Синдром Маха (Mach) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Синдром Марфана: причины появления, симптомы, диагностика и способы лечения.
Определение
Синдромом Марфана, или Марфана-Ашара, называют наследственное заболевание соединительной ткани с преимущественным поражением сердечнососудистой системы, скелета и органа зрения. Частота синдрома в популяции составляет от 1:3000 до 1:15000. Впервые его описали французские врачи Антонин Бернард Марфан (1896) и Эмиль Шарль Ашар (1902).
Причины появления синдрома Марфана
Синдром Марфана имеет аутосомно-доминантный тип наследования (то есть дети получают патологический ген от родителей, которые страдают данным заболеванием). Молекулярной основой синдрома Марфана является нарушение синтеза одного из белков соединительной ткани - фибриллина, который в норме придает ей эластичность и сократительную способность.
При СМ наблюдается дефицит фибриллина или его аномальное строение, поэтому соединительная ткань обладает повышенной растяжимостью и теряет способность выдерживать физиологические нагрузки.
Классификация заболевания
В зависимости от количества пораженных систем организма выделяют несколько форм синдрома Марфана:
- стертую - со слабо выраженными изменениями в 1-2 системах;
- выраженную - со слабо выраженными изменениями в 3 системах; значительно выраженными изменениями хотя бы в 1 системе; выраженными изменениями в 2-3 и более системах.
Принципиальную роль в определении прогноза болезни играет характер ее течения.
Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, с каждым годом жизни пациента возрастают риски фатальных осложнений.
Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Симптомы синдрома Марфана
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ весьма разнообразна.
Симптомы патологии сердечно-сосудистой системы
- Изменения сердечно-сосудистой системы отмечаются у большинства больных. Их основная причина - потеря способности стенок артерий и клапанных структур сердца выдерживать естественные гемодинамические нагрузки. Наиболее частая сердечная патология при синдроме Марфана -недостаточность митрального клапана, когда наблюдается поражение эластических структур створок и сухожильных нитей клапана с развитием его дисфункции. Со временем у многих больных эта дисфункция переходит в умеренную или тяжелую митральную недостаточность, требующую хирургической коррекции. Реже можно наблюдать аортальную и трикуспидальную недостаточность. Стенозы клапанов для СМ не характерны. При небольшой или умеренной хронической митральной недостаточности жалобы обычно отсутствуют (особенно при медленном нарастании недостаточности). Со временем появляются одышка, ощущение быстрой утомляемости и учащенное сердцебиение.
Клапанные пороки нередко осложняются инфекционным эндокардитом (воспалением внутренней оболочки сердца) - внезапно повышается температура, появляются озноб, боль в суставах, бледность кожных покровов и слизистых).
Кроме того, формируется геморрагическая сыпь, деформируются фаланги пальцев и ногтевые пластины (симптом «барабанных палочек» и «часовых стекол»).
Самую большую опасность представляют, пожалуй, патологические изменения аорты. Поскольку внутренний слой стенки сосудов также состоит из волокон соединительной ткани, сосуды постепенно изнашиваются. Принимая во внимание тот факт, что давление крови в аорте выше, чем на других участках сосудистого русла, это приводит к постепенному ее расширению, патологическому скоплению крови между сосудистыми стенками и формированию аневризмы или спонтанному разрыву. Основные симптомы аневризмы аорты следующие: осиплость голоса; нехватка воздуха; боль в плече; кровохарканье, боль в спине.
Симптомы поражения опорно-двигательного аппарата
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести заболевания и особенностей организма пациента. Люди с синдромом Марфана обычно отличаются высоким ростом. У детей с СМ очень длинные, непропорциональные росту руки и патологически удлиненные, тонкие пальцы, так называемые пальцы паука (арахнодактилия).
Лицо у людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Для пациентов характерны деформации грудной клетки, которые могут быть двух вариантов: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь).
Часто определяются различной степени выраженности сколиоз (отклонения позвоночного столба в сторону) или кифоз (формирование горба).
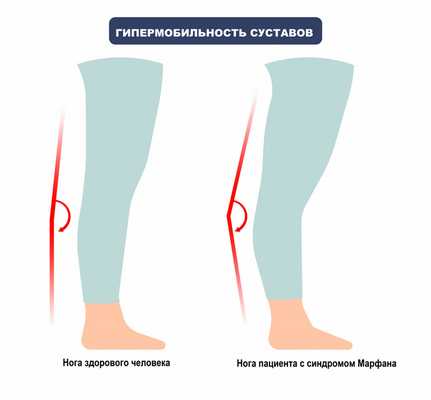
Кроме того, у пациентов с СМ нередко диагностируют плоскостопие, повышенную подвижность суставов, слабость связочного аппарата, неразвитость мышечных структур и подкожно-жирового слоя.
Симптомы поражения кожи
Высокий темп роста и нарушение выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется повышенной растяжимостью кожного покрова с образованием растяжек (стрий).
Симптомы поражения органа зрения
Чаще всего повреждения глаз у пациентов с синдромом Марфана включают:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Симптомы поражения органов дыхания
В легких пациентов с СМ может разрастаться может патологически разрастаться соединительная ткань, приводя к сужению бронхов и легочному фиброзу. Нередко на фоне генетической мутации манифестирует бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет риск развития спонтанного пневмоторакса — неотложной ситуации, при которой в плевральную полость попадает воздух, и легкое спадается: у пациента внезапно появляется одышка, резкая или ноющая боль в груди, сухой кашель.
Симптомы поражения желудочно-кишечного тракта
У людей с FBN1 мутацией нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз. Пациент испытывает привкус горечи во рту, тяжесть в области правого подреберья, в надчревной области. Часто беспокоит изжога, отрыжка, вздутие живота.
Симптомы поражения мочевыделительной системы
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Признаком нефроптоза легкой степени являются периодические одно- или двусторонние боли в проекции почек. Как правило, они проходят после непродолжительного пребывания в горизонтальном положении. Боль обычно тупая, ноющая, слабая или умеренная. Усугубление опущения ведет к нарушению оттока мочи и создает условия для развития инфекционных процессов и гидронефроза (скопления жидкости в почке). При присоединении инфекции может повышаться температура тела, появляться слизь и гной в моче.
По мере снижения функции почек у больного развиваются отеки, гипертензия и другие системные нарушения.
Симптомы поражения нервной системы
Расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. У таких пациентов развивается синдром хронической усталости, проявляющийся астенией и депрессивными расстройствами.
Интеллектуальная деятельность в большинстве случаев не нарушена, наоборот - среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Диагностика синдрома Марфана
Диагностика генетической аномалии включает комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7-18 лет — измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку 10 см.
- Лабораторное обследование.
Для подтверждения нарушений развития соединительной ткани и оценки степени выраженности мутации гена FBN1 пациентам с подозрением на синдром Марфана назначают:
Электрокардиография (ЭКГ) - повсеместно распространенный метод изучения работы сердца, в основе которого лежит графическое изображение электрических импульсов с�.
Синдром Стокса-Адамса
Синдром Морганьи-Адамса-Стокса (с-м МАС) - патологическое состояние, сопровождающееся внезапными приступами потери сознания, развивающимися вследствие острого нарушения сердечного ритма, приводящего к ишемии головного мозга. Согласно проведенным исследованиям, синдром МАС наблюдается у 70% пациентов, страдающих атриовентрикулярными блокадами и у 40% больных с мерцательной аритмией и нарушениями сердечного ритма.
Частота и тяжесть приступов могут быть совершенно различными. Некоторые приступы протекают с легким помутнением сознания, при других возникает аритмия, фибрилляция желудочков, отсутствие пульса и АД. Продолжительность приступа может составлять несколько секунд или минут. Как только частота сердечных сокращений нормализуется, человек сразу приходит в себя, но часто не помнит случившегося. В тяжелых случаях, может наблюдаться до 100 припадков в сутки.
Если вам нужен опытный кардиохирург, которому можно доверить самое ценное, собственную жизнь - обращайтесь в ОН КЛИНИК!
Как лечат синдром стокса-адамса
Синдром МАС - опасное состояние, которое требует серьезного лечения, поскольку в случае наступления аритмии, любой приступ может стать фатальным для человека. Медикаментозное лечение синдрома Стокса-Адамса результативно лишь в том случае, если наблюдаемые нарушения сердечного ритма, хорошо контролируются лекарствами. Во всех остальных случаях, пациентам необходима установка кардиостимулятора.
Показания и противопоказания
Основным показанием к установке кардиостимулятора, является аритмия, протекающая по типу бради- или тахикардии, которая сопровождается синдромом Адамса-Стокса, атриовентрикулярными блокадами и т.д. Абсолютных противопоказаний к имплантации водителя ритма нет. К относительным, относятся острые инфекционно-воспалительные болезни, лихорадка, обострение хронической патологии, психические проблемы.
Преимущества обращения в ОН КЛИНИК
В медицинском центре ОН КЛИНИК работают лучшие кардиологи Москвы, врачи высшей категории, кандидаты и доктора медицинских наук, имеющие огромный опыт успешного лечения синдрома МАС. Комплексный подход к проблеме позволяет решить ее наиболее результативно.
В качестве водителя ритма мы применяем широкий спектр комплексных сложных электронных систем, со строенной возможностью наблюдения ЭКГ. Электрокардиостимуляторы, изготовленные из биосовместимых материалов, обладают высокой степенью надежности и широкими возможностями модуляции импульсов по форме, амплитуде и частоте, что позволяет настроить прибор непосредственно под индивидуальные требования организма пациента. Для лекарственной профилактики приступов назначается противоаритмическая терапия препаратами нового поколения.

Перебои в работе сердца. Аритмия. Ответы на вопросы кардиолога ОН КЛИНИК
Лечение синдрома МАС в ОН КЛИНИК
Кислородное голодание мозга - это мощный удар по интеллекту и центральной нервной системе. Итогом любого приступа может стать смерть. Поэтому крайне важно своевременно диагностировать заболевание и провести грамотное лечение. Кардиологи ОН КЛИНИК проблему МАС решают с помощью таких оперативных методов:
- установка электрокардиостимулятора;
- лазерная деструкция альтернативных путей прохождения импульсов.
Имплантация кардиостимулятора, автоматически включающегося в момент замедления сердечных сокращений, позволяет обеспечить устойчивую поддержку сердечного ритма и правильную частоту сокращений миокарда (сердечной мышцы).
Подготовка к операции
Перед операцией необходимо пройти комплексное обследование и сдать ряд необходимых анализов. Если нет никаких противопоказаний для установки электрокардиостимулятора, проводится операция.
Как проводится установка кардиостимулятора
Имплантация электрокардиостимулятора в ОН КЛИНИК относится к микрохирургическим операциям и не требует общего наркоза. Операция проводится под местной анестезией и занимает меньше часа.
Процедура имплантации заключается в следующем:
- специальный электрод из тонкопроводящего сплава, под визуальным контролем, проводится через подключичную вену в правое предсердие, таким образом, чтобы он находился в непосредственном контакте с эндокардом;
- второй конец электрода подсоединяется к кардиостимулятору, который имплантируется под кожу в подключичной области, под малую грудную мышцу или (редко) в надчревной области.
Фиксация прибора выполняется таким образом, чтобы доставлять наименее дискомфорта пациенту. Тип кардиостимулятора и режим его работы подбирается строго индивидуально для каждого клинического случая.
Чего ожидать после операции
После имплантации кардиостимулятора пациент несколько суток проводит в стационаре, под врачебным наблюдением, а затем выписывается домой. Обязательно посещать кардиохирурга каждые три месяца в первый год после операции, в дальнейшем, раз в полгода и раз в год. Более подробно о периоде реабилитации и дальнейшем режиме жизни расскажет кардиохирург.
Техническое оснащение ОН КЛИНИК и высокая квалификация специалистов позволяет нам оказывать полный спектр услуг по консервативному и оперативному лечению кардиологических заболеваний. Собственный комфортный стационар, полностью оснащенные операционные, постоянный врачебный контроль и круглосуточная качественная медицинская помощь - основные преимущества обращения в наш медицинский центр.
Расстройство аутистического спектра: «дети дождя»

Обзор
Кадр из фильма «Человек дождя», самой известной истории о человеке с аутизмом в поп-культуре
Автор
Редакторы
Статья на конкурс «био/мол/текст»: Они видят мир по-другому, не любят контактировать с обществом, имеют «странности» в поведении и нарушения речи. Родители и воспитатели часто принимают их за одаренных детей со своими особенностями, но врачи уже давно определили их диагноз — «расстройство аутистического спектра». В этой статье вы узнаете о том, что такое расстройство аутистического спектра и что известно о причинах его развития.
Конкурс «био/мол/текст»-2018
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2018.
Генеральный спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Спонсором приза зрительских симпатий выступил медико-генетический центр Genotek.
Если вы знаете одного человека с аутизмом,
то вы знаете одного человека с аутизмом.
Стивен Шор,
профессор Университета Адельфи (США),
имеет диагноз «аутизм»
У простого обывателя при упоминании термина «расстройство аутистического спектра» (РАС) в голове, скорее всего, всплывет образ главного героя фильма «Человек дождя», и на этом, пожалуй, всё. На постсоветском пространстве тема РАС не освещается в достаточной степени, а диагностика в большинстве случаев далека от совершенства [1]. Ежегодно в мире увеличивается количество детей с расстройствами аутистического спектра. Медики говорят о разных причинах: улучшенная система диагностики, подозрение влияния ранней вакцинации, вредное воздействие пресловутых ГМО и даже старший возраст будущих пап. Так что такое РАС и что ученым уже удалось узнать о причинах его развития?
Расстройство аутистического спектра (РАС) — это расстройство нервной системы, которое характеризуется дефицитом в социальных взаимодействиях и коммуникацией с наличием стереотипий (повторяющихся действий) [2], и, по данным Соединенных Штатов Америки за 2014 год, оно диагностируется у одного из 59 детей [3]. В России распространенность составляет один случай на 100 детей, но официальный диагноз получают гораздо меньшее количество людей [1]. РАС диагностируется во всех расовых, этнических и социально-экономических группах, в пять раз чаще встречается у мальчиков, чем у девочек [4]. На данный момент причины болезни не известны, но предполагается, что оно возникает вследствие сложного взаимодействия между генетическими, эпигенетическими и экологическими факторами [5], [6] (рис. 1).
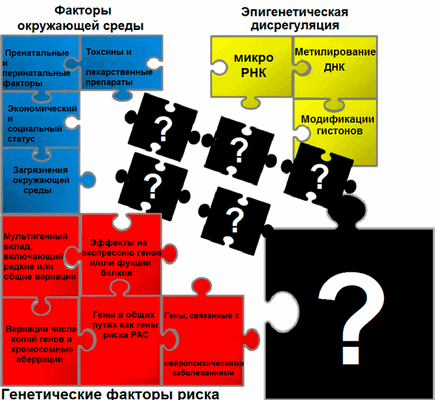
Рисунок 1. Причины РАС
До мая 2013 года в число официальных диагнозов аутистического спектра в американском «Диагностическом и статистическом руководстве по психическим расстройствам» (Diagnostic and statistical manual of mental disorders, DSM) входили: аутистическое расстройство, первазивное расстройство развития без дополнительных уточнений (ПРР-БДУ), синдром Аспергера, детское дезинтегративное расстройство и синдром Ретта. Сегодня в последнем, пятом издании DSM, существует только один диагноз — «расстройство аутистического спектра» с тремя степенями тяжести, но многие терапевты, клиницисты, родители и организации продолжают использовать такие термины, как ПРР-БДУ и синдром Аспергера [2].
Симптомы
Расстройство аутистического спектра часто характеризуется проблемами в социальных, коммуникативных и интеллектуальных способностях пациентов. В зависимости от возраста и интеллекта, у детей с аутизмом заметна различная степень дефицита коммуникации. Эти дефициты проявляются в речевых задержках, монотонной речи, эхолалии (неконтролируемом автоматическом повторении слов, услышанных в чужой речи), а также варьируют от плохого понимания до полного отсутствия устной речи. Невербальная коммуникация также нарушена и может включать трудности в установлении зрительного контакта, сложности в понимании выражений лица и жестов. Еще одной важной особенностью людей с РАС является дефицит социально-эмоциональной взаимности (рис. 2) [7].
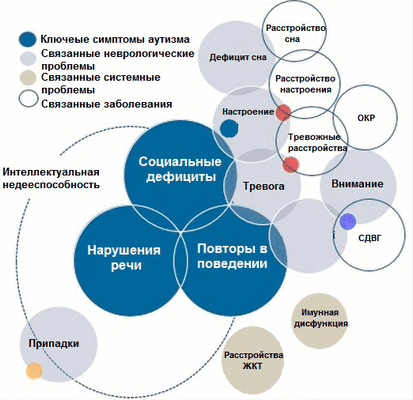
Рисунок 2. Симптоматика РАС
Проще говоря, дети с расстройством аутистического спектра не интересуются общением с людьми, плохо их понимают, любят придерживаться различных ритуалов, склонны к повторяющимся движениям тела, могут иметь языковые проблемы и задержки в интеллектуальном развитии. Различные симптомы приводят к значительному ухудшению во многих областях адаптивного функционирования. Одновременно с этим, дети с РАС часто имеют и множество сильных сторон: усидчивость, внимание к деталям, хорошая зрительная и механическая память, склонность к однообразной работе, что может быть полезно в некоторых профессиях.
История заболевания
Термин «аутизм» используется уже более 100 лет (с 1908 года). Впервые он был озвучен Эйгеном Блейлером, швейцарским психиатром, и использовался для описания пациентов с шизофренией, которые были особенно поглощены сами собой. Термин аутизм, который использовал Блейлер, происходит от греческого слова autós, что означает «сам». Оно предназначалось для описания «изолированного я», которое он увидел у людей с шизофренией [8]. На самом деле, эти диагнозы различны, так как у ребенка с аутизмом нет галлюцинаций, иллюзий, они не пользуются речью, чтобы передать свои иррациональные мысли, потому что они часто вообще не используют речь. К тому же дети с РАС имеют стабильные симптомы на протяжении жизни, а диагноз «шизофрения» обычно подразумевает периоды ремиссии.
В 1943 году доктор по имени Лео Каннер проводил наблюдения групп детей, которые ранее считались умственно отсталыми. Он отмечал, что у детей были трудности в социальном взаимодействии, тревожность при отклонении от привычного уклада жизни, эхолалия, ограниченность репертуара спонтанной активности, но при этом хороший интеллектуальный потенциал, неплохая память, гиперчувствительность к сенсорным воздействиям. Каннеру принадлежит введение термина «ранний детский аутизм» (РДА) для описания совокупности симптомов у детей, которых он изучал [8].
Немецкий ученый Ганс Аспергер в 1944 году описал «более мягкую» форму аутизма, которая до сегодняшнего времени была известна как синдром Аспергера. Он описывал случаи с мальчиками, которые были очень умными, но имели проблемы с социальными взаимодействиями. Он отмечал у детей трудности со зрительным контактом, стереотипные слова и движения, а также сопротивление изменениям, но при этом они не имели недостатков в речевом и языковом образовании. В отличие от Каннера, Аспергер отмечал также проблемы с координацией у этих детей, но при этом больше способностей к абстрактному мышлению. К сожалению, исследование Аспергера было обнаружено лишь три десятилетия спустя, когда люди начали подвергать сомнению используемые в то время диагностические критерии. Только в 1980-х годах работа Аспергера была переведена на английский язык, опубликована и получила известность [8], [9].
В 1967 году психиатр Бруно Беттельгейм писал, что аутизм не имеет органической основы, но является результатом воспитания матерей, которые сознательно или бессознательно не хотели своих детей, что в свою очередь приводило к сдержанности в отношениях с ними. Он утверждал, что основной причиной заболевания было отрицательное родительское отношение к младенцам на критических ранних стадиях их психологического развития [10].
Бернард Римланд, психолог и отец ребенка с аутизмом, не соглашался с Беттельгеймом. Он не мог смириться с мыслью, что причиной аутизма его сына были либо его родительские ошибки, либо ошибки его жены. В 1964 году Бернард Римланд опубликовал работу «Инфантильный аутизм: синдром и его последствия для нейронной теории поведения», которая указала направление для дальнейших исследований в то время [8].
Аутизм стал лучше известен в 1970-х годах, но на тот момент многие родители всё еще путали аутизм с умственной отсталостью и психозом. Ученые же начали вносить ясность в этиологию заболевания: исследование 1977 года на близнецах показало, что аутизм в значительной степени обусловлен генетикой и биологическими различиями в развитии мозга [10]. В 1980 году диагноз «инфантильный аутизм» впервые включен в «Диагностическое и статистическое руководство по психическим расстройствам» (DSM); болезнь также официально отделена от детской шизофрении. В 1987 году DSM заменил «инфантильный аутизм» более широким определением «аутистическое расстройство» и включил его в пересмотренную третью редакцию. Тогда же психолог и доктор философии Ивар Ловаас опубликовал первое исследование, которое показало, как интенсивная поведенческая терапия может помочь детям с аутизмом, что подарило родителям новую надежду (рис. 3) [8], [9]. В 1994 году синдром Аспергера добавили в DSM, расширяя диагнозы аутистического спектра, включая, таким образом, более «мягкие» случаи [10].

Рисунок 3. Ученые, которые внесли свой вклад в учение о расстройствах аутистического спектра

Рисунок 4. Фрагмент рисунка о связи РАС и вакцин
Наконец, в 2013 году DSM-5 объединяет все подкатегории состояния в один диагноз «расстройства аутистического спектра», а синдром Аспергера больше не считается отдельным состоянием 8.
Причины РАС
Точная причина расстройства аутистического спектра (РАС) в настоящее время неизвестна. Оно может возникать в результате генетической предрасположенности, экологических или неизвестных факторов, то есть РАС не является этиологически однородным. Вероятно, существует множество подтипов РАС, каждый из которых имеет различное происхождение.
Генетика
Предполагается, развитие РАС во многом связано с влиянием генетических факторов. В поддержку генетики как причины можно добавить результаты исследований, показывающие, что РАС чаще встречается у мальчиков, чем у девочек, что, скорее всего, вызвано генетическими различиями, связанных с Y-хромосомой [14]. В пользу теории говорят также исследования близнецов с РАС, которые определили показатели конкордантности (конкордантность — наличие определённого признака у обоих близнецов) для монозиготных (60-90%) и дизиготных (0-10%) близнецов [15]. Высокая конкордантность в парах монозиготных близнецов и существенно более низкая конкордантность в парах дизиготных близнецов свидетельствуют о значительной роли генетических факторов. В исследовании, проведенном в 2011 году, почти 20% младенцев со старшим биологическим братом с РАС также имели РАС, а если таких старших братьев было несколько, то вероятность иметь диагноз РАС была еще выше [16].
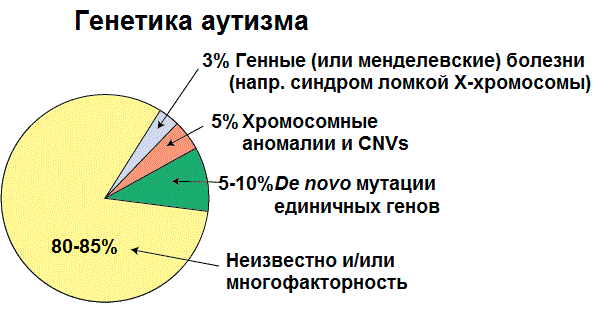
Рисунок 5. Генетические нарушения, ассоциированные с РАС
Нейробиологические факторы
Генетические аномалии могут приводить к аномальным механизмам развития мозга, что в свою очередь приводит к его структурным и функциональным, а также когнитивным и нейробиологическим нарушениям. Нейробиологические различия, связанные с диагнозом РАС, включают структурные и функциональные патологии головного мозга, в том числе:
- увеличенное серое вещество в лобной и височной долях [25];
- уменьшенное белое вещество по сравнению с серым веществом в подростковом возрасте [26];
- анатомические и функциональные различия в мозжечке и лимбической системе [26].
Исследователи в 2018 году обнаружили, что мальчики с РАС имеют меньшую фрактальную размерность (мера структурной сложности объекта) в правой части мозжечка, чем здоровые дети [27].
Некоторые исследования сосредоточены на гипотезе о том, что нарушенные взаимодействия между областями мозга являются главной причиной развития РАС [28], в то время как другие исследователи изучают молекулярные причины, такие как нарушения работы определенных типов нейронов (например, зеркальных нейронов) или нарушения нейротрансмиссии (передачи сигнала между нейронами) [29].
Другие причины
Все больше и больше исследователей пишут об экологических причинах, которые могут вносить свой вклад в аутизм. В исследованиях был выявлен ряд потенциально опасных веществ, которые могут быть связаны с развитием РАС: свинец, полихлорированные дифенилы (ПХД), инсектициды, автомобильные выхлопы, углеводороды и антипирены [25], однако пока ни для одного из этих веществ не было доказано наличие триггерного влияния на возникновение РАС.
Также возрастает интерес к роли иммунной системы в этиологии болезни. В июне 2018 сообщили, что 11,25% детей с РАС имеют пищевые аллергии, что значительно выше, чем 4,25% детей, страдающих аллергией без данного диагноза, что можно добавить к растущему набору доказательств, указывающих на иммунологическую дисфункцию, как возможный фактор риска для РАС [30].
Также недавно выходили исследования, которые связывали недостатки в диете беременных матерей и наличие повышенного уровня пестицидов в крови, с наличием диагноза РАС у их детей 31.
Диагностика
Осматривать ребенка с задержками развития должен врач с целью найти причину задержки развития. Если ребенок проявляет какие-либо симптомы расстройства аутистического спектра, то его, скорее всего, направят к специалистам для консультации, к примеру, к детскому психиатру, детскому психологу, педиатрическому неврологу.
Для правильного диагностирования надо учитывать полную историю пациента, физический осмотр, неврологическое обследование и прямую оценку социального, языкового и когнитивного развития ребенка. Необходимо предоставить достаточное время для стандартизированных интервью родителей относительно текущих проблем и истории поведения, а также структурированного наблюдения за социальным и коммуникативным поведением, игрой.
Согласно новому исследованию 2018 года, новый анализ крови может обнаружить около 17% детей с РАС. Ученые идентифицировали группу метаболитов крови, которые могли бы помочь обнаружить некоторых детей с расстройством аутистического спектра. Как часть проекта «Метаболом детского аутизма» (CAMP), крупнейшего исследования метаболомики РАС, эти результаты являются ключевым шагом на пути к разработке биомаркерного теста на РАС [34].
В августе 2018 года исследователи сообщили о различиях в экспрессии генов бактерий в ротовой области, которые могут отличать детей с РАС от их здоровых сверстников. Исследование предполагает, что нарушения микробиома ЖКТ, ранее выявленные у детей с РАС, могут распространяться на рот и горло [35].
Исследователи из Медицинской школы Университета Миссури и Центра аутизма и неврологических расстройств им. М. У. Томпсона в июне 2018 года выявили связь между дисбалансом нейромедиаторов и характеристикой соединений между регионами мозга, которые играют роль в социальной коммуникации и языке. В исследовании было описано два теста, которые могли бы привести к более точному лечению [36].
Лечение
Лечение, используемое в 60-х и 70-х годах, состояло из приема ЛСД, применения электрического тока и жесткого контроля поведения больного, которое часто включало боль и наказания. Только в 80-х и 90-х годах врачи начали внедрять более современные методы лечения детей с аутизмом, такие как поведенческая терапия с упором на положительное подкрепление и обучение под постоянным контролем [9].
Сегодня лечение может включать в себя как психотерапию, так и медикаментозное лечение. Многие люди с аутизмом имеют дополнительные симптомы, такие, как нарушение сна, судороги и проблемы с ЖКТ. Лечение этих симптомов может улучшить внимание пациентов, их обучение и связанное с этим поведение. Некоторые лекарства, используемые для других состояний, помогают с определенными симптомами: антипсихотики (рисперидон и арипипразол), антидепрессанты, стимуляторы, противосудорожные препараты [37]. Сейчас рисперидон и арипипразол являются единственными лекарствами, одобренными FDA (Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов, США) для симптомов, связанных с расстройствами аутистического спектра, с учетом раздражительности, часто наблюдаемой при этом диагнозе. Дети и подростки с расстройством аутистического спектра, по-видимому, более восприимчивы к побочным эффектам при использовании лекарств, поэтому рекомендуется использование небольших доз [38].
Среди немедикаментозного лечения в настоящее время используют прикладной анализ поведения, когнитивно-поведенческую терапию, обучение социальным навыкам, сенсорную интеграционную терапию, трудотерапию, логопедию [35].
Дети с расстройствами аутистического спектра могут иметь и сильные стороны. Их уникальные взгляды на мир дают возможность другим людям увидеть мир с другой стороны, дети с РАС могут вырасти в талантливых и успешных людей, которые сделают замечательные открытия для улучшения нашего мира. Новые исследования в области диагностики и лечения «детей дождя» дают этим необычным детям надежду на более успешную социальную адаптацию и даже выздоровление.
Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
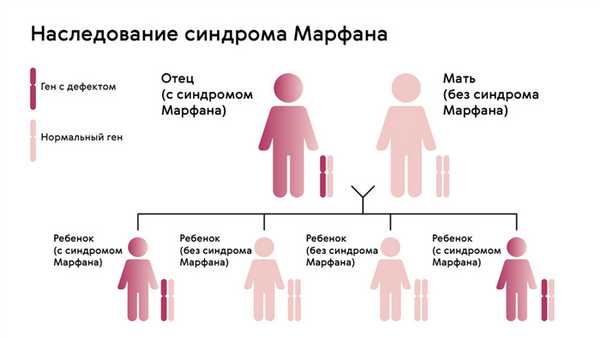
Рисунок. 1. Схема наследования синдрома Марфана. Источник: МедПортал
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5-10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25-50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
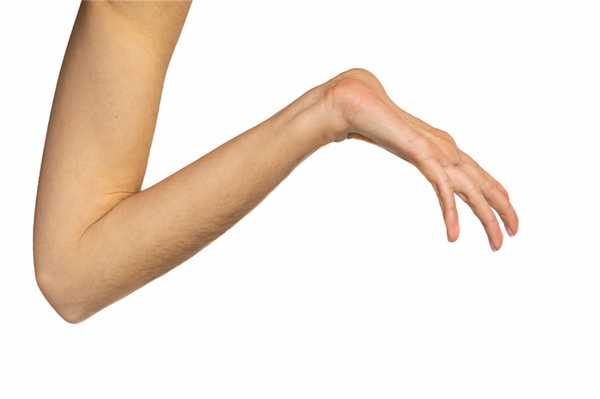
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
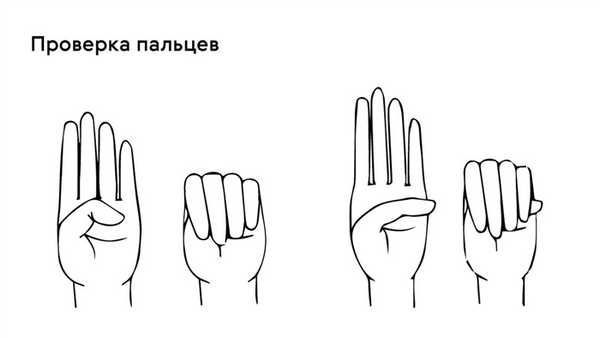
Рисунок 4. Проверка на арахнодактилию. Источник
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65-100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7-18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Синдром МАС

Века до XVII в Европе, если человек терял сознание и это сопровождалось судорогами, то его лечили, как правило, представители церкви, и основными средствами в их «арсенале» лечения были вера и молитва. Они не проводили дифференциацию диагноза между эпилепсией, истерией или синдромом Морганьи — Адамса — Стокса (МАС).
Со временем пациенты в обмороке и с судорогами начали чаще доставаться врачам, которые уже тогда имели привычку «щупать пульс». Открытие той или иной патологии могло продолжаться больше ста лет, как это было с синдромом МАС.
Стоит добавить, что никто из светил медицины того времени не стремился увековечить свою фамилию в названии описываемого страдания. Широкое применение термина «синдром Морганьи — Адамса — Стокса» приписывается выдающемуся русскому терапевту Дмитрию Дмитриевичу Плетневу (первое документальное выступление — доклад «Der Morgagni-Adams-Stokessche Symptomenkomplex» в 1908 г. в Берлине). Вероятно, благодаря его школе название прочно укоренилось в головах «наших» медиков, в то время как в остальном мире обычно говорят и пишут «синдром Адамса — Стокса» (или «Стокса — Адамса»).
Синдром Морганьи — Адамса — Стокса (МАС) — приступы потери сознания, сопровождающиеся нарушениями дыхания и судорогами, возникающие вследствие острой гипоксии головного мозга, обусловленной внезапным падением сердечного выброса.
Причины синдрома — нарушения ритма и проводимости, приводящие как к чрезмерному урежению, так и учащению ритма:
- неполная и полная атриовентрикулярная (АВ) блокада (II ст. II типа и III ст.);
- синдром слабости синусового узла;
- желудочковая тахикардия;
- трепетание и фибрилляция предсердий.
К указанным состояниям, в свою очередь, приводят ИБС (в том числе инфаркт миокарда), миокардиты разной этиологии, кардиомиопатии, пороки сердца и, потенциально, множество других патологий, от системных аутоиммунных заболеваний до алкоголизма, в исходе которых есть поражение проводящей системы сердца.
В основе проявлений синдрома МАС лежит отсутствие сердечного выброса в периоды асистолии желудочков. Изменения стенок сосудов головного мозга, обычно вследствие атеросклероза, повышают риск развития данной патологии.
Чаще всего такое происходит при АВ-блокаде (причина 50-60 % случаев синдрома МАС). Особенно опасны длительные периоды асистолии желудочков, возникающие в результате перехода АВ-блокады II степени в полную АВ-блокаду. При этом импульсы из синусового узла до миокарда желудочков не доходят, а новый водитель ритма желудочков начинает функционировать с некоторой задержкой.
Если полная блокада выше АВ-узла («проксимальная»), то роль водителя ритма берут на себя пейсмекерные клетки АВ-соединения (способны генерировать около 40 импульсов в минуту). Если ниже («дистальная», «трехпучковая» блокада) — импульсы вынуждены генерировать клетки пучка Гиса (25-30 в минуту). Развитие неврологических симптомов обычно происходит при уменьшении частоты сокращений желудочков
При синдроме слабости синусового узла (в том числе и при синоаурикулярной блокаде, 30-40 % случаев развития синдрома Морганьи-Адамса-Стокса) главный водитель ритма в силу разных причин начинает генерировать импульсы с неадекватно низкой частотой. При этом временно перестают сокращаться и предсердия, и желудочки. При достаточно длительном периоде асистолии мозг опять‑таки недополучает своего кислорода и случается приступ МАС.
Возможно впадение в другую крайность. При трепетании предсердий (до 5 % случаев синдрома МАС) АВ-узел обычно не пропускает импульсы с частотой 1:1, и желудочки сокращаются с более или менее приемлемой скоростью ( <150 уд./мин). Однако при улучшении проведения по АВ-узлу (при эмоциональном или физическом напряжении, приеме лекарств) или при наличии дополнительных проводящих путей каждый импульс начинает достигать миокарда желудочков, что приводит к их частым неэффективным сокращениям (>200-250 уд./мин). Сердечный выброс падает, уменьшается поступление кислорода к головному мозгу, что приводит к представленной ниже клинической картине. Этот же механизм лежит в основе развития синдрома МАС при трепетании или фибрилляции желудочков.
Распространенность МАС никто не считал. Согласно Рекомендациям Европейского общества кардиологов 2009 года аритмогенные обмороки составляют 7-11 % в структуре всех синкопальных состояний. Вероятность появления синдрома МАС закономерно увеличивается вместе с распространением заболеваний сердечно-сосудистой системы. У детей синдром МАС встречается реже, чем у взрослых и в основном связан с полной АВ-блокадой, которая может быть приобретенной и врожденной.
Приступ МАС, при наличии свидетелей, не остается незамеченным и мало кого оставляет равнодушным. Больной бледнеет, чувствует внезапное резкое головокружение, общую слабость. Пожаловаться и что‑то предпринять уже не успевает, так как через несколько секунд теряет сознание (вследствие асистолии желудочков дольше 10 секунд).
Через 10-30 секунд после потери сознания появляются тонико-клонические судороги, при этом часто происходит непроизвольное мочеиспускание и, реже, дефекация. Через 30-60 секунд от начала приступа прекращается самостоятельное дыхание (до этого дыхание либо аритмичное, либо периодическое с паузами), развивается цианоз, расширяются зрачки.
Если среди окружающих найдется человек, решившийся оказать первую помощь, то первым делом ему стоит пощупать пульс на сонных артериях. Пульс во время приступа не определяется или очень редкий. Артериальное давление не определяется, тоны сердца не выслушиваются.
Приступ может продолжаться от 10 секунд до 4-5 минут. Если длится недолго, до судорог дело может не дойти. После восстановления сердечной деятельности — что можно оценить по появлению пульса или тонов сердца — к больному практически сразу возвращается сознание. Возможна ретроградная амнезия. Расслабляться не стоит, так как приступы могут повторяться несколько раз в сутки.
Во время приступа окружающим обычно не до инструментальных методов обследования, разве что это удачно произойдет в момент записи ЭКГ. Синдром МАС позволяет заподозрить в первую очередь отсутствие пульса во время приступа (если его кто‑нибудь пытался определить). Среди других факторов: указания в анамнезе на подобные потери сознания, аритмии и блокады сердца, синдром Вольфа — Паркинсона — Уайта, вообще любое кардиологическое заболевание (обычно тяжелое), прием антиаритмических препаратов, сердечных гликозидов. Мониторирование ЭКГ, при необходимости длительное (от суток до месяцев, в этом случае возможна подкожная имплантация аппарата для пролонгированной записи ЭКГ), позволяет зарегистрировать сердечный ритм в момент приступа и снимает вопросы о кардиальном происхождении синкопе.
Синкопальные состояния хотя бы раз в жизни встречаются примерно у трети населения планеты и у половины кардиологических больных. Если больной выжил, то дифференциальный диагноз проводят с любыми обмороками (в том числе при истерии), с приступами при эпилепсии, вазовагальным обмороком, преходящими нарушениями мозгового кровообращения или инсультом, обмороках при врожденных и приобретенных пороках сердца.
Главное отличие заключается в том, что при перечисленных состояниях определяется нормальный или учащенный пульс. АД низкое, высокое или нормальное, но тоже определяется. Для обмороков не характерен цианоз. При вазовагальном обмороке возможна брадикардия, но в целом картина менее драматична, пульс определяется, судороги маловероятны, человек приходит в сознание через 1-2 минуты, особенно если приподнять ему ноги. При пороках сердца аускультация хорошим врачом позволяет заподозрить изменения клапанного аппарата, а эхокардиография — подтвердить их. При нарушениях мозгового кровообращения после восстановления сознания обычно регистрируются очаговые изменения чувствительности и двигательные расстройства.
Приступ МАС может купироваться самостоятельно, а может закончиться смертью больного, поэтому выжидательная тактика недопустима. Объем неотложной помощи при синдроме Морганьи-Адамса-Стокса соответствует таковой при остановке сердца.
Во-первых, нужно убедиться в отсутствии пульса на сонных артериях. Затем нанести прекардиальный удар кулаком в область грудины, немного выше мечевидного отростка, «с силой 3-5 кг». После удара — проверить пульс, если отсутствует, то можно нанести еще 1-2, но со следующим этапом не затягивать.
Непрямой массаж сердца — только при отсутствии пульса на сонных артериях, при наличии сердечной деятельности есть неплохой шанс убить больного своей реанимацией, особенно при условии правильного выполнения. Искусственная вентиляция легких. Вызов кардиобригады «скорой» или транспортировка в ОРИТ.
Лечение синдрома Морганьи-Адамса-Стокса
Даже однократный приступ МАС при АВ-блокаде или синдроме слабости синусового узла — это абсолютное показание для имплантации электрокардиостимулятора.
Современные искусственные водители ритма имеют размеры менее 6 см, срок службы до 10 лет и являются не просто испускателями электрического тока, но весьма сложными вычислительными приборами с большим количеством функций. Электрокардиостимулятор (ЭКС) работает в ждущем режиме и генерирует импульсы в ответ на изменения ритма сердца, способен увеличивать частоту стимуляции в соответствии с физической активностью человека, хранит в своей «памяти» данные о работе сердца за период времени, после чего врач может их считать и проанализировать.
Прибор устанавливается в большинстве случаев под местной анестезией, электроды вводятся через подключичную вену в полости сердца и фиксируются к стенкам, ЭКС обычно укладывается в подключичной области подкожно или подмышечно. Оценка функции и при необходимости перепрограммирование осуществляются дистанционно. По данным Международного общества по лечению аритмии (2011 г.), ежегодно имплантируется до миллиона аппаратов.
Прогноз без установки ЭКС: однолетняя выживаемость после первого приступа МАС составляет, по некоторым данным, менее 50 %. При имплантации и нормальной работе искусственного водителя ритма прогноз определяется тяжестью патологии, которая привела к поражению проводящей системы сердца.
В анамнезе: отравление комплексом токсических веществ при тушении пожара, после чего появились спонтанные приступы синкопе, кардиалгии.
Больной внезапно почувствовал резкую общую слабость, головокружение, затем потерял сознание на 4-5 минут. Со слов жены (фельдшер кардиологической бригады скорой помощи), кожа резко побледнела, стала влажной, самостоятельное дыхание отсутствовало, пульс на сонных артериях не определялся, зрачки широкие, на свет не реагировали, произошло непроизвольное мочеотделение.
Жена выполнила прекардиальный удар кулаком, после чего через 5-6 секунд восстановилось спонтанное дыхание, сердечная деятельность, сознание.
В стационаре диагностирована токсическая миокардиодистрофия, синдром слабости синусового узла, приступ МАС.
От имплантации ЭКС больной отказался. Наблюдение в течение года показало, что на фоне регулярного приёма беллоида и ограничения нагрузок больной периодически испытывает пресинкопальные состояния, однако приступы МАС не отмечались.
Читайте также:
- Мы во сне. Что делать, если не спиться?
- Нормотензивная гидроцефалия на КТ, МРТ головного мозга
- Варианты течения рассеянного склероза их диагностика и лечение
- Нечеткость контуров органов брюшной полости. Закрытые травмы живота
- Цитологическое исследование материала из легких. Цитология эпителия пищеварительного тракта.