учащение этих симптомов неоднократно фиксировалось по фармакодинамическим свойствам руфинамида во всех ранних исследованиях [3, 5, 6].
ЧИЛ в основной группе была выше, чем ЧИК, что отразилось на значениях параметров ПАП, ПОП и ЧБНЛ, которые подтверждали клиническую и статистическую значимость адъювантной терапии руфинамидом. В противоположность этому, частотные характеристики неблагоприятных эффектов в этих же группах сравнения, повышение их абсолютных и относительных рисков, а также ЧБНЛ до проявления неблагоприятного эффекта подчинялись другим закономерностям. По совокупности значений показатели-маркеры неблагоприятных побочных эффектов (ЧИЛ, ЧИК, ПАР, ПОР, ЧБНЛ), за исключением показателей частоты тошноты/рвоты и головной боли/головокружения, были не сопоставимы с уровнем статистической значимости позитивного эффекта терапии, и более того, иногда принимали отрицательные значения.
Таким образом, представлены результаты первого сравнительного анализа сопоставления параметров эффективности и безопасности руфинамида в репрезентативных, но гетерогенных выборках пациентов с разными формами эпилепсии (в общей сложности в массивы сравнения вошли более 1800 человек). Приведенная выше характеристика групп сравнения и гетерогенность включенных в них контингентов в наибольшей степени соответствуют критериям реальной практики и, на наш взгляд, адекватны популяции нуждающихся в приеме ПЭП больных. Полученные результаты с высокой степенью надежности могут быть экстраполированы на обширные контингенты страдающих эпилепсией пациентов, среди которых по организационным и финансовым причинам не всегда возможны дифференциально-диагностическая проработка и тщательный мониторинг эффективности лечения.
Установлено, что руфинамид эффективен не только у больных с СЛГ старше 4 лет (в группах сравнения их насчитывалось 466 из 1847 участников исследования). Пациентов с парциальными формами эпилепсии в обеих группах сравнения оказалось больше, чем страдающих СЛГ (1268 и 466). Еще 113 больных страдали недифференцированными формами генерализованной эпилепсии, синдромом Драве либо сложными формами эпилепсии, в которых не удавалось однозначно делать выводы о первично-парциальной природе приступов, несмотря на их вторичную генерализацию. Таким образом, руфинамид был эффективен при лечении достаточного большого перечня форм эпилепсии, включая и устойчивую к лечению парциальную эпилепсию.
Руфинамид имеет благоприятный профиль безопасности и переносимости. Среди значимых побочных явлений выделяются тошнота, рвота и реже головная боль/головокружение. Руфинамид может применяться в неврологической практике как препарат второй линии при неэффективности стандартных ПЭП.
Синдром Леннокса — Гасто - симптомы и лечение
Что такое синдром Леннокса — Гасто? Причины возникновения, диагностику и методы лечения разберем в статье доктора Поздняковой А. А., детского невролога со стажем в 5 лет.
Над статьей доктора Поздняковой А. А. работали литературный редактор Вера Васина , научный редактор Сергей Макаров и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Синдром Леннокса — Гасто (Lennox — Gastaut Syndrome) — это тяжёлая форма эпилепсии, которая начинается в детстве.
Заболевание проявляется выраженным снижением интеллекта и частыми приступами трёх видов:
- тоническими — внезапное напряжение всех мышц, основной признак заболевания;
- атоническими — кратковременное расслабление мышц с потерей сознания;
- атипичными абсансами — сокращение мышц, потеря сознания, автоматические действия; атипичные абсансы длятся дольше типичных, более постепенно начинаются и заканчиваются, их сложнее контролировать и лечить [14] .
Приступы при синдроме Леннокса — Гасто вызваны внезапными вспышками аномальной электрической активности в головном мозге и с трудом поддаются лечению противосудорожными препаратами.
Распространённость синдрома Леннокса — Гасто
Синдром Леннокса — Гасто — это редкое заболевание. Его выявляют у 2 из 100 000 детей, чаще у мальчиков. Среди всех форм эпилепсии на этот синдром приходится 2-5 % случаев [4] .
Заболевание проявляется в возрасте от года до 9 лет, чаще в 2-4 года [1] .
Причины синдрома Леннокса — Гасто
Выделяют криптогенный и симптоматический варианты синдрома.
Примерно у 40 % детей причина болезни неясна, в таких случаях синдром называют идиопатическим, или криптогенным [4] .
Вторичный, или симптоматический, вариант синдрома Леннокса — Гасто — это проявление диффузного поражения головного мозга (диффузные поражения, в отличие от очаговых, затрагивают весь мозг, а не отдельный участок). Причинами такого поражения могут быть:
- тяжёлые генетические заболевания;
- энцефалит и менингит;
- черепно-мозговые и родовые травмы;
- пороки развития коры головного мозга;
- метаболические нарушения (митохондриальные заболевания);
- опухоли головного мозга; .
При симптоматическом варианте прогноз хуже, чем при идиопатическом. У 30 % детей развитию заболевания предшествуют инфантильные спазмы (синдром Веста) [4] . Спазмы не являются причиной синдрома Леннокса — Гасто, но у таких детей болезнь протекает тяжелее.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Леннокса — Гасто
Для синдрома Леннокса — Гасто характерны различные типы приступов: тонические и атонические припадки, атипичные абсансы.
При тонических приступах напрягаются мышцы всего тела, рук и ног, чаще с разгибанием, чем со сгибанием. Эти приступы непродолжительны: не более полминуты, обычно около 10 секунд. Часто они возникают по ночам и могут остаться незамеченными для родителей ребёнка.
Атонические приступы состоят из миоклонического и атонического компонентов. При миоклоническом приступе пациент непроизвольно вздрагивает, выбрасывает руки вперёд и подгибает ноги. Затем следует атонический приступ, при котором слабеют мышцы, человек теряет сознание и падает, что нередко приводит к травмам. Как правило, атонический приступ длится всего несколько секунд.
Атипичные абсансы проявляются кратковременной потерей сознания, не больше чем на несколько минут. Во время приступа подёргиваются мышцы лица, пациент высовывает язык, кивает и часто моргает. Также человек может совершать автоматические действия, например включить свет, о чём в дальнейшем забывает.
У взрослых пациентов могут возникать генерализованные тонико-клонические приступы. Они проявляются потерей сознания, судорогами по всему телу, прикусом языка, непроизвольным мочеиспусканием. У детей таких приступов, как правило, не бывает.
Бессудорожный эпилептический статус часто встречается при синдроме Леннокса — Гасто. Это состояние может длиться от пары часов до нескольких недель. Проявляется в двух основных формах: спутанным сознанием и тоническими приступами. У пациента снижается психическая и двигательная активность, вплоть до ступора, лицо становится похожим на маску, слабеют мышцы, возникает слюнотечение, временами теряется ясность сознания. Такое состояние трудно распознать при тяжёлых когнитивных нарушениях — эпизоды спутанного сознания можно принять за нарушения интеллекта. Вероятно, бессудорожный статус усугубляет уже имеющиеся интеллектуальные нарушения [12] .
Умственная отсталость тоже относится к классическим проявлениям синдрома Леннокса — Гасто. У некоторых пациентов она наблюдается ещё до начала болезни. В течение пяти лет после начала заболевания умственная отсталость развивается у 90 % пациентов, но в литературе описаны редкие случаи нормального интеллекта при синдроме Леннокса — Гасто.
Также у многих пациентов наблюдается агрессивность, импульсивность и аутичные черты (задержка речи, повторяющиеся стереотипные движения, трудности в общении, ограниченный круг интересов) [3] .
Патогенез синдрома Леннокса — Гасто
Патогенез синдрома Леннокса — Гасто до конца не изучен. Это связано с разнообразием симптомов и причин заболевания, а также с ограниченными данными о генетических причинах болезни.
В основе синдрома Леннокса — Гасто лежит повышенная возбудимость коры головного мозга. Предполагается, что у детей с этим заболеванием нарушено формирование синапсов между нервными клетками, из-за чего в дальнейшем повышается их возбудимость. К такому нарушению может приводить повреждение как корковых, так и подкорковых структур (кортикоретикулярных связей и таламуса).
Нарушения в работе этих структур могут быть вызваны различными заболеваниями: гипоксически-ишемической энцефалопатией, менингоэнцефалитом, нейрокожными синдромами, опухолями и пороками развития головного мозга. Синдром Леннокса — Гасто может развиться при любом типе повреждения головного мозга.
Обычно в начале заболевания возникают тонические приступы, затем присоединяются атипичные абсансы, миоклонические и тонико-клонические приступы. Тонический компонент связан с поражением мезэнцефальной ретикулярной формации, а клонический — с поражением переднего мозга. Отсюда следует, что присоединение атипичных абсансов и клонических приступов вызвано возрастающим участием коры головного мозга.

При некоторых состояниях центральной нервной системы, например при кровоизлиянии или дефиците кислорода, повышается возбудимость в коре головного мозга. Неконтролируемая электрическая активность, возникающая при повышенной возбудимости, проявляется симптомами синдрома Леннокса — Гасто [5] .
Классификация и стадии развития синдрома Леннокса — Гасто
Согласно классификации Международной противоэпилептической лиги (ILAE), синдром Леннокса — Гасто относится к генерализованной криптогенной или симптоматической формам эпилепсии, т. е. болезнь вызвана аномальной электрической активностью в обоих полушариях, но причина может быть как выявлена, так и неизвестна.
Согласно Проекту классификации ILAE 2001 года, синдром Леннокса — Гасто входит в группу детских эпилептических энцефалопатий. К тяжёлым детским энцефалопатиями также относят синдромы Веста, Драве и Дуза [2] .
В Международной классификации болезней (МКБ-10) синдром Леннокса — Гасто кодируется как G40.4 Другие виды генерализованной эпилепсии и эпилептических синдромов.
Осложнения синдрома Леннокса — Гасто
Синдром Леннокса — Гасто часто приводит к развитию эпилептического статуса, из-за чего значительно ухудшаются умственные способности. Пациент может утратить необходимые для жизни навыки, и в дальнейшем ему потребуется постоянный уход.
Во время эпистатуса слабеют и беспорядочно подёргиваются мышцы лица и рук, снижается двигательная и психическая активность. Контакт с пациентом затруднён, он может лежать неподвижно или его движения становятся замедленными. Чаще всего приступы происходят по утрам. Эпистатус свидетельствует о неблагоприятном течении заболевания и может угрожать жизни.
У многих пациентов с синдромом Леннокса — Гасто развиваются экстрапирамидные и мозжечковые расстройства, которые проявляются слюнотечением, нарушением походки, координации, глотания и речи (дисфагией и диазартрией). При нарастающей дисфагии трудно есть и принимать лекарства, может потребоваться установка гастростомы [1] .
Нарушение походки и атонические приступы часто приводят к падениям и черепно-мозговым травмам. Нередко пациентам приходится пользоваться инвалидной коляской.
Также синдром Леннокса — Гасто может осложняться нарушением сна и психозом.
Диагностика синдрома Леннокса — Гасто
Диагностика синдрома Леннокса — Гасто включает сбор анамнеза, электроэнцефалографию (ЭЭГ), магнитно-резонансную или компьютерную томографию (МРТ или КТ). Может потребоваться генетическое тестирование.
Сбор жалоб и истории болезни
Для постановки диагноза важна подробная история болезни: как протекала беременность, роды и перинатальный период, какие симптомы беспокоят.
Неврологические нарушения при идиопатическом синдроме Леннокса — Гасто обычно отсутствуют. При симптоматических формах могут возникать нарушения со стороны нервной системы: паралич, неустойчивость при ходьбе, микроцефалия, нарушения речи, косоглазие. Также на синдром Леннокса — Гасто будет указывать задержка в развитии или потеря уже приобретённых навыков и устойчивость судорог к противоэпилептическим препаратам.
Магнитно-резонансная и компьютерная томография (МРТ и КТ)
Характер нарушений зависит от формы заболевания. Идиопатический вариант синдрома Леннокса — Гасто протекает без структурных изменений головного мозга. У многих пациентов на КТ и МРТ выявляют диффузную атрофию головного мозга, при которой отмирают нейроны и разрушаются нервные связи. При симптоматической форме наблюдаются очаговые поражения коры головного мозга.
Электроэнцефалография (ЭЭГ)
При диагностике синдрома Леннокса — Гасто проводится ЭЭГ с записью во время ночного сна и бодрствования. ЭЭГ позволяет выявить эпилептиформную активность — острые волны и пики. Для записи и оценки различных типов приступов может проводиться видео-ЭЭГ-мониторинг.

В самом начале заболевания на ЭЭГ заметно только изменение основной биоэлектрической активности. Кроме того, судороги и клинические симптомы развиваются с течением времени, отставание в развитии также не всегда проявляется в начале болезни. Поэтому часто диагноз устанавливают после нескольких лет наблюдения за пациентом [3] .
Синдром Леннокса — Гасто проявляется характерным паттерном ЭЭГ: медленными спайк-волновыми комплексами между приступами и генерализованной пароксизмальной быстрой активностью во сне (т. е. изменениями потенциала в форме острых волн, пиков и др.). Спайк-волна — это комплекс, который имеет высокую амплитуду и возникает при комбинации спайка с медленной волной.
Чтобы установить диагноз, необходимо наличие минимум двух типов генерализованных приступов и медленных спайк-волн на ЭЭГ в состоянии бодрствования.
![ЭЭГ при синдроме Леннокса — Гасто [15]](https://probolezny.ru/media/bolezny/sindrom-lennoksa-gasto/eeg-pri-sindrome-lennoksa-gasto-15_s.jpg)
Генетическое тестирование
Иногда проводится тестирование для выявления генетических нарушений: дефекта транспортёра глюкозы (SLC2A1), позднего детского нейронального липофусциноза цероидов (CLN2) и туберозного склероза (TSC 1,2). У детей с такими нарушениями часто наблюдаются эпилептические приступы.
Дифференциальная диагностика
Синдром Леннокса — Гасто следует отличать от других ранних эпилептических энцефалопатий:
- от синдромов Драве, Дуза, Ландау — Клеффнера, Веста и Ангельмана;
- атипичной эпилепсии детства с центро-темпоральными спайками;
- эпилептической энцефалопатии с продолженной спайк-волновой активностью во сне;
- синдрома псевдо-Леннокса (при такой эпилепсии во время засыпания или пробуждения быстро сокращаются мышцы лица и плеч, нарушается речь и усиливается потливость).
Также проводится дифференциальная диагностика с некоторыми наследственно-дегенеративными заболеваниями, например нейрофиброматозом 1-го типа [3] [12] .
Лечение синдрома Леннокса — Гасто
При синдроме Леннокса — Гасто применяют противосудорожные препараты, кортикостероды, кетогенную диету, стимуляцию блуждающего нерва и хирургическое лечение. Не доказано, что какой-то определённый препарат помогает лучше, чем другой. Поэтому терапию для каждого пациента подбирают индивидуально [8] .
Медикаментозное лечение
Судороги при синдроме Леннокса — Гасто устойчивы к лечению — полностью устранить их не получится, но можно уменьшить частоту.
Наиболее эффективны противосудорожные препараты широкого спектра: Вальпроат натрия, Клобазам, Ламотриджин, Топирамат и Зонисамид. Вальпроат натрия — это препарат первой линии в лечении синдрома Леннокса — Гасто. Часто сочетают два лекарства, например Вальпроат натрия с Ламотриджином или Клобазамом. Ламотриджин хорошо помогает при атонических припадках. Также могут быть эффективны Леветирацетам и Перампанел.
Если противосудорожные препараты не помогают, то назначают кортикостероиды: адренокортикотропный гормон, Преднизолон, Метилпреднизолон. При приёме кортикостероидов часто возникают побочные эффекты, например отёки, высокое давление, ожирение.
Для экстренного лечения частых атипичных абсансов, бессудорожного эпистатуса или других тяжёлых приступов обычно назначают бензодиазепины. При бессудорожном эпилептическом статусе может применяться короткий курс Клобазама или бензодиазепинов, высокие дозы кортикостероидов или вальпроевой кислоты. В некоторых случаях бензодиазепины могут увеличить частоту тонических судорог.
Кетогенная диета
Кетогенная диета показана, если лечение противосудорожными препаратами неэффективно. При такой диете потребляют много жиров, умеренное количество белков и мало углеводов. В рацион входят орехи, сыр, масло, жирная рыба. Исключают конфеты, картофель, выпечку, мёд, шоколад [13] .
Стимуляция блуждающего нерва
Стимуляцию блуждающего нерва проводят с помощью вживлённого электрода, подключённого к генератору импульсов. Этот метод рекомендован пациентам с атоническими приступами, которые могут привести к травмам и инвалидности. Стимуляция блуждающего нерва позволяет уменьшить частоту приступов.

Хирургическое лечение
При синдроме Леннокса — Гасто может проводиться мозолотомия и гемисферэктомия. Операции позволяют облегчить симптомы и в некоторых случаях уменьшить частоту приступов.
Мозолотомия — это рассечение мозолистого тела, т. е. пучка нервных волокон, соединяющих полушария мозга. После операции припадки не могут распространяться из одного полушария в другое.

Гемисферэктомия — это частичное или полное удаление полушария головного мозга. Проводится в редких случаях, когда остальные методы лечения неэффективны.
Прогноз. Профилактика
Судороги при синдроме Леннокса — Гасто часто не поддаются лечению. Они вызывают у пациентов страх перед физической травмой, при постоянных и плохо контролируемых приступах ухудшаются умственные способности. У пациентов часто возникают трудности в учёбе, социальной и личной жизни.
Практически во всех случаях судороги продолжаются и во взрослом возрасте. При этом заболевание сопровождается слюнотечением, нарушением походки, речи и глотания.
Прогноз лучше, если до начала приступов не было поражения мозга и отклонений в интеллектуальном развитии. Обычно если синдрому Леннокса — Гасто предшествовали инфантильные спазмы, то судороги тяжелее контролировать. Умственные способности у таких пациентов ухудшаются сильнее.
Как правило, частота припадков снижается в период полового созревания. Однако у 2/3 пациентов спустя десять лет после начала болезни приступы по-прежнему происходят ежедневно.
Атипичные абсансы иногда сменяются фокальными приступами: двигательными нарушениями, ощущением онемения и ударов током в руках, ногах или лице, покраснением кожи, дискомфортом в верхней части живота.
Гиперактивность, агрессия и аутистические черты могут сохраняться во взрослом возрасте, но чаще развивается медлительность и апатия. Большинство пациентов нуждаются в посторонней помощи, лишь немногие могут жить самостоятельно [7] .
Профилактика заключается в раннем выявлении заболевания, что позволит облегчить симптомы болезни [8] .
Юношеская миоклоническая эпилепсия
Юношеская миоклоническая эпилепсия — это форма генерализованной эпилепсии, основу клинической картины которой составляют миоклонические приступы — асинхронные мышечные сокращения, кратковременно возникающие в симметричных участках тела, преимущественно в руках и плечевом поясе. Наряду с миоклоническими эпизодами в клинике могут наблюдаться абсансы и клонико-тонические генерализованные эпиприступы. Юношеская миоклоническая эпилепсия диагностируется на основании клиники заболевания и результатов электроэнцефалографии, при исключении органической церебральной патологии по данным неврологического осмотра и МРТ. Лечение проводится преимущественно препаратами вальпроевой кислоты. Как правило, необходимо пожизненное наблюдение эпилептолога.
МКБ-10

Общие сведения
Юношеская миоклоническая эпилепсия (ЮМЭ) составляет до 12% от всех форм этого заболевания и около 23% случаев идиопатической генерализованной эпилепсии. ЮМЭ является одной из разновидностей миоклонической эпилепсии — генерализованной эпилепсии, протекающей с миоклоническими приступами. В эту группу заболеваний также входят: детская доброкачественная миоклония, синдром Веста (миоклоническая энцефалопатия детского возраста с церебральной гипсаритмией), болезнь Лафоры и др.
Первое описание ЮМЭ датировано 1867 г. Однако в качестве отдельной нозологической единицы юношеская миоклоническая эпилепсия была выделена лишь в 1955 г. по предложению группы германских врачей во главе с Янцем, после чего она стала упоминаться как синдром Янца. В научной литературе по неврологии и эпилептологии можно встретить также термин «импульсивный petit mal».

Причины
Юношеская миоклоническая эпилепсия является наследственной. Случаи развития этой формы эпилепсии вследствие органического поражения головного мозга не зафиксированы. У половины пациентов в семейном анамнезе имеются родственники 1-й или 2-й линии, у которых происходят различного типа эпиприступы. Гены, ответственные за развитие заболевания, пока точно не установлены. Предполагают несколько возможных вариантов - хромосома 15q, один из локусов короткого плеча хромосомы 6, гены C6orf33 и BRD2 и др. Большинство генетиков склоняются к мнению о полигенном механизме наследования миоклонической эпилепсии. Специфический патогенез ЮМЭ не идентифицирован.
Симптомы
Юношеская миоклоническая эпилепсия манифестирует в возрасте от 8 до 24 лет. Наиболее часто дебют заболевания приходится на возрастной отрезок 12-18 лет. Патогномоничным симптомом ЮМЭ выступают миоклонические приступы — короткие, внезапно возникающие, непроизвольные мышечные сокращения, имеющие асинхронный характер.
Как правило, в начале заболевания пароксизмы отмечаются в утренние часы при пробуждении больного. Сокращения мышц происходят симметрично в обеих половинах тела, чаще охватывают только плечевой пояс и руки, реже — распространяются на нижние конечности или на все тело. Во время пароксизма пациенты могут автоматически отбрасывать или ронять удерживаемые в руках предметы, при вовлечении нижних конечностей происходит падение.
Пароксизмы ЮМЭ могут иметь одиночный характер или возникать кластерами. В редких случаях наблюдается т. н. миоклонический эпилептический статус. Отличительной чертой является полная сохранность сознания пациента во время миоклонического пароксизма, даже в тех случаях, когда речь идет о миоклоническом статусе.
В 3-5% случаев юношеская миоклоническая эпилепсия протекает с наличием только миоклонических пароксизмов. В подавляющем большинстве наблюдений (около 90%) через некоторое время после дебюта заболевания (в среднем через 3 года) у пациента возникают генерализованные тонико-клонические эпиприступы. Они могут начинаться с серии нарастающих миоклонических подергиваний, затем переходящих в клонико-тонические судороги. Примерно у 40% пациентов отмечаются абсансы — кратковременные эпизоды «выключения» сознания.
Диагностика
Установить диагноз ЮМЭ в ее начальном периоде весьма затруднительно. Зачастую возникающие на фоне пробуждения миоклонические эпизоды расцениваются как нервозность ребенка, а сами дети обычно не обращают внимания на подобные мелкие симптомы. Как правило, родители обращаются к неврологу, когда у ребенка появляются тонико-клонические приступы. Исследование неврологического статуса не определяет каких-либо нарушений. Инструментальные методы включают:
- МРТ головного мозга. Проводят для исключения церебральной патологии (внутримозговой опухоли, абсцесса мозга, церебральной кисты, энцефалита, внутримозговой гематомы) и органического происхождения эпиприступов. При необходимости исключения аневризмы сосудов головного мозга пациента направляют на МР-ангиографию.
- Электроэнцефалография. В 75% ЭЭГ выявляет наличие интериктальных эпилептиформных паттернов. Регистрируются билатерально-симметричные пароксизмальные разряды, состоящие из комплексов полиспайк-волна с частотой 4-6 Гц. В 17% случаев наблюдаются комплексы частотой 3 Гц. Иктальная ЭЭГ выявляет высокие и среднеамплитудные спайки частотой 10-16 Гц, после которых регистрируются медленные волны нерегулярного характера. Число спайков одного пароксизма варьирует от 5 до 20 и зависит скорее не от длительности, а от интенсивности приступа. Для ранней диагностики ЮМЭ может потребоваться проведение ЭЭГ при пробуждении, суточный ЭЭГ-видеомониторинг, провоцирующие пробы (депривация сна, фотостимуляция).
Массивность и билатеральная синхронность отличает пароксизмы ЮМЭ от неэпилептического миоклонуса, эпизоды которого носят спорадический и фокальный характер. Юношеская миоклоническая эпилепсия также требует дифференцировки от других форм эпилепсии, протекающих с миоклоническими эпизодами. Так, в отличии от ЮМЭ, при эпилепсии с генерализованными судорожными пароксизмами пробуждения или при юношеской абсанс эпилепсии миоклонические пароксизмы не являются доминирующими в клинической картине заболевания.
Эпилепсия с миоклонически-астатическими пароксизмами, синдром Леннокса-Гасто и эпилепсия с миоклоническими абсансами дебютируют в более раннем детском возрасте и сопровождаются задержкой психического развития. Последняя характеризуется приступами, в которых миоклонические судороги сочетаются с абсансами, в то время как пароксизмы ЮМЭ протекают без нарушения сознания.
Лечение юношеской миоклонической эпилепсии
Режимные мероприятия
Важное значение имеет не только фармакотерапия эпилепсии, но и соблюдение пациентом некоторых жизненных норм, позволяющих избегать провоцирования приступов. Как и при других видах эпилепсии, при ЮМЭ приступы могут быть вызваны нарушением режима, психической и физической перегрузкой, стрессом, недосыпанием, приемом содержащих алкоголь напитков. Поэтому пациенту следует избегать подобных провоцирующих факторов. Положительно влияет на течение заболевания спокойный, простой и неторопливый уклад жизни, пребывание на природе, вдали от городской суеты. В связи с этим некоторые семьи, где у ребенка диагностирована ЮМЭ, переезжают и живут в сельской местности.
Фармакотерапия
Медикаментозная терапия ЮМЭ проводится вальпроатами. Монотерапия данными препаратами оказалась эффективной в отношении всех видов приступов, сопровождающих клинику ЮМЭ, — миоклонических, тонико-клонических и абсансов. При недостаточности монотерапии возможно комбинированное лечение. Купирование резистентных абсансов достигается сочетанием вальпроатов с этосуксимидом, резистентных клонико-тонических приступов — сочетанием вальпроатов с примидоном или фенобарбиталом.
Для контроля миоклонических пароксизмов эффективен клоназепам, однако его действие не распространяется на тонико-клонические генерализованные приступы. При этом полное купирование миоклонических приступов лишает пациента возможности заранее знать о приближающемся тонико-клоническом приступе по возникающим перед ним миоклоническим проявлениям. Поэтому назначение клоназепама оправдано лишь при стойких миоклонических пароксизмах и должно сочетаться с препаратом вальпроевой кислоты.
Результаты лечения ЮМЭ противоэпилептическими препаратами нового поколения (леветирацетамом, ламотриджином, топираматом) пока проходят проверку в клинических условиях. Отмечена высокая перспективность леветирацетама.
Прогноз
Юношеская миоклоническая эпилепсия считается хроническим заболеванием, которое продолжается в течение всей жизни пациента. Случаи спонтанной ремиссии редки. У 90% больных, по различным причинам прервавших лечение антиэпилептическим препаратом, отмечалось возобновление эпиприступов. Однако имеются указания на то, что в отдельных случаях у пациентов после отмены препарата наблюдалась длительная ремиссия.
В целом, при правильно подобранной терапии приступы контролируются у большинства больных, хотя у половины из них на фоне лечения могут наблюдаться отдельные пароксизмы. Относительно резистентное к терапии течение наблюдается редко, в основном в случаях, когда у пациента отмечаются все 3 разновидности пароксизмов ЮМЭ.
Синдром Леннокса-Гасто
Синдром Леннокса-Гасто — отдельная форма эпилепсии детского возраста, характеризующаяся наличием полиморфных пароксизмов (миоклонических, атонических, тонических и абсансов) и задержкой нейро-психического развития. Может иметь криптогенный характер или выступать синдромом других патологических состояний (церебральных аномалий, генетических обменных заболеваний, перинатальной патологии). Синдром Леннокса-Гасто диагностируется по типичной вариативной картине эпиприступов и характерному паттерну электроэнцефалограммы. Дополнительно проводится МРТ и КТ головного мозга. Антиконвульсантная терапия синдрома малоэффективна, проводится поиск альтернативных методов лечения. Прогноз вариабельный, но в большинстве случаев неблагоприятный.

Синдром Леннокса-Гасто (СЛГ) — вариант эпилепсии детского возраста, для которого характерно сочетание атонических, миоклонических, тонических эпиприступов и атипичных абсансов, медленный островолновой паттерн ЭЭГ. В 1950 г. СЛГ был выделен в качестве отдельного эпилептического синдрома, а в 1964-1966 гг. неврологическое сообщество признала его самостоятельной нозологической формой. Синдром Леннокса-Гасто по различным данным составляет от 3% до 10% всех случаев детской эпилепсии. Его распространенность колеблется в пределах 1-2,8 случаев на 10 тыс. Несколько чаще встречается у мальчиков. Типичный возраст начала заболевания от 2 до 5 лет, реже — 6-8 лет. Сегодня СЛГ является тяжелым заболеванием с прогрессирующим течением, эффективное лечение которого пока является предметом надежд многих специалистов в области детской неврологии и эпилептологии.

Причины синдрома Леннокса-Гасто
Синдром Леннокса-Гасто относится к заболеваниям, этиологические факторы которых пока точно не установлены. Известно, что во многих случаях синдром носит симптоматический характер и формируется на фоне генетической патологии, последствий различных неблагоприятных факторов, действующих в перинатальном периоде и на 1-ом году жизни. Однако в большинстве случаев морфологический субстрат заболевания остается не выявленным. К этиофакторам, способным спровоцировать развитие СЛГ, относят гипоксию плода, внутриутробные инфекции (краснуху, цитомегалию, герпес, токсоплазмоз), родовые травмы новорожденных (в первую очередь внутричерепные), недоношенность, асфиксию новорожденных, тяжелые инфекционные заболевания постнатального периода (менингит, энцефалит), аномалии развития головного мозга (гидроцефалию, кортикальную дисплазию, гипоплазию мозолистого тела и др.), метаболические нарушения с поражением ЦНС, отдельные генетические заболевания (например, туберозный склероз).
В 25-40% случаев синдром Леннокса-Гасто возникает у детей с отягощенным по эпилепсии семейным анамнезом. Кроме того, существует гипотеза об этиологической роли иммунных нарушений, в т. ч. возникающих вследствие вакцинации. Примерно в 30% случаев СЛГ является следствием эволюции синдрома Веста. Когда синдром Леннокса-Гасто манифестирует на фоне полного благополучия в здоровье ребенка и отсутствия в его анамнезе вышеперечисленных факторов, говорят о криптогенной (не имеющей вероятной причины) форме заболевания. Криптогенный вариант СЛГ встречается в 10-20% случаев и отличается более благоприятным течением.
Симптомы синдром Леннокса-Гасто
Симптоматический синдром Леннокса-Гасто, как правило, дебютирует на фоне уже имеющегося отставания в умственном и психическом развитии. При криптогенной форме развитие ребенка на момент манифестации синдрома соответствует норме. СЛГ отличается большой вариативностью приступов, их различной продолжительностью и частотой.
Атонические пароксизмы обусловлены кратковременной утратой мышечного тонуса. При их генерализованном характере происходит падение ребенка, т. н. «дроп-атака». Локальные пароксизмы могут иметь вид внезапного подгибания коленей, выпадения предметов из рук, кивков головой и т. п. Отличительной чертой атонических эпизодов при СЛГ является их молниеносность и кратковременность (до 5 сек.). Генерализованные атонические пароксизмы СЛГ требуют дифференцировки от приступов миоклонически-астатической эпилепсии, обмороков, ОНМК.
Миоклонические пароксизмы представляют собой локальные мышечные подергивания. Чаще охватывают мышцы-сгибатели проксимальных отделов рук, при распространении на нижние конечности происходит падение. Характеризуются симметричным серийным возникновением в обеих конечностях и стереотипностью. Нуждаются в дифференцировке с миоклониями при клещевом энцефалите и токсических поражениях ЦНС; миоклонусом неэпилептического характера, для которого типичны нерегулярные асимметричные миоклонии, возникающие в ответ на различные сенсорные раздражители (звук, свет, прикосновение) и не сопровождающиеся изменениями ЭЭГ.
Тонические пароксизмы СЛГ часто возникают в период сна и отличаются своей кратковременностью (средняя длительность 10 сек.). Сопровождаются отключением сознания. Могут иметь генерализованный характер или проявляться в виде тонического напряжения отдельных мышечных групп (заднешейных, спинных, мышц брюшного пресса, плечевого пояса и пр.). Тонические пароксизмы сопровождаются тахикардией, цианозом лица, слезотечением, апноэ, гиперсаливацией. Минимальные локальные пароксизмы тонического характера иногда с трудом можно отдифференцировать от зевоты или потягивания.
Атипичные абсансы связаны с частичным нарушением сознания. Проявляются временным «оцепенением», отсутствием любой двигательной активности. При малой продолжительности абсансы зачастую не распознаются окружающими ребенка людьми. При СЛГ абсансы могут сопровождаться мышечной гипотонией (атонические абсансы) и гипертонусом мышц спины (ретропульсивные абсансы). Чаще, чем другие виды эпилепсии, синдром Леннокса-Гасто сопровождается статусом абсансов — непрерывно следующими друг за другом абсансами. Такой бессудорожный эпистатус обычно возникает при пробуждении, может длиться несколько часов и дней.
Задержка психомоторного развития (ЗПР) отмечается почти во всех случаях СЛГ. Ее выраженность зависит от формы синдрома (криптогенная или симптоматическая), характера фоновой патологии ЦНС, тяжести и частоты эпилептических пароксизмов. Как правило, на первый план выходят проблемы с обучением владению новыми навыками и с усвоением новой информации. Зачастую наблюдается агрессивность, гиперактивность, эмоциональная нестабильность, характерные для аутизма особенности характера. Около 50% подростков, имеющих синдром Леннокса-Гасто, не владеют навыками самообслуживания. Еще 25% социально и эмоционально дезадаптированы по причине выраженной олигофрении. Особенности поведения и характера не дают возможность нормально адаптироваться в социуме даже тем пациентам, у которых олигофрения имеет легкую степень выраженности. Нормальная социальная адаптация наблюдается лишь в 15% случаев.
Диагностика синдрома Леннокса-Гасто
Синдром Леннокса-Гасто устанавливается на основании типичной клинической картины, состоящей из полиморфных эпиприступов и симптомов отставания нейро-психического развития. Учитывается также возраст начала пароксизмов и семейный эпилептический анамнез. Большую диагностическую роль играет электроэнцефалография. Межприступная (интериктальная) ЭЭГ в бодрствующем состоянии регистрирует плохую структурированность и замедленность основного ритма. ЭЭГ-паттерн имеет картину гипсаритмии с большим количеством спайков различной амплитуды. Наиболее высокие пики регистрируются в лобной области. ЭЭГ-паттерн в период приступов зависит от их формы.
Методы нейровизуализации (МРТ и КТ головного мозга) выявляют преимущественно неспецифичные патологические изменения: внутреннюю гидроцефалию, атрофию подкорковых областей и корковых структур преимущественно лобной зоны, гипоплазию лобных долей. Попытки проанализировать при помощи ПЭТ головного мозга степень утилизации глюкозы церебральными тканями дали противоречивые сведенья: в одних случаях были выявлены зоны гиперметаболизма, в других — гипометаболизма; у части пациентов метаболизм глюкозы был в пределах нормы.
По причине большой вариативности пароксизмов, синдром Леннокса-Гасто следует дифференцировать с целым рядом других форм эпилепсии, дебютирующих в детском возрасте: с миоклонической эпилепсией, доброкачественной роландической эпилепсией, синдромом Веста, детской абсансной эпилепсией, дисметаболической эпилепсией при болезни Гоше, Краббе, Ниманна-Пика и др.
Лечение синдрома Леннокса-Гасто
Терапия проводится противоэпилептическими средствами. Применяются вальпроевая к-та, этосуксимид, карбамазепин, ламотриджин и др. В большинстве случаев проводится комбинированное лечение одним из указанных фармпрепаратов и вальпроатом натрия. Однако до 90% случаев синдрома Леннокса-Гасто являются резистентными к антиконвульсантной терапии. В связи с этим основной целью лечения является уменьшение числа эпиприступов и улучшение качества жизни ребенка и его семьи в межпароксизмальный период.
Неврологами и эпилептологами ведется поиск новых способов терапии. Доказанной является положительная роль кетогенной диеты, заключающейся в резком ограничении употребления углеводов и повышении содержания жиров в пище. Рядом клиницистов отмечен положительный эффект лечения синдрома Леннокса-Гасто большими дозировками иммуноглобулина. Наблюдалась эффективность применения АКТГ и глюкокортикоидов. В случаях, когда синдром Леннокса-Гасто сопровождается частыми и тяжелыми эпипароксизмами с падением и угрозой травматизации ребенка, совместно с нейрохирургом может быть рассмотрен вопрос о проведении хирургической операции рассечения мозолистого тела — каллозотомии. Подобное вмешательство не избавляет пациентов от приступов, но существенно уменьшает их интенсивность.
К новым способам лечения относится имплантация стимулятора блуждающего нерва и RNS-стимулятора. В первом случае прибор устанавливается подкожно в область ключицы, а его электрод проводят к проходящему в шее блуждающему нерву. По данным проведенных в США и Европе исследований, в 60% случаев данное устройство позволяет снизить количество эпиприступов. Во втором случае прибор вшивается под кожу головы, а его электроды имплантируются в зону эпилептогенного очага. С их помощью, подобно ЭЭГ, устройство постоянно регистрирует электрическую активность мозга. При получении сигналов, свидетельствующих о начинающемся пароксизме, прибор генерирует ответные импульсы, обеспечивающие супрессию эпилептической активности.
Прогноз синдрома Леннокса-Гасто
Синдром Леннокса-Гасто имеет в основном неблагоприятный прогноз. До 10% случаев заканчивается гибелью детей в течение первого десятилетия жизни. Летальные исходы связаны преимущественно с тяжелой травматизацией во время эпиприступов с падением. Прогностически неблагоприятными критериями считаются: манифестация синдрома в более раннем возрасте, начало судорог на фоне ЗПР, предшествующий синдром Веста, высокая частота и интенсивность пароксизмов. Невозможность медикаментозного купирования эпиприступов приводит к прогрессирующей ЗПР. Практически у всех пациентов наблюдается выраженная в различной степени умственная отсталость, половина больных не способны к самообслуживанию.
Синдром Веста (Уэста) - симптомы и лечение
Что такое синдром Веста (Уэста)? Причины возникновения, диагностику и методы лечения разберем в статье доктора Аграновича Андрея Олеговича, эпилептолога со стажем в 12 лет.
Над статьей доктора Аграновича Андрея Олеговича работали литературный редактор Вера Васина , научный редактор Роман Люкманов и шеф-редактор Маргарита Тихонова
Синдром Веста (West syndrome) — это одна из форм эпилептической энцефалопатии. Заболевание проявляется судорогами, задержкой психического и моторного развития. Также для синдрома Веста характерна гипсаритмия — хаотическая активность головного мозга, которая наблюдается на энцефалографии между приступами [1] .

Cиндром Веста ещё называют синдромом инфантильных спазмов, а иногда просто инфантильными или эпилептическими спазмами.
Заболевание встречается примерно в 0,25 случаях на 1000 новорождённых [2] .
Причины синдрома Веста
К развитию заболевания могут приводить различные факторы:
- Иммунные — в большинстве случаев болезнь Веста возникает из-за аутоиммунного процесса, например аутоиммунного энцефалита.
- Генетические — согласно последним исследованиям, генетические факторы приводят к развитию заболевания у 14,4 % пациентов. Чаще всего болезнь связывают с мутациями в генах GRIN1, GRIN2A, MAGI2, MEF2C, ARX, CDKL5, KCNQ2, FOXG1. Генетические мутации при синдроме Веста могут быть как случайными, так и наследоваться от родителей. Эти аномалии могут воздействовать не только напрямую, формируя патологическую клеточную структуру, но и приводить к структурным, сосудистым и метаболическим нарушениям, которые создают условия для развития эпилептических спазмов.
- Структурные — изменения головного мозга из-за врождённых пороков развития, нейрокожных синдромов и внутриутробных инфекций (например, цитомегаловирусной и герпетической инфекции), гипоксии плода и преждевременных родов. Генетически-структурные факторы приводят к развитию заболевания в 10 % случаев, структурно-врождённые — в 10,8 %, структурно-приобретённые — в 22,4 % случаев.
- Метаболические причины — нарушение обмена электролитов и витаминов, например биотина, фолиевой кислоты, витамина В12 и пиридоксина, низкий уровень кальция, нарушение обмена аминокислот, например фенилкетонурия и ацидемия — аномально высокая кислотность крови. Также синдром Веста может развиваться при дефиците сульфитоксидазы и фруктозо-1-6-дифосфатазы. Из-за метаболических причин заболевание возникает у 4,8 % пациентов.
- Инфекционные — инфекции центральной нервной системы приводят к развитию синдрома Веста у 2 % пациентов [3] .
Симптомы синдрома Веста
Основные симптомы синдрома Веста: инфантильные спазмы и задержка или регресс психомоторного развития.
Эпилептические спазмы — это приступы, при которых внезапно сгибаются или разгибаются мышцы плечевого пояса. Если такой приступ происходит у детей младше года, то его называют инфантильным спазмом.
Эпилептические спазмы иногда называют «салаамовыми поклонами», сравнивая с характерным жестом восточного приветствия: поднятием рук вверх и наклоном головы вперёд.
Инфантильные спазмы длятся 0,5-1 секунду. Они могут повторяться от нескольких раз в день до нескольких раз в минуту.
Задержка психомоторного развития может появиться как у детей с изначально нормальным развитием, так и при тяжёлой органической патологии. Ребёнок с синдромом Веста отстаёт от сверстников по развитию интеллекта, речи и моторных навыков. В норме ребёнок начинает разговаривать и постепенно развивать речь от 1 до 4 лет, к 6 месяцам он уже может садиться, а к году — ходить.
Задержка развития может усугубляться нарушением зрения или слуха, что дополнительно затрудняет обучение.
Патогенез синдрома Веста
Патогенез синдрома Веста до конца не изучен. Например, неясно, почему заболевание может развиться как при патологии всей коры головного мозга, так и при локальном очаге поражения.
Выделяют несколько теорий развития заболевания:
- Повышение в крови кортикотропин-рилизинг-гормона. Эта гипотеза основывается на эффективности адренокортикотропного гормона (АКТГ), который используют при лечении болезни. Данный гормон подавляет выработку кортикотропин-рилизинг-гормона.
- Десинхрония развития головного мозга. Предполагается, что в основе эпилептических спазмов лежит неравномерное развитие коры головного мозга.
- Активация глутаматных рецепторов. Известно, что глутамат оказывает на нейроны возбуждающее действие. Как показали эксперименты, проведённые на животных, именно введённые агонисты глутаматных рецепторов стали причиной развития инфантильных спазмов и гипсаритмии.
- Нарушение взаимодействия между корой головного мозга и подкорковыми структурами. В эксперименте на животных нарушение корково-подкорковых взаимодействий приводило к эпилептическим спазмам, значительной задержке развития и аутистическому поведению.
- Активация рецепторов ГАМК-Б. Эта гипотеза также была подтверждена экспериментально.
Разнородность гипотез позволяет предположить, что единого механизма развития синдрома Веста нет, и его патогенез проявляется на нескольких уровнях [1] .
Классификация и стадии развития синдрома Веста
Эпилептические спазмы подразделяют на генерализованные, фокальные и спазмы с неизвестным началом. Первично генерализованные формы эпилепсии имеют генетическую природу заболевания и моментально вовлекают оба полушария в патологическое возбуждение. Сокращение мышц может быть как симметричным, так и асимметричным. Во время приступа пациент может потерять сознание.
Фокальные приступы исходят из локального очага. При этом могут сокращаться как отдельные мышцы, так и группы мышц.

Синдром Веста может дебютировать как с генерализованными, так и с фокальными приступами, поэтому его относят к группе сочетанных генерализованных и фокальных эпилепсий.
В зависимости от того, какая группа мышц вовлечена в патологический процесс, выделяют три вида инфантильных спазмов:
- флексорные — активируются мышцы сгибания и возникают салаамовы судороги, при которых ребёнок будто обнимает себя;
- экстензорные — вовлечены разгибательные мышцы;
- смешанные [15] .
Осложнения синдрома Веста
К основным осложнениям синдрома Веста можно отнести интеллектуальные нарушения, а также задержку или регресс психоречевого развития, которые также являются симптомами болезни.
К основным осложнениям относятся интеллектуальные нарушения и задержка или регресс психоречевого развития.
Кроме того, у детей с синдромом Веста могут возникать психические нарушения:
- шизофреноподобные расстройства — сопровождаются галлюцинациями и бредовыми идеями, например манией преследования, ревности, навязчивыми мыслями о болезни или смерти близкого человека;
- аутистикоподобное поведение — проявляется сложностями в общении, неспособностью устанавливать тёплые эмоциональные отношения, речевыми расстройствами, сильной реакцией на сенсорные стимулы, ограниченными интересами, часто повторяющимися действиями (например, мычанием, прыжками, раскачиваниями, похлопываниями ладонями и постукиваниями пальцами) [18] .
Диагностика синдрома Веста
Основными критериями установления диагноза являются гипсаритмия, инфантильные спазмы и задержка психомоторного развития.
Диагностику проводит невролог или эпилептолог.
Врач обращает внимание на признаки регресса или задержки психомоторного развития. Доктор постарается установить временную связь между этими нарушениями и появлением инфантильных спазмов. Также он спросит, как протекала беременность и роды и есть ли в семье дети с подобным заболеванием.
Чтобы подобрать лечение, врач уточнит, какие противоэпилептические препараты уже применялись и были ли они эффективны.
Неврологический осмотр
Врач отмечает, есть ли признаки дисэмбриогенеза — малых врождённых дефектов, каждый из которых существенно не влияет на работу организма, но в совокупности могут служить маркерами наследственных патологий. Также доктор оценивает зрительный и слуховой контакт с ребёнком, изменение мышечного тонуса и наличие врождённых рефлексов, например хватательного, рефлекса опоры и автоматической походки новорождённых. Большинство таких рефлексов в норме должны исчезнуть к первому году жизни.
Если спазмы у ребёнка случаются часто, то врач сможет зарегистрировать это состояние. Если во время приёма приступа не было, то его описывают со слов родственников.
Энцефалография (ЭЭГ)
Для большинства эпилепсий предпочтителен видео-ЭЭГ-мониторинг с обязательным включением записи сна [16] . Длительность мониторинга индивидуальна — от 2 до 24 часов. Исследование чаще всего проводится амбулаторно.
При проведении видео-ЭЭГ-мониторинга врач обращает внимание на следующие признаки:
- Гипсаритмия — повторяющаяся картина активности головного мозга, при которой медленные волны высокой амплитуды чередуются с острыми волнами, т. е. точками высокой активности мозга.
- Иктальные эпилептические паттерны — острые волны, подавление активности, комплексы «острая-медленная волна». Эти паттерны подтверждает эпилептический характер приступа.
- Чёткий очаг — может указывать на наличие патологии, которую лечат хирургически (например, опухоль).
Также ЭЭГ позволяет оценить эффективность лечения. Как часто и как долго её нужно проводить, зависит от частоты приступов и эффективности лечения, т. е. всё индивидуально.

Принципиально рутинная электроэнцефалограмма не отличается от видео-ЭЭГ-мониторинга. В обоих случаях используется электроэнцефалограф, который регистрирует биоэлектрическую активность головного мозга и выводит эту информацию в виде графиков. Но видео-ЭЭГ-мониторинг сопровождает эту запись видео-контролем двигательной активности, который синхронизирован по времени с ЭЭГ. Также видео-ЭЭГ-мониторинг проводится дольше, от часа до нескольких суток, что значимо повышает качество и информативность исследования.
Магнитно-резонансная томография (МРТ)
Если есть подозрения на фокальную кортикальную дисплазию, т. е. на локальное нарушение в строении коры головного мозга, рекомендуется выполнить МРТ головного мозга высокого разрешения (не менее 1,5 Тс) [16] . МРТ проводят по эпилептологическому протоколу, что подразумевает меньшую толщину среза, большую длительность исследования и использование специальных программ. Детям желательно проводить МРТ под наркозом: в течение всего исследования важно сохранять неподвижность, без наркоза ребёнок вряд ли сможет это сделать.
Генетические исследования
При подозрении на генетическую причину заболевания врач-генетик может назначить специальные исследования: секвенирование отдельных генов, кариотипирование, микроматричный хромосомный анализ и полное экзомное секвенирование [16] .
Синдром Веста следует отличать от доброкачественного миоклонуса раннего младенческого возраста, доброкачественной миоклонической эпилепсии младенцев и гиперкинезов.
Лечение синдрома Веста
При развитии инфантильного спазма нужно обратиться к врачу. После установки диагноза лечение нужно начинать немедленно: чем дольше протекают инфантильные спазмы и гипсаритмия, тем более выраженными будут когнитивные нарушения [19] .
Гормональные препараты
Лечение инфантильных спазмов начинают с гормональной терапии. Как правило, для этого применяется аденокортикотропный гормон (АКТГ) или его синтетический аналог Тетракозактид.
АКТГ назначают для снижения частоты приступов и улучшения картины ЭЭГ. В среднем длительность курса составляет от 1 до 3 месяцев.
Противоэпилептические препараты
- обострениях язвенной болезни желудка и двенадцатиперстной кишки; ;
- первичной надпочечниковой недостаточности;
- устойчивой к лечению сердечной недостаточности; ;
- инфекционных заболеваниях (если одновременно не применяются антибиотики);
- адреногенитальном синдроме;
- беременности и грудном вскармливании;
- повышенной чувствительности к АКТГ.
Также препарат рекомендован пациентам с туберозным склерозом [19] .
Приём Вигабатрина более нескольких месяцев может сопровождаться побочными эффектами: токсическим воздействием на сетчатку глаза и базальные ганглии головного мозга, что проявляется неврологическими и психическими нарушениями. В этом случае необходимо постепенно прекратить приём препарата или изменить дозы.
Также возможно использование других противоэпилептических препаратов, иногда сразу нескольких: Вальпроевой кислоты, Леветирацетама, Топирамата, Зонисамида, Клоназепама, Клобазама. Вигабатрин является препаратом второй линии именно при синдроме Веста. При других формах эпилепсии он практически не используется, в отличие от перечисленных антиэпилептических препаратов.
Иногда у людей, которые долго страдали от приступов и наконец, благодаря медикаментозному лечению, достигли ремиссии может развиться феномен «насильственной нормализации энцефалограммы», или синдром Ландольта. Этот синдром проявляется бессонницей, психозом и аффективными нарушениями: продолжительными периодами грусти, веселья или их сочетанием.
Иммуноглобулины
Иммуноглобулины используются в терапии, когда другие препараты не помогают. Чаще всего эффективность антиэпилептической терапии оценивается через несколько недель или месяцев лечения. Если иммуноглобулины тоже не помогли, при этом синдром не связан с метаболическим или структурным нарушением, рассматривают различные комбинации противоэпилептических препаратов и применение кетогенной диеты [19] .
При кетогенной диете в рационе практически отсутствуют углеводы, содержится умеренное количество белков и много жиров. Диету подбирает команда из диетолога, эпилептолога и педиатра при неэффективности других методов лечения.
Эффективность диеты оценивают через несколько месяцев. Обычно её применяют не более нескольких лет, при большем сроке нужно контролировать биохимические показатели и кетоновые тела в крови.
При наличии чётких изменений на МРТ или выраженных и явных изменениях на ЭЭГ может быть предложено хирургическое лечение: резекционная хирургия или имплантация стимулятора блуждающего нерва [20] .

Реабилитация
ЛФК, занятия с логопедами, дефектологами и психологами нужны только в тех случаях, когда есть соответствующие нарушения.
При синдроме Веста очень важно своевременное и адекватное лечение. В последние годы достигнут значительный прогресс в подборе методов терапии, но всё равно встречаются случаи, устойчивые к лечению.
Согласно исследованиям, от синдрома Веста погибает 22 % пациентов, 34 % — имеют психомоторные нарушения и 16 % детей развиваются нормально. У 55 % детей в дальнейшем развиваются другие виды эпилептических приступов [21] .
Ещё в одном исследовании было показано, что без лечения, при позднем его начале или устойчивости к препаратам умственная отсталость развивается у 90 % детей. Синдром Веста также может трансформироваться в другие формы эпилепсии, чаще всего в фокальные или мультифокальные формы, а так же в синдром Леннокса — Гасто [16] .
Профилактики синдрома Веста не существует. Исключением, вероятно, является туберозный склероз, при котором у большинства пациентов развиваются эпилептические спазмы. В таких случаях, несмотря на невысокий уровень доказательности, считается целесообразным для профилактики принимать Вигабатрин [1] .
Читайте также: