Синдром Лоренса (Lawrence) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Неврология:
Популярные разделы сайта:
Синдромы Де Санктиса-Кахионе, Лоренса-Муна-Бидля, Лоу. Особенности
Синдром Де Санктиса-Кахионе. 1. Аутосомное рецессивное наследование, приводящее к нарушению восстановления ДНК.
2. Общие признаки синдрома Де Санктиса-Кахионе. Пигментозная ксеродерма.
3. Неврологические признаки синдрома Де Санктиса-Кахионе. Микроцефалия, припадки, умственная отсталость, варьирующая спастичность, гипотала-мические нарушения, атрофия коры головного мозга и мозжечка.
4. Продолжительность жизни укорочена вследствие частых случаев малигнизаций и дисфункции ЦНС.
Синдром Лоренса-Муна-Бидля. 1. Аутосомно рецессивный.
2. Общие признаки синдрома Лоренса-Муна-Бидля. Ожирение, гипогонадизм, гипогенита-лизм, полидактилия и пигментная ретинопатия.
3. Неврологические признаки синдрома Лоренса-Муна-Бидля. Умственная отсталость и прогрессирующее снижение зрения.
4. Продолжительность жизни нормальная, но ранняя смерть может наступать вследствие поражения почек или сердца.
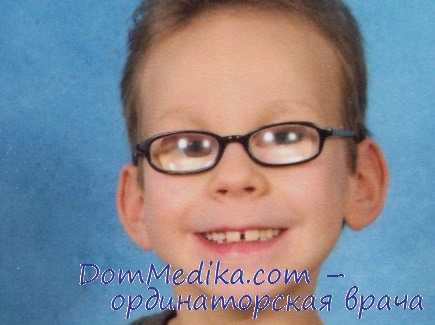
Синдром Лоу. 1. Рецессивный, сцепленный с половыми хромосомами.
2. Общие признаки синдрома Лоу. Глаукома, катаракта, аминоацидурия и органическая ацидурия.
3. Неврологические признаки синдрома Лоу. Хореоатетоз, гипотония, умственная отсталость и припадки.
4. Значительное отставание в развитии.
5. Лечение с помощью цитрата калия и цитрата натрия корректирует ацидоз, улучшает минерализацию костей и ускоряет рост.
ЛОРЕНСА-МУНА-БИДЛЯ СИНДРОМ
Лоренса-Муна-Бидля синдром (J. Z. Laurence, английский офтальмолог, 1830—1874; В. Ch. Moon, американский офтальмолог, 1844 — 1914; A. Biedl, чешский терапевт, 1869—1933) — нейроэндокринное заболевание, проявляющееся пигментным ретинитом, ожирением, полидактилией, гипогенитализмом и умственной отсталостью. Синдром описан в 1866 г. Лоренсом и Муном как сочетание пигментного ретинита с гипогенитализмом, задержкой роста и олигофренией. В 1920 г. Барде (G. Bardet) обратил внимание на полидактилию при этом синдроме, а в 1922 г. Бидль описал другие пороки развития при этом синдроме. Описано немногим более 400 больных. Заболевание часто имеет семейный характер, чаще встречается у лиц мужского пола.
Содержание
Этиология и патогенез
Этиология и патогенез изучены недостаточно. Наибольшее значение придается генетическим факторам, однако тип наследования не уточнен. Допускается, что синдром является следствием внутриутробного повреждения плода, напр, при токсоплазмозе (см.), краснухе (см.) у беременных. Наряду с врожденными пороками развития скелета, глаз, мозга, внутренних органов, прогрессирующими дистрофическими изменениями (напр., сетчатки глаз, почек) придается значение вторичным расстройствам, связанным с нарушением функции гипоталамических центров.
Патологическая анатомия
Морфологические изменения в мозге не специфичны для Лоренса-Муна-Бидля синдрома и у многих больных вообще отсутствуют. Описаны дистрофические изменения ядер гипоталамуса с уменьшением числа ганглиозных клеток и замещение их глиозными элементами, а также атрофия мозговых извилин, врожденное отсутствие мозолистого тела и др. Часто наблюдаются дефекты развития почек — фетальная дольчатость, гипоплазия, дисплазия; микроскопически в почках обнаруживается широкий спектр изменений — от небольшого разрастания мезенхимальных элементов до выраженной мезангиальной пролиферации, кистозного расширения канальцев с образованием кортикальных и медуллярных кист, перигломерулярный и интерстициальный фиброз, хроническая воспалительная клеточная инфильтрация. Врожденные пороки сердца и сосудов выявлены при аутопсии у 2/3 умерших. Половые органы, как правило, недоразвиты.
Клиническая картина
Ожирение (см.) встречается у 81—95% больных, чаще начинается уже на первом году жизни и увеличивается с возрастом. Пигментная дистрофия сетчатки, или пигментный ретинит (см. Тапетopетинальные дистрофии), хотя и относится к кардинальным симптомам заболевания, описана лишь у 15% больных; нарушения зрения наблюдаются у 92—93% больных (более 70% больных слепнут). Причиной прогрессирующей потери зрения наряду с пигментным ретинитом являются атрофия зрительного нерва (см.), глаукома (см.), катаракта (см.), близорукость (см.); описаны пороки развития глаз: микрофтальмии (см. Глаз), анофтальмия (см.), аниридия (см.), колобома радужной оболочки (см. Колобома).
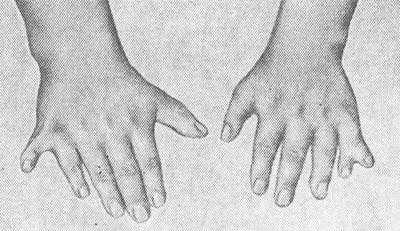
Полидактилия, обычно шестипалость (рис. 1), встречается у 70—80% больных; у некоторых больных имеется синдактилия (см.), иногда в сочетании с полидактилией, брахидактилия (см. Кисть), плоскостопие (см.). Описаны пороки развития черепа: микроцефалия (см.), гидроцефалия (см.), брахицефалия (см.), лобный гиперостоз (см. Морганьи синдром), деформация турецкого седла, асимметрия лица, а также дефекты позвонков и ребер. Больные обычно малого роста, созревание скелета замедлено.
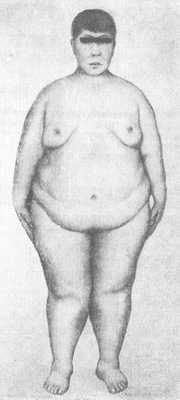
Гипогонадизм (см.) наблюдается у 74—85% больных мужчин и у 45—53% женщин; он может быть связан как с первичной недостаточностью половых желез, так и с понижением продукции гонадотропинов. У мужчин отмечается характерный евнухоидный вид, резкое ожирение, нередко гинекомастия (рис. 2), малые размеры яичек и наружных половых органов, рост волос на лобке по женскому типу. У женщин могут полностью отсутствовать вторичные половые признаки, наблюдаться аменорея и гипоплазия половых органов, вместе с тем в ряде случаев возможно нормальное половое развитие со способностью к деторождению. У некоторых больных имелись нарушения толерантности к глюкозе вплоть до развития сахарного диабета; иногда развивается несахарный диабет, отмечается артериальная гипертензия.
Врожденные пороки сердца, дефекты развития аорты и коронарных сосудов при жизни диагностируются редко.
На фоне врожденных дефектов почек (поликистоз, гипоплазия, гломерулярная дисплазия) возникают воспалительные процессы (хронический гломерулонефрит, пиелонефрит, абсцессы почек), которые обнаруживаются при урологическом обследовании с использованием радиорентгенологических методов и биопсии почек.
Клейн и Амманн (D. Klein, F. Ammann, 1969) предложили выделять полную форму синдрома (все пять кардинальных симптомов), неполную (один или два симптома отсутствуют), абортивную (один-два симптома или нечеткие проявления всех), атипичную (пигментного ретинита нет, но отмечаются другие поражения глаз) и экстенсивную форму (наряду с пятью основными симптомами имеются другие пороки развития). Полная форма встречается относительно редко: из 132 случаев, собранных по литературе Теленом (E. Thelen, 1958), она выявлена лишь в 26.
Диагноз
Диагноз при полной форме синдрома не представляет затруднений. Полидактилия выявляется уже при рождении. Нередко на первом году жизни развивается ожирение. В дальнейшем выявляются другие симптомы. Дифференциальный диагноз проводится с адипозо-генитальной дистрофией (см.), к-рой не свойственны поражения глаз, полидактилия и другие пороки развития, олигофрения, а также с синдромом Альстрема — Халлгрена, характеризующимся сочетанием пигментного ретинита с ожирением, глухотой, сахарным диабетом (иногда психические расстройства) при отсутствии полидактилии и гипогонадизма.
Лечение и Прогноз
Прогноз неблагоприятный. Обычно больные слепнут, ожирение и почечная недостаточность усиливаются. Больные чаще умирают в молодом возрасте; старшей из умерших было 50 лет. У трети умерших причиной смерти была уремия.
Библиография Жуковский М. А. Детская эндокринология, с. 316, М., 1971; Потемкин В. В. Эндокринология, М., 1978; Руководство по клинической эндокринологии, под ред. В. Г. Баранова, Л., 1977; Bardet G. Sur un syndrome d’obesite congenitale avec polydactilie et retinite pigmentaire, P., 1920; Biedl A. Ein Geschwisterpaar mit adiposogenitaler Distrophie, Dtsch, med. Wschr., S. 1630, 1922; Laurence J. C. a. Moon R. С. Four cases of «retinitis pigmentosa», occuring in the same family and accompanied by general imperfections of development, Ophthal. Rev., v. 2, p. 32, 1866; Me Loughlin T. G. a. Shanklin D. R. Pathology of Laurence — Moon — Bardet — Biedl syndrome, J. Path. Bact., v. 93, p. 65, 1967; Rimoin D. L. a. Schimke R. N. Genetic disorders of the endocrine glands, St Louis, 1971; Textbook of endocrinology, ed. by R. H. Williams, Philadelphia, 1974.
Синдром Лоренса-Муна-Барде-Бидля
Синдром Лоренса-Муна-Барде-Бидля — это аутосомно-рецессивная нейроэндокринная болезнь, для которой характерно ожирение, дистрофические поражения сетчатки глаз, интеллектуальные расстройства. Патология также проявляется разнообразными деформациями скелета, врожденными пороками развития внутренних органов, неврологическими осложнениями. Для диагностики заболевания требуется проведение нейровизуализации, осмотра глазного дна, комплексного лабораторного обследования. Поддерживающая терапия включает медикаменты (ноотропы, гормоны), соблюдение диеты, логопедическую и дефектологическую коррекцию.
МКБ-10

Общие сведения
Первые сведения о патологии появились в 1866 г., когда английский офтальмолог Д. Лоренс и американский офтальмолог Р. Мун описали семейный случай ожирения, умственной отсталости и развившегося впоследствии тетрапареза. В 20-х гг. ХХ века французский врач Дж. Барде и венгерский специалист А. Бидль заявили о сходных клинических наблюдениях, которые в 1925 г. были объединены в одно заболевание — синдром Лоренса-Муна-Барде-Бидля. В Европе и США болезнь встречается с частотой 1 случай на 140-160 тыс. населения, а в азиатских и африканских странах — в 2-3 раза чаще.
Причины
На сегодня описано более 15 вариантов генных дефектов, при которых возникает синдром Лоренса-Муна-Барде-Бидля. 45% случаев связаны с мутацией в локусе BBS1 (11q13), также часто встречается поражение BBS2 (16q22), BBS3 (3р13), BBS4 (15q21). Наследование заболевания происходит по аутосомно-рецессивному типу, однако четкий семейный характер прослеживается только у 40% пациентов. Патология имеет вариабельную пенетрантность.
Патогенез
По механизму развития синдром принадлежит к категории цилиопатий, которые характеризуются нарушением строения и функционирования ресничек. В норме эти образования находятся на поверхности клеток различных тканей, причем они делятся на первичные (неподвижные) и вторичные (подвижные). Для патологии Лоренса-Муна-Барде-Бидля типично поражение первичных ресничек, состоящих из 9 пар периферических микротрубочек, одной пары центральных микротрубочек.
Несмотря на свою неподвижность, реснички участвуют во многих физиологических процессах в клетке: помогают в транспорте клеточных сигналов, влияют на особенности протекания биохимических реакций, способствуют правильной ориентации плоскости деления. В физиологических условиях они содержатся в фоторецепторах, киноцилиях, остеоцитах, других клетках внутренних органов, поэтому у страдающих синдромом Лоренса-Муна-Барде-Бидля наблюдается полиморфизм симптоматики.
Симптомы
Первые клинические проявления синдрома возникают уже в раннем детстве, однако вследствие разнообразия симптоматики и отсутствия патогномоничных признаков они не всегда диагностируются своевременно. Наиболее типичные органы-мишени: глаза, трубчатые кости, органы эндокринной системы, головной мозг. Некоторые ученые предлагают выделять отдельно синдромы Лоренса-Муна и Барде-Бидля, но эта теория пока не получила должной поддержки.
Для синдрома Лоренса-Муна-Барде-Бидля характерна избыточная масса тела, у 72-92% пациентов формируется ожирение по центральному типу — максимальное количество жировых отложений сосредоточено в области живота. В результате метаболических расстройств у половины больных развивается сахарный диабет 2-го типа. Выраженный гипогонадизм (недоразвитие половых органов) выявляется у 74-85% мужчин, 45-53% женщин.
Еще одна типичная группа симптомов при болезни Лоренса-Муна-Барде-Бидля — психоневрологические нарушения. В раннем детском возрасте у 90% пациентов отмечается задержка речевого развития разной степени тяжести, а впоследствии более 80% больных имеют речевые дефекты. Умственная отсталость встречается в 75-90% случаев. Неврологические нарушения представлены расстройствами координации движений, снижением функции обонятельного нерва.
Осложнения
Течение синдрома Лоренса-Муна-Барде-Бидля нередко осложняется разнообразными соматическими заболеваниями. В 68% случаев развивается почечная недостаточность, обусловленная гидронефрозом, поликистозом или другими аномалиями почек. Более половины больных сталкиваются со стоматологическими проблемами, обширным кариозным поражением зубов. К более редким осложнениям наследственной болезни относят фиброз печени, врожденные пороки сердца.
Патология характеризуется быстрым набором веса, поэтому ожирение наблюдается, начиная с дошкольного возраста, и быстро достигает 3-4 степени. На фоне этого возникают тяжелые метаболические расстройства, в разы повышается риск эндокринных заболеваний, сердечно-сосудистых болезней — у 50% пациентов старше 34 лет проявляется артериальная гипертензия. При тяжелом поражении церебральных структур возможны судорожные приступы, спастическая параплегия или тетраплегия, экстрапирамидные нарушения.
Диагностика
При развернутой клинической картине диагностика не представляет затруднений, постановка диагноза синдрома Лоренса-Муна-Барде-Бидля возможна после клинического обследования у педиатра или врача-генетика. Однако неполные формы заболевания зачастую упускаются детскими врачами, из-за чего поддерживающая терапия назначается с опозданием. Для подтверждения диагноза применяются следующие методы:
- Рентгенография скелета. Исследование выполняется для детального изучения строения пальцев кистей и стоп, оценки степени костных деформаций, подготовки к хирургической коррекции полидактилии и синдактилии.
- Офтальмоскопия. По всей поверхности глазного дна определяются скопления пигмента в сочетании со сужением артерий сетчатки, признаками дегенерации в макулярной зоне. Такая картина указывает на возникновение пигментного ретинита.
- Нейровизуализация. По результатам КТ или МРТ головного мозга у большинства пациентов обнаруживается агенезия мозолистого тела, аномалии структур задней черепной ямки. Больным с судорожным синдромом диагностика дополняется ЭЭГ.
- Обследование почек. Как скрининговый метод диагностики используется УЗИ почек, по данным которого выявляются врожденные структурные аномалии органа. Функциональная активность почек оценивается с помощью реносцинтиграфии.
- Лабораторные методы. Пациентам рекомендован расширенный биохимический анализ крови, липидограмма, анализ на глюкозу натощак и глюкозотолерантный тест. Также проводится исследование уровней инсулина, С-пептид, содержания половых гормонов.
Лечение синдрома Лоренса-Муна-Барде-Бидля
Специфическое лечение заболевания отсутствует. Оказанием медицинской помощи занимается мультидисциплинарная команда, которая состоит из эндокринолога, невролога, нейропсихолога, коррекционных педагогов и других специалистов. С учетом ведущих клинических симптомов у конкретного пациента и используются следующие направления терапии:
- Диетотерапия. Для контроля массы тела, предупреждения сахарного диабета и гиперлипидемии подбирается питание с уменьшенным количеством простых углеводов, насыщенных жиров. Больным рекомендуется сбалансированный по калорийности рацион с повышенным содержанием витаминов, микроэлементов.
- Гормонотерапия. Заместительная терапия гонадотропинами применяется для коррекции гипогонадизма, возможного восстановления репродуктивной функции. При метаболических нарушениях может потребоваться введение инсулина.
- Защита глаз от ультрафиолета. Чтобы предупредить пигментный ретинит, пациентам показано ношение качественных солнцезащитных очков, в подростковом возрасте рассматривается вариант контактных линз с УФ-фильтрами.
- Ноотропная терапия. Специфические препараты назначаются для стимуляции мозгового кровообращения, улучшения метаболических процессов в нервной ткани. Лечение позволяет корректировать интеллектуальные нарушения, задержку развития речевых навыков.
- Нейропсихологическая реабилитация. Больным со сниженным интеллектом требуется помощь дефектологов, подбор специальных обучающих программ. Для ликвидации речевых нарушений необходима логопедическая коррекция.
- Хирургическое лечение. Помощь хирургов необходима для устранения врожденных аномалий пальцев рук и ног для улучшения их функциональности, достижения максимально возможного косметического результата.
Учитывая вероятные осложнения, пациентам с синдромом Лоренса-Муна-Барде-Бидля требуется пожизненное диспансерное наблюдение у офтальмолога, кардиолога, нефролога. Чтобы контролировать состояние внутренних органов, регулярно проводится лабораторно-инструментальное тестирование, при выявлении отклонений в показателях здоровья назначается соответствующее лечение.
Прогноз и профилактика
Синдром Лоренса-Муна-Барде-Бидля относится к неизлечимым патологиям, но при раннем начале терапии удается значительно повысить качество жизни больных, предупредить угрожающие жизни осложнения. Менее оптимистичный прогноз при наличии тяжелых врожденных пороков, результирующих полиорганной недостаточностью. Для профилактики болезни парам с отягощенной наследственностью показано медико-генетическое консультирование при планировании семьи.
1. Синдром Барде-Бидля/ Е.А. Потрохова, М.Л. Баабаян, Л.С. Балева// Российский вестник перинатологии и педиатрии. — 2020. — №6.
2. Синдром Барде-Бидля с врожденной аномалией развития почек у девочки 14 лет/ А.В. Бурлуцкая, Н.В. Савельева// Кубанский научный медицинкий вестник. — 2019. — №3.
3. Синдром Лоренса-Муна-Барде-Бидля/ С.Д. Касымова, М.А. Мирахмедова// Вестник последипломного образования в сфере здравоохранения. 2017. — №3.
4. Ранняя диагностика синдрома Барде-Бидля, ассоциированного с ожирением/ Н.Н. Волеводз, И.А. Еремина, Т.В. Семичева// Ожирение и метаболизм. — 2008. — №1.
Другие уточненные синдромы врожденных аномалий, не классифицированные в других рубриках
Термин «синдром» используется для обозначения устойчивых сочетаний патологических симптомов, как уточненной этиологии, так и этиологически неясных. В клинической генетике и тератологии термин «синдром» имеет несколько значений. Он часто применяется как синоним «болезни», когда речь идет о единой нозологической форме с определенной этиологией. При этом синдром чаще называют - врожденные нарушения.
Протокол "Другие уточненные синдромы врожденных аномалий, не классифицированных в других рубриках"
Код по МКБ-10: Q87.8

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
1. Умственная отсталость.
2. Легкая умственная отсталость.
3. Умеренная умственная отсталость.
4. Тяжелая умственная отсталость.
5. Глубокая умственная отсталость.
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, зрения, слуха, ожирение, расторможенность, врожденные пороки развития со стороны органов зрения, почек, опорно-двигательного аппарата; отягощенная наследственность, перинатальная патология.
Физикальное обследование
Синдром Альпорта - наследственная болезнь, характеризующаяся снижением остроты зрения (катаракта, пигментный ретинит, лентиконус, сферофакия), глухотой в связи с недоразвитием слуховых анализаторов, признаками нефрита (протеинурия, гематурия) и развитием в возрасте 16-35 лет почечной недостаточности. Наследуется по аутосомно-рецессивному типу, сцепленному с полом.
Синдром Лоренса-Муна-Бидля (фенотипические проявления) - основным симптомом заболевания является ожирение, пигментный ретинит, гипогенитализм, полидактилия, умственная отсталость. Полидактилия обнаруживается у 60-70% больных. Пигментная дегенерация сетчатки ведет к снижению зрения, обычно проявляющемуся к 6-7 годам и неуклонно прогрессирующее. Ожирение появляется еще раньше - на 1-2 году жизни и быстро прогрессирует. Среди других нарушений у больных отмечаются отставание в росте, глухота (5% случаев), врожденные пороки сердца, различные аномалии глаза (микрокорнеа, катаракта, микрофтальмия, макулярная дегенерация и другие). Часто имеются аномалии в строении черепа и лица (акроцефалия, широкое переносье, эпикант). Умственная отсталость от легкой до тяжелой.
Синдром Целвегера (церебро-гепато-ренальный синдром) - множественные признаки дисплазии (аномалии развития мозгового и лицевого черепа, глаз, ушей, половых органов), расстройство глотания, выраженная гипотония мышц, сухожильная арефлексия, нарушение психомоторного развития в сочетании с врожденной патологией печени и почек. Возможно в основе - расстройство аминокислотного обмена. Тип наследования - аутосомно-рецессивный.
Лабораторные исследования: общий анализ крови и мочи, кал на яйца глист, кариотип.
Инструментальные исследования
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза головного мозга.
2. Электромиография (ЭМГ) - снижение амплитуды интерференционной кривой при произвольном мышечном сокращении.
Тяжелый врожденный синдром Лоренса: критерии оценки, лечение
Автор статьи: Алена Арико (эксперт по эндокринологии).
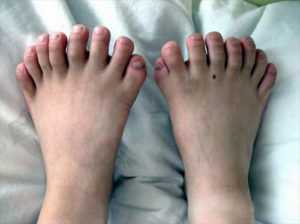
Синдром Лоренса считается врожденным, относится к редким заболеваниям, которые передаются ребенку в том случае, если оба родителя имеют патологические гены, при этом они сами могут быть полностью здоровыми. Чаще болеют мужчины.
При изучении структуры тканей мозга выявлены такие изменения:
- в гипоталамусе функционирующие клетки замещаются на неактивные;
- мозговые извилины атрофируются;
- отсутствует мозолистое тело, теряется связь между полушариями головного мозга.
Почки бывают не полностью сформированными, канальцы расширяются с формированием кисты, разрастается соединительная ткань. Такие изменения поддерживают хронический воспалительный процесс. У большинства пациентов есть пороки строения сердца, недоразвитие половых органов.
Гормональная недостаточность бывает периферической (нарушено образование стероидов в семенниках или яичниках) либо центрального происхождения (поражен гипоталамус, гипофиз). У части больных нарушена толерантность к глюкозе, выявляют сахарный и несахарный диабет, артериальную гипертензию.
У детей обычно обнаруживают несколько групп признаков. При классическом варианте имеется ожирение, нарушение зрения, полидактилия (больше 5-ти пальцев), гипогонадизм (недоразвитие половых желез), поражение почек и умственная отсталость.
Повышенная масса тела выявляется почти у 90% пациентов, проявляется уже на первых месяцах жизни, а по мере взросления степень ожирения увеличивается. Аппетит может быть не изменён или даже снижен. Жир откладывается на туловище и конечностях, особенно в области бёдер и плеч.
Потеря зрения связана с пигментным ретинитом. Он характеризуется нарушением адаптации к темноте, ночью пациент полностью не видит. По мере прогрессирования происходят следующие изменения:
- сужаются поля зрения;
- наступает атрофия (разрушение волокон) глазного нерва;
- присоединяется глаукома (высокое внутриглазное давление) и катаракта (помутневший хрусталик);
- близорукость.
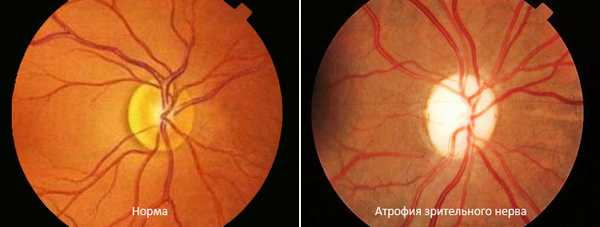
Снижение чёткости контуров предметов возникает в школьном возрасте, а к 20-ти годам большинство больных полностью слепнет.
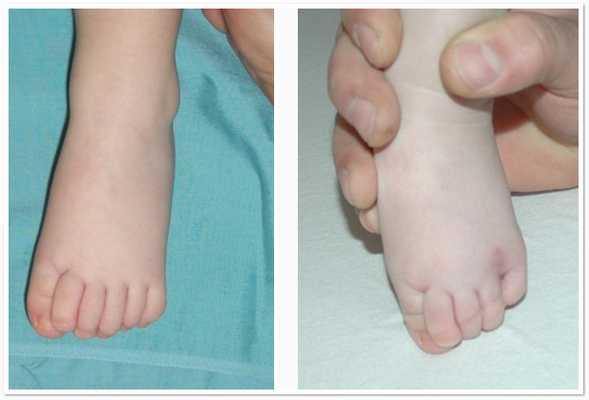
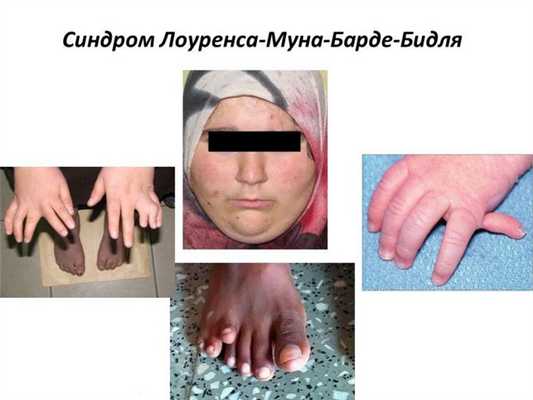
Полидактилию выявляют у 73% больных, пальцы могут срастаться. Не всегда шестой палец виден, иногда его находят только на рентгене. Также к костным аномалиям относятся:
- пороки черепа: уменьшенный или увеличенный в области лобных долей, асимметричный, деформированное «турецкое седло»;
- искривление рёбер, позвоночника;
- плоскостопие;
- низкий или высокий рост.
Умственная отсталость встречается часто, но не у всех больных. Описаны случаи олигофрении с раннего возраста и внезапная потеря умственных способностей с 7-ми лет. Психические нарушения в основном проявляются заторможенностью, быстрой утомляемостью при умственных нагрузках, снижением памяти. Нередко дети жалуются на головные боли, головокружения, прослеживается судорожный синдром и слабость в нижних конечностях.
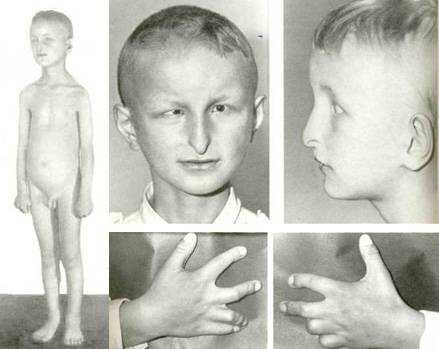
Синдром Лоренса-Муна-Бидля
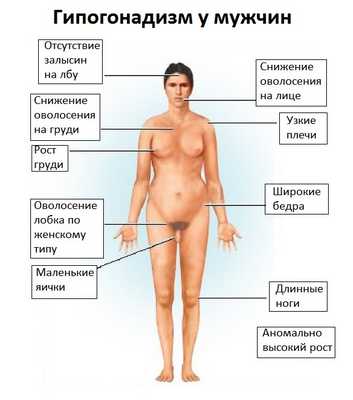
Проявления гипогонадизма: у мужчин не опущены или уменьшены яички, недостаточно развит половой член, нарушается образование сперматозоидов, обычно низкая потенция и слабое половое влечение. На лобке волосы растут по женскому типу, молочные железы увеличены. Женщины могут иметь детей, но встречаются пациентки с отсутствием месячных, недоразвитием матки и придатков.
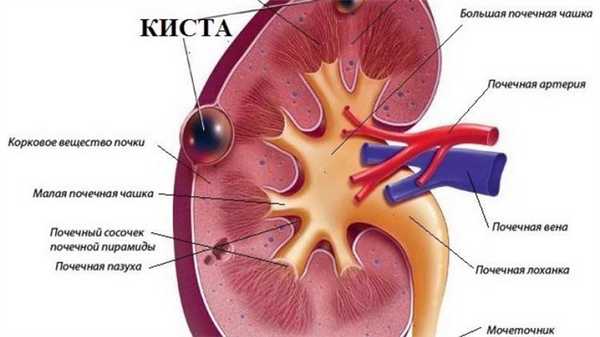
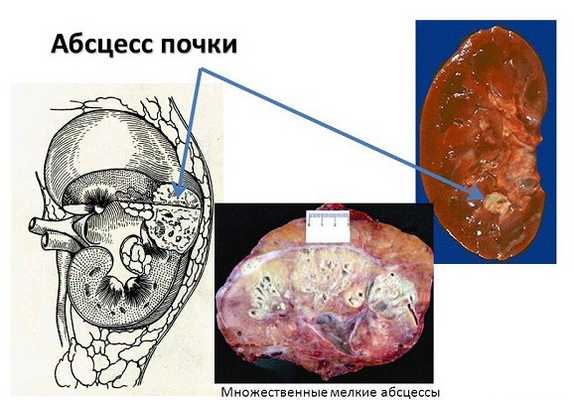
Аномальное строение почек: кисты, недоразвитие, нехватка клубочков провоцируют хронические, рецидивирующие воспалительные процессы. У пациентов диагностируют:
- пиелонефрит;
- абсцесс (нагноение) почечной ткани;
- гломерулонефрит.
При тяжелом течении возникает недостаточная фильтрация мочи, больные часто погибают от отравления организма продуктами азотистого обмена (уремии).
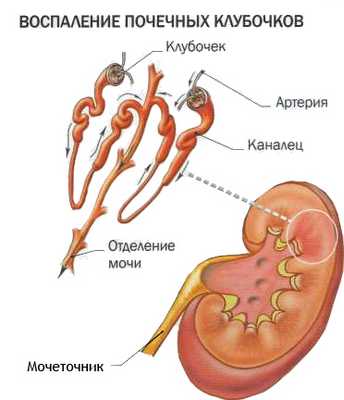
Гломерулонефрит
При наличии хотя бы четырех или пяти основных признаков диагноз можно считать подтвержденным. Сложнее ситуация, когда имеется 2-3 группы симптомов. Критериями, которые подтверждают предположение о наличии синдрома Лоренса, являются:
- наличие ожирения в семье;
- непропорциональное развитие тела с широкой грудной клеткой;
- беременность у матери протекала с угрозой выкидыша;
- ребенок родился раньше или позже срока;
- двигательные навыки появились с запозданием;
- интеллект снижен;
- ЭЭГ - грубые отклонения от возрастной нормы;
- ЭХО ЭГ - расширены желудочки мозга;
- глазное дно - ретинопатия, пигментный ретинит;
- рентгенография костей скелета показывает множественные аномалии;
- анализ крови на половые гормоны - снижен уровень;
- УЗИ почек диагностирует выраженные анатомические изменения.
Воздействовать на причину болезни невозможно, поэтому показана симптоматическая терапия. Наиболее убедительные результаты в лечении ожирения получены при комбинации низкокалорийной диеты и лечебной физкультуры. Детям рекомендуется исключение жирных продуктов животного происхождения и сладостей, мучных изделий.
При нарушении работы почек в рационе ограничивают белок. При нарушенной толерантности к углеводам или сахарном диабете показан «Метформин». Замедлить разрушение сетчатой оболочки помогает ношение солнцезащитных очков, использование «Тауфона», витаминных капель. Полидактилию и сращение пальцев устраняют хирургическим путем. В репродуктивном периоде показана коррекция дефицита половых гормонов путем заместительной терапии.
В целом синдром имеет прогрессирующее течение. С возрастом нарастает ожирение, недостаточность функции почек, исчезает зрение. Каждый третий пациент умирает от уремической комы. Она вызвана повреждением головного мозга невыведенными продуктами обмена. Большинство больных не доживает до 40 лет. При наличии 1-3-х проявлений (без поражения почек) бывали случаи, когда продолжительность жизни достигала 50-53-х лет.
Читайте подробнее в нашей статье о синдроме Лоренса.
Особенности синдрома Лоренса
О существовании этой врожденной патологии было известно еще с 1866 года, когда было описано офтальмологами Лоренсом и Муном сочетание задержки роста, полового развития, умственной отсталости со слабым зрением. В дальнейшем терапевтом Бидлем симптоматика была дополнена другими аномалиями.
Синдром относится к редким заболеваниям, которые передаются ребенку в том случае, если оба родителя имеют патологические гены, при этом они сами могут быть полностью здоровыми. Чаще болеют мужчины.
Рекомендуем прочитать статью о причинах гигантизма. Из нее вы узнаете о причинах гигантизма у детей его классификации, симптомах и внешние проявлениях, а также о диагностике состояния и лечении людей.
А здесь подробнее о синдроме Прадера.
Причины развития
- в гипоталамусе функционирующие клетки замещаются на неактивные;
- мозговые извилины атрофируются;
- отсутствует мозолистое тело, поэтому теряется связь между полушариями головного мозга.
Проявления синдрома Лоренса
Ожирение
Повышенная масса тела выявляется почти у 90% пациентов. Ее выявляют уже на первых месяцах жизни, а по мере взросления степень ожирения увеличивается. Аппетит может быть не изменен или даже немного снижен. Жир откладывается на туловище и конечностях, особенно в области бедер и плеч.
Нарушение зрения

Снижение четкости контуров предметов возникают в школьном возрасте, а к 20-ти годам большинство больных полностью слепнет.
Полидактилия
Шестипалость выявляют у 73 % больных, пальцы могут срастаться. Не всегда шестой палец виден, иногда его находят только на рентгене. Также к костным аномалиям относятся:
Умственная отсталость
Низкий интеллект встречается часто, но не у всех больных. Описаны случаи олигофрении с раннего возраста и внезапная потеря умственных способностей с 7-ми лет. Психические нарушения в основном проявляются заторможенностью, быстрой утомляемостью при умственных нагрузках, снижением памяти. Нередко дети жалуются на головные боли, головокружения, бывает судорожный синдром и слабость в нижних конечностях.
Гипогонадизм
У мужчин не опущены или уменьшены яички, недостаточно развит половой член, нарушается образование сперматозоидов, обычно низкая потенция и слабое половое влечение. На лобке волосы растут по женскому типу, молочные железы увеличены. Женщины могут иметь детей, но встречаются пациентки с отсутствием месячных, недоразвитием матки и придатков.
Поражение почек
Аномальное строение почек - кисты, недоразвитие, нехватка клубочков ̶ провоцируют хронические, рецидивирующие воспалительные процессы. У пациентов диагностируют:
Диагностика больных
При наличии хотя бы четырех или пяти основных признаков диагноз можно считать подтвержденным. Сложнее ситуация, когда имеется 2-3 группы симптомов. В таком случае показано дополнительное обследование. Критериями, которые подтверждают предположение о наличии синдрома Лоренса, являются:
- наличие ожирения в семье;
- непропорциональное развитие тела с широкой грудной клеткой;
- беременность у матери протекала с угрозой выкидыша;
- ребёнок родился раньше или позже срока;
- двигательные навыки появились с запозданием;
- интеллект снижен;
- ЭЭГ - грубые отклонения от возрастной нормы;
- ЭХО ЭГ - расширены желудочки мозга;
- глазное дно - ретинопатия, пигментный ретинит;
- рентгенография костей скелета показывает множественные аномалии;
- анализ крови на половые гормоны - снижен уровень;
- УЗИ почек диагностирует выраженные анатомические изменения.
Воздействовать на причину болезни невозможно, поэтому показана симптоматическая терапия. Были предприняты попытки снижения массы тела при помощи медикаментов, но наиболее убедительные результаты получены при комбинации низкокалорийной диеты и лечебной физкультуры. Детям рекомендуется исключение жирных продуктов животного происхождения, сладостей, мучных изделий.
При нарушении работы почек в рационе ограничивают белок. При нарушенной толерантности к углеводам или сахарном диабете показан «Метформин».
Замедлить разрушение сетчатой оболочки помогает ношение солнцезащитных очков, использование «Тауфона», витаминных капель. Полидактилию и сращение пальцев устраняют хирургическим путем. В репродуктивном периоде показана коррекция дефицита половых гормонов путем заместительной терапии.
Прогноз для больных
Рекомендуем прочитать статью о синдроме Уотерхауса-Фридериксена. Из нее вы узнаете о причинах развития, патогенезе синдрома Уотерхауса-Фридериксена при менингококковой инфекции, а также о симптомах патологии, диагностике и лечении данного синдрома.
А здесь подробнее о болезни гипопитуитаризм.
Синдром Лоренса относится к генетическому заболеванию, при котором у пациентов имеется ожирение, нарушение функций почек, половых органов, зрения, умственная отсталость и полидактилия. Фактором риска являются родственные браки, носительство дефектных генов родителями.
При диагностике учитывают наличие нескольких патологий одновременно и критерии, подтверждающие заболевание. Проводится симптоматическое лечение на фоне диеты и физкультуры. Прогноз чаще неблагоприятный.
Полезное видео
Смотрите на видео о наследственный заболеваниях:

Провоцировать нарушение работы гипофиза могут многие факторы. Признаки не всегда явные, а симптомы больше схожи на проблемы со стороны эндокринологии у мужчин и женщин. Лечение комплексное. Какие нарушения связаны с работой гипофиза?

Встречается гигантизм, причины которого - нарушение выработки гормона роста, у взрослых и детей. Симптомы - жажда, умственное отставание, высокий рост, нарушения формирования вторичных половых признаков. Лечение зависит от степени прогрессирования заболевания.

Возникает субклинический токсикоз преимущественно в неблагоприятных по количеству йода районах. Симптомы у женщин, в том числе при беременности, смазаны. Лишь нерегулярные месячные могут указать на проблему узлового зоба.
Развиться синдром Уотерхауса Фридериксена может по причине менингококковой инфекции. Преимущественно проявляется у детей. Каковы патогенез и лечение заболевания?

Основные причины, почему может развиться подострый тиреодит, - инфекция или вирус. Симптомы проявляются болью в горле, температурой. В зависимости от стадии они отличаются. Без лечения осложнения могут привести к рецидиву и нарушению работы щитовидки. Диагностику и лечение тиреоидита де Кервена нужно начинать сразу.
Читайте также:
- Системная красная волчанка. Лекарственные средства для лечения остеопороза
- УЗ-признаки заболеваний сосудов околоушной железы
- Синдром дефицита внимания и гиперактивности (СДВГ): синонимы, определение
- Показания к операции при неспецифическом язвенном колите. Виды операций при язвенном колите.
- Рентгенограмма с патологической тенью сердца при митральном стенозе: описание, заключение