Синдром миоклонический - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 22.01.2026
ФГБОУ ВО «Московский государственный университет им. М.В. Ломоносова», Москва, Россия;
ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, Москва, Россия
Московский государственный университет им. М.В. Ломоносова
ФГБОУ ВО «Московский государственный университет им. М.В. Ломоносова», Москва, Россия
Российская детская клиническая больница (РДКБ), Москва
Российский национальный исследовательский медицинский университет им. Н.И. Пирогова, Москва
Кафедра неврологии и нейрохирургии педиатрического факультета Российского государственного медицинского университета, Москва
Психическое развитие детей с опсоклонус-миоклонус-синдромом с учетом различий в возрасте, времени дебюта и срока течения заболевания
Опсоклонус-миоклонус-синдром (ОМС), возникающий чаще всего в детском возрасте, является редким и малоизученным в отношении этиологии и патогенеза заболеванием. Актуальность исследования — недостаточность данных о психическом развитии детей при ОМС. Цель исследования. Анализ особенностей психического развития детей с ОМС при разном возрасте начала и разных типах течения заболевания. Материал и методы. Обследовали 26 детей с ОМС (10 мальчиков, 16 девочек) в возрасте от 1 года 7 мес до 13 лет. В процессе клинико-неврологического и психологического обследования анализировали данные историй болезни, проводили беседы с родителями, оценивали эмоциональное состояние детей, поведенческие двигательные проявления болезни, когнитивное развитие больных и особенности их межличностных отношений. Результаты и заключение. Установлено, что при начале заболевания в наиболее ранний этап онтогенеза (до 3 лет) отмечаются более выраженные нарушения психического развития. Выделен возраст пациентов (от 3 до 5 лет), в котором наблюдается максимальное количество задержек психического развития с возможностями компенсации и развитием, близким к нормативному. Обсуждаются вопросы возможной роли психотравматического и психосоциальных факторов в развитии нарушений психического развития у детей с ОМС с учетом возраста. Подчеркивается значение возрастного фактора для планирования лечебно-реабилитационных мероприятий и оценки прогноза заболевания.
Опсоклонус-миоклонус-синдром (ОМС) в настоящее время является редким и малоисследованным как с медицинской, так и с клинико-психологической точки зрения тяжелым неврологическим заболеванием [1, 2].
Среди неврологических проявлений ОМС центральное место занимают несистемное головокружение, шаткость походки, частые падения, тремор головы, туловища, рук и ног, нарушение мышечного тонуса. Характерные признаки заболевания — явления опсоклонуса и миоклонуса. Опсоклонус характеризуется непроизвольными, хаотичными, разнонаправленными саккадическими движениями глаз с горизонтальным и вертикальным компонентами. Под миоклонусом подразумеваются полиморфные миоклонии в виде коротких, отрывистых движений мышц небольшой амплитуды. Эти проявления наблюдаются в основном в мышцах век, губ, туловища, в проксимальных отделах конечностей. Заболевание может быть слабой, средней и сильно выраженной степени тяжести, но в любом случае довольно быстро происходит регресс достигнутых ребенком до болезни двигательных навыков. Примерно у 2/3 пациентов течение болезни является хроническим и рецидивирующим.
У больных имеются и психологические нарушения [3, 4]. Речь идет об особенностях когнитивного и эмоционального развития детей с ОМС, особенностях их поведения и трудностях в становлении речи. Возникающие нарушения в психическом развитии могут быть выражены в разной степени. В одних случаях это дефицитарность некоторых функций, которая позволяет ребенку оставаться в границах нормативного развития, в других — высокая степень их нарушения, приводящая к тотальному недоразвитию.
Выявлены определенные сложности и в отношении своевременного диагностирования ОМС в связи с тем, что как минимум в 20% случаев манифестация заболевания отличается специфической и нетипичной картиной. Во многих случаях оно ошибочно квалифицируется как одно из заболеваний с похожей клинической картиной (энцефалит, синдром Гийена—Барре и др.). Существуют сложности, связанные с определением нужной схемы лечения, оценкой динамики течения и прогноза заболевания.
Начало болезни, как правило, происходит постепенно, но в течение относительно короткого времени: у ребенка появляются и усиливаются атаксия и тремор во всем теле, возникают редкие некоординированные движения. Он довольно быстро утрачивает возможность удерживать равновесие и самостоятельно ходить, пользоваться ложкой и другими привычными предметами, пить из кружки (если до начала болезни он умел это делать). Начинают периодически отмечаться проявления хаотичного движения глазных яблок (опсоклонус). Наряду с этим нередко возникают подергивание языка, нарушение возможности полноценного произнесения уже имеющихся в активе ребенка слов. Для психического состояния пациента в этот период характерны незатихающий сильный плач, который не удается успокоить обычными средствами, нарушения сна и недостаток периодов спокойного бодрствования. Иногда наблюдается апатичность, потеря интереса к окружению, игровой деятельности и общению. Характерна дисфория в отношении к близким, прежде всего к матери.
Болезненные ощущения создают для ребенка ситуацию крайней дестабилизации, дезинтеграции окружающей действительности, вынуждают его искать физическую опору, лежать. Болезнь погружает его в состояние крайнего телесного дискомфорта, регрессивной беспомощности и переполненности тревогой.
При катамнестических исследованиях, направленных на оценку развития заболевания, были отмечены трудности в обучении и поведении у таких детей. Так, по результатам проведенного в Великобритании катамнестическое исследования [6], лишь 1/3 из 101 находившегося под наблюдением пациента не имели сложностей в когнитивном функционировании, что позволило некоторым из них даже получить высшее образование. У большей же части пациентов в отдаленном периоде были отмечены речевые и интеллектуальные расстройства разной степени выраженности. Статистически подкрепленные исследования [7—9] показали, что неврологическая симптоматика в сочетании с отклонениями в когнитивном развитии отмечается примерно у ½ больных с ОМС. При этом нарушения в когнитивном развитии и поведении чаще имеют место в случаях более раннего начала заболевания и большого количества его рецидивов.
Число рецидивирующих эпизодов может оказаться значимым фактором для когнитивного, эмоционального и психического развития детей в целом, поскольку указывает на тяжесть заболевания (выраженность неврологической симптоматики) и длительность воздействия патогенной причины [10]. При этом важно учитывать, что наряду с основными нарушениями развития формируются и специфические функциональные перестройки, в структуру которых включаются вторичные и третичные компенсаторные компоненты [11—14].
В аспекте изложенных выше особенностей заболевания весьма актуальной задачей является анализ характера психического развития ребенка в зависимости от его возраста при дебюте болезни. Период онтогенеза, на который приходится начало болезни в детской неврологии, имеет особое значение, поскольку каждый возраст имеет свои характеристики в отношении устойчивости, сенситивности и ранимости по отношению к вредоносным воздействиям. Изучение этого вопроса в клинических и психологических исследованиях традиционно для ряда неврологических заболеваний, однако применительно к пациентам с ОМС результатов пока недостаточно, учитывая невысокую частоту заболевания в популяции.
Цель настоящего исследования — изучение психического развития детей разного возраста с ОМС в зависимости от возраста дебюта заболевания.
Материал и методы

В исследование были включены 26 детей (10 мальчиков, 16 девочек) с ОМС в возрасте от 1 года 7 мес до 13 лет. Средний возраст детей, в котором возник дебют болезни составил 22±11 мес (табл. 1). Таблица 1. Распределение детей с ОМС по полу и возрасту
Все пациенты проходили лечение в отделении психоневрологии Российской детской клинической больницы. Некоторые дети в дальнейшем посещали отделение для консультаций с лечащим врачом-неврологом и психологом.
В большинстве случаев обследование детей проводилось в присутствии матери, поскольку часто попытки разделения больного ребенка с матерью приводили к сильной тревоге и у больного, и у матери (это наблюдалось в основном в диадах с детьми до 5-летнего возраста, но также имело место и у детей более старшего возраста). В отдельных случаях, помимо психолога, ведущего ребенка, при обследовании присутствовал врач-невролог либо еще один психолог.
В зависимости от возраста дети были разделены на две группы: 1-я группа — пациенты до 4 лет 11 мес, 2-я группа — больные старше этого возраста.
Для обследования обеих групп были использованы следующие методы исследования: анализ историй болезни пациентов; полуструктурированная беседа с родителями, направленная на выяснение жалоб, касающихся развития ребенка, а также значимых для оценки развития достижений и трудностей в познавательном развитии, эмоционально-поведенческом реагировании и социальной коммуникации; наблюдение за поведением, эмоциональными реакциями ребенка.
Структура психологического обследования для выявления особенностей когнитивного и эмоционально-личностного развития различалась в зависимости от возраста ребенка: патопсихологическое обследование детей 1-й группы выстраивалось в соответствии с традицией московской школы патопсихологии и психологии аномального развития [11].
Для детей 2-й группы применялась схема проведения нейропсихологического тестирования по Лурия, адаптированного для детского возраста и направленного на качественную оценку состояния отдельных психических функций. В процессе тестирования оценивались: 1) уровень познавательного развития (что включало исследование общей ориентации ребенка в автобиографических данных и пространстве, работоспособности, анализ уровня развития сенсомоторных навыков, зрительного и зрительно-пространственного восприятия, памяти, внимания, мышления, речи, графических навыков); 2) сформированность навыков, обеспечивающих социальную коммуникацию (анализировались особенности контакта и поведения ребенка в процессе беседы, реакции на предложенные задания, готовность к сотрудничеству с психологом, особенности реагирования на помощь взрослого, успех или неудачу, также принималась во внимание специфика взаимодействия с родителями (чаще с матерью), а также игры с психологом); 3) состояние произвольной активности и эмоционально-волевой сферы (анализировалось при наблюдении за поведением ребенка).
Обследование проводилось после дневного сна и удовлетворения физиологических потребностей детей, в спокойном состоянии ребенка, когда не было обострения неврологической симптоматики и выраженной эмоциональной лабильности, которые могли бы явиться препятствием к работе с ним.
Результаты
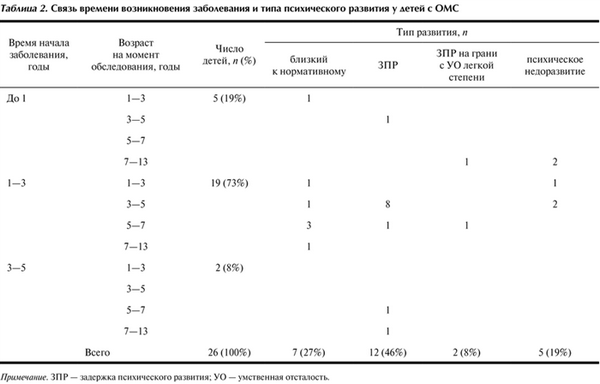
Возраст детей на момент обследования сильно варьировал, что вносило коррективы в анализ связи между временем возникновения заболевания, возрастом пациентов на момент обследования и типом психологического (психического) развития у детей с ОМС. Периоды от последнего ухудшения до момента обследования у детей также различались, что так или иначе влияло и на результаты обследования, поскольку периоды восстановления и периоды возможной компенсации нарушений в когнитивном и эмоциональном развитии у всех детей были разными (табл. 2). Таблица 2. Связь времени возникновения заболевания и типа психического развития у детей с ОМС Примечание. ЗПР — задержка психического развития; УО — умственная отсталость.
Основной возрастной диапазон возникновения ОМС (76%) — от 1 года до 3 лет. Реже (19%) заболевание возникает в возрасте у детей до 1 года, еще реже (8%) — в период от 3 до 5 лет.
Были выявлены разные варианты дизонтогенеза — от близкого к норме развития до выраженного психического недоразвития. Близкое к нормативному развитие наблюдалось в 27% случаев — у 7 больных из 26. Задержки психического развития разной степени были отмечены у 14 детей. При этом у 12 (46%) они были относительно легкими, а в 2 (8%) случаях находились на границе с умственной отсталостью. Выраженное психическое недоразвитие имело место у 5 (19%) детей. В целом результаты обследования свидетельствуют о том, что большинство (73%) детей развивались ненормативно.
При сопоставлении характера психического развития со временем появления первого эпизода заболевания было установлено, что опасность выраженных нарушений психического развития выше при дебюте ОМС до 3 лет. Отмечается тенденция увеличения случаев задержанного развития (включая грубые формы задержки) и психического недоразвития — 65% (17 из 26). Наиболее выраженные формы нарушений развития, которые встречаются у детей с дебютом заболевания в возрасте до 3 лет и не встречаются при начале заболевания в возрасте старше 3 лет, указывают на то, что именно этот возрастной период является периодом максимальной сенситивности нервной системы к действию патогенного фактора, что обусловливает развитие когнитивных, речевых и эмоционально-личностных отклонений, когда манифестация заболевания может нанести наиболее тяжелый вред. В силу особой уязвимости ребенка в этот период болезненный эпизод способен вызвать не только ухудшение в работе всего организма, но и существенное изменение его развития.
Вместе с тем сделанный выше вывод имеет некоторые ограничения, поскольку наблюдались случаи близкого к нормативному развития при начале заболевания и до 1 года (4%; 1 из 26), и в период от 1 года до 3 лет (23%; 6 из 26). Таким образом, раннее начало заболевания хотя и является фактором риска в отношении развития тяжелых нарушений психического развития, все же напрямую, видимо, его не обусловливает. Здесь могут иметь значение тяжесть первого приступа заболевания, длительность и особенности периода до начала лечения, количество и массивность перенесенных рецидивов, которые влияют на возможности компенсаторной перестройки в сложном и многоэтапном процессе развития психических функций в онтогенезе.
Как показало настоящее исследование, в возрастном диапазоне от 3 до 5 лет наблюдается максимальное количество (31%) задержек психического развития, способных к компенсации (редукции) при соответствующем лечении и проведении реабилитационных мероприятий. В связи с этим следует отметить, что существенный вклад в возникновение и усугубление задержек развития могут вносить третичные, психосоциальные по своему генезу симптомы. Одним из психосоциальных факторов может быть излишняя опека больного ребенка со стороны родителей. Если сначала такая гиперопека оправдана болезненным состоянием ребенка, то затем, при улучшении его состояния, она часто реализуется по инерции, в силу приобретенного близкими психотравматического эмоционального опыта и длительного переживания ими состояния неизвестности и беспомощности в сочетании со страхом возможного возвращения регресса поведенческих нарушений. Это может стать фокусом для психологической коррекции с целью минимизации негативного вклада психосоциальной составляющей в психическое развитие ребенка и создания условий для организации оптимальной для него среды.
Таким образом, в психическом развитии детей с ОМС проявляется сложное соотношение обусловленного болезнью психофизиологического отставания и компенсаторных функционально-психологических явлений в процессе онтогенеза, которые требуют дальнейшего изучения. Необходимо исследование психических особенностей ребенка с ОМС, особенностей его мотивации и переживаний, что крайне важно для организации психокоррекции и реабилитации. Нельзя недооценивать психотравмирующий характер детских переживаний в связи с потерей привычной для него стабильности предметов в условиях выраженных проявлений опсоклонуса, невозможностью совершения безопасных перемещений и реализации приобретенных навыков. Простые и привычные для здорового ребенка дошкольного возраста ситуации для больного становятся предельно драматичными. Можно предположить, что в зависимости от возраста ребенка неожиданно наступающие массивные нарушения движений и невозможность поддержания позы в условиях тяжелого начала ОМС способны вызывать ощущения не только чуждости и враждебности мира, но и неподконтрольности собственного тела, зачастую сопряженные со страхами фрагментации «Я» и его внутренней дезинтеграции. Наблюдаемые в таких случаях сложные эмоционально-поведенческие реакции ребенка проявляются прежде всего дисфоричным настроением, неудовлетворенностью общением даже с близкими и общей эмоциональной разбалансированностью, что может указывать на психотравмирующий фактор у ребенка, связанный с пережитой (в некоторых случаях — неоднократно) ситуацией. Это может быть самостоятельным источником более низкого психического функционирования и затяжных психических перестроек у детей с ОМС.
В проведенном исследовании была подтверждена предполагаемая связь времени начала заболевания по отношению к этапу онтогенеза с выраженностью последующих нарушений психического развития. Однако требуют дальнейшего изучения целый ряд факторов (тяжесть перенесенного манифеста заболевания, количество и тяжесть эпизодов ухудшений и др.), роль и место психотравматического и психосоциальных факторов в аспекте нарушений психического развития у детей с ОМС. Но независимо от дальнейшего изучения ОМС при оценке прогноза болезни и планировании психокоррекционных мероприятий уже сейчас важно учитывать ряд факторов: возраст ребенка к периоду начала заболевания и его возраст на момент обследования.
Миоклоническая дистония
Миоклоническая дистония - генетически гетерогенное состояние, которое приводит к нарушению работы мышц (миоклоническим гиперкинезам), а также дистонии мускулатуры верхней части тела - шеи, пояса верхних конечностей. Симптомами данного состояния являются резкие мышечные подергивания (рук, шеи, изредка ног), особенно при выполнении тонких движений. Затем присоединяется дистония, которая может проявляться кривошеей и необычной позой больного. Диагностика миоклонической дистонии производится на основании данных настоящего статуса пациента и молекулярно-генетического анализа. Лечение заболевания симптоматическое, включает в себя бензодиазепины и противосудорожные средства, иногда применяют ботулотоксин для устранения спастических нарушений.

Общие сведения
Миоклоническая дистония (миоклонус-дистония, этанол-чувствительная миоклония) - генетическое состояние различной природы, характеризующееся нарушением работы мышц с развитием гиперкинезов и дистонии. Впервые было описано еще в 1940-м году Бенедеком, однако он не смог идентифицировать патологию как отдельный тип миоклонии, это сделали Даубетс и Петерс в 1966-м году. В настоящее время врачи-генетики определили, что это состояние является гетерогенным, то есть за его развитие отвечают мутации различных генов, многие из которых пока не удалось идентифицировать. По этой причине неясен механизм наследования миоклонической дистонии. В большинстве случаев отмечается аутосомно-доминантная передача патологии, при этом имеются указания на наличие материнского импринтинга, при котором дефектный ген, передающийся от матери, не приводит к развитию заболевания. Встречаемость миоклонической дистонии оценивается цифрами 1:500000.

Причины миоклонической дистонии
Достоверно известно, что миоклоническая дистония имеет генетическую природу, при этом встречаются как наследственные семейные случаи, так и спорадические мутации генов. До недавнего времени был известен лишь один ген, мутации которого вызывают развитие данного заболевания - SGCE, локализованный на 7-й хромосоме. В европейской популяции дефекты этого гена встречаются у 20-30% больных миоклонической дистонией. Продуктом его экспрессии является белок саркогликан-эпсилон, относящийся к группе трансмембранных протеинов, встречающийся в скелетных мышцах, миокарде и нейронах ряда структур центральной нервной системы.
Совсем недавно появились указания на то, что у части больных миоклонической дистонией на фоне отсутствия дефектов в SGCE наблюдались мутации гена DRD2, расположенного на 11-й хромосоме. Ген кодирует последовательность одного из рецепторов к дофамину (D2), преобладающего в некоторых базальных ядрах головного мозга. В основном при миоклонической дистонии выявляется миссенс-мутация Val154Ile гена DRD2, приводящая к изменению структуры рецептора. В результате этого меняются многие процессы контроля над мышечным тонусом, что клинически проявляется гиперкинезами и дистонией. Кроме того, при этом заболевании возникают фобии, навязчивые состояния и панические атаки.
Указания на повышенную предрасположенность лиц с мутациями вышеуказанных генов к алкоголизму лишь отчасти имеют под собой основания. Напитки на основе этилового спирта способны ослаблять симптомы миоклонической дистонии, по этой причине больные в ряде случаев употребляют алкоголь в умеренных дозах для облегчения своего состояния, со временем это может стать причиной серьезной зависимости.
Симптомы миоклонической дистонии
При рождении ребенка и в первые годы жизни миоклоническая дистония ничем себя не проявляет, заметить наличие этого состояния не представляется возможным. Первые признаки заболевания возникают в возрасте 15-30 лет, симптоматика патологии нарастает достаточно быстро, но в дальнейшем ее склонность к прогрессированию не выявляется. На начальных этапах миоклонической дистонии развиваются гиперкинезы мышц верхней половины туловища - плечевого пояса, рук, шеи, изредка лица. Иногда подергивания перерастают в судороги, нередко имеющие скручивающий характер и различную частоту возникновения. Гиперкинезы и судороги при миоклонической дистонии редко проявляются в состоянии покоя, в основном их появление связано с мелкими и точными движениями - письмом или рисованием. Мышечные подергивания при этом достаточно резкие, часто описываются больными как «молниеносные».
Еще одним проявлением миоклонической дистонии являются нарушения мышечного тонуса, которые нередко поражают шею, верхние конечности, иногда - мышцы гортани и ног. Наиболее часто отмечается фокальная и спастическая дистония, имеющая асимметричный характер, при развитии на мышцах шеи в подобных случаях возникает кривошея. Как правило, после возникновения дистония не усиливается и не поражает новые участки мышц. В некоторых случаях одним из проявлений миоклонической дистонии может выступать тремор рук. Возможна также дизартрия, обусловленная нарушением функций мышц гортани. Других мышечных симптомов при этом заболевании обычно не определяется.
При миоклонической дистонии нередко выявляются разнообразные расстройства психики: фобии, навязчивые состояния, панические атаки, депрессии. Этанол способен значительно уменьшать выраженность гиперкинезов и дистонии, поэтому многие больные употребляют различные количества алкогольных напитков. При отсутствии врачебного контроля это приводит к стойкой алкогольной зависимости и сопутствующим нарушениям. Поэтому психические симптомы при миоклонической дистонии могут иметь различную природу - первичную, обусловленную нарушением биохимических процессов в нервной системе из-за генетической мутации, и вторичную, вызванную неумеренным употреблением алкоголя.
Диагностика и лечение миоклонической дистонии
Для определения миоклонической дистонии используют данные результатов осмотра больного и молекулярно-генетических анализов. При осмотре выявляют тремор рук, клонические гиперкинезы мышц, плечевого пояса, рук и шеи, которые усиливаются при физической нагрузке или выполнении движений, требующих сложной согласованной работы пучков мышечных волокон (например, рисование). Также может обнаруживаться кривошея, обусловленная спастической дистонией мускулатуры, в более тяжелых случаях определяется необычное положение тела пациента. В анамнезе больного обнаруживаются случаи судорожных припадков и другие неврологические симптомы - все это также косвенно указывает на наличие миоклонической дистонии.
К процессу диагностики миоклонической дистонии зачастую подключают психиатра или нарколога, поскольку при этом состоянии нередко выявляются расстройства психики и проблемы с наркотическими веществами. При обследовании пациента может регистрироваться депрессивное или подавленное состояние, навязчивые идеи и фобии. Панические атаки часто связаны с судорожными припадками, после которых больных миоклонической дистонией начинает одолевать страх смерти, боязнь за свое здоровье или жизнь. Обследование у нарколога иногда обнаруживает признаки алкогольной или (намного реже) бензодиазепиновой зависимости - последняя нередко возникает при неправильном лечении или нарушении схемы приема препаратов.
Молекулярно-генетическая диагностика миоклонической дистонии в настоящее время не получила широкого распространения. В значительной степени это обусловлено тем, что генетические дефекты, приводящие к данному состоянию (мутации генов SGCE и DRD2) достоверно установлены всего лишь примерно для трети случаев заболевания. Однако ряд клиник все же предлагает определение миоклонической дистонии генетическими методами путем прямого секвенирования последовательностей вышеуказанных генов. Пренатальная диагностика необходима в тех случаях, когда миоклонической дистонией страдает отец ребенка - из-за импринтинга передача дефектного гена от матери потомству происходит крайне редко. Дифференциальную диагностику производят с семейной миоклонией (болезнью Унферрихта-Лундборга), синдромом Туретта, болезнью Вильсона, другими первичными и вторичными формами дистонии.
Специфического лечения миоклонической дистонии не существует, применяют симптоматическую терапию. При наличии тремора и выраженных гиперкинезов хорошим эффектом обладают препараты из группы бензодиазепинов, однако назначать их можно только после консультации нарколога - важно, чтобы у больного не было алкогольной или бензодиазепиновой зависимости. Судорожные припадки, возникающие при миоклонической дистонии, устраняются при помощи традиционной противосудорожной терапии. Спастические дистонии мышц и в особенности кривошея могут быть значительно ослаблены инъекциями малых доз ботулинового токсина для частичной химической денервации мышечной ткани. Эффективность перечисленных терапевтических мероприятий неодинакова у различных больных миоклонической дистонией, что обусловлено значительной генетической и фенотипической гетерогенностью данного состояния.
Прогноз и профилактика миоклонической дистонии
Прогноз миоклонической дистонии относительно выживаемости пациентов довольно благоприятный - состояние не имеет склонности к прогрессированию симптомов, не поражает жизненно важные системы и органы. Однако качество жизни больных может в значительной степени снижаться из-за гиперкинезов, которые нередко делают невозможными выполнение тонких движений (например, письмо), дистонической кривошеи и других симптомов заболевания. Кроме того, прогноз миоклонической дистонии ухудшает повышенный риск развития алкогольной зависимости - даже благополучный в социальном плане человек может пристраститься к спиртосодержащим напиткам из-за того, что они облегчают неврологические симптомы. Еще больше повышают риски психоэмоциональные расстройства пациента, которые увеличивают вероятность развития алкогольной зависимости и могут стать причиной серьезного невроза. Специфической профилактики миоклонической дистонии не существует, при возникновении заболевания следует выполнять все предписания врача.
Энцефалопатия Кинсбурна, или синдром опсоклонуса-миоклонуса, в детском возрасте
Опсоклонус-миоклонус-синдром (ОМС) был впервые описан Paul Sandifer (1962), в том же году Kinsbourne (1962) сообщил о шестерых пациентах в возрасте 9-20 мес с некоординированными, нерегулярными движениями туловища и конечностей, миоклонусом и хаотичным
Опсоклонус-миоклонус-синдром (ОМС) был впервые описан Paul Sandifer (1962), в том же году Kinsbourne (1962) сообщил о шестерых пациентах в возрасте 9-20 мес с некоординированными, нерегулярными движениями туловища и конечностей, миоклонусом и хаотичными движениями глазных яблок. Автор предположил наличие у этих пациентов миоклонической энцефалопатии. Четверо из них получали адренокортикотропный гормон (АКТГ) с хорошим терапевтическим эффектом. Несколько похожих случаев были представлены в литературе и ранее (Cоgan, 1954; Arthuis и др., 1960). Благодаря большому количеству публикаций в 1970-80-е гг. это заболевание стало известно под разными названиями — синдром «танцующих глаз» (Ford, 1966), «детская полимиоклония» (Dyken and Kolar, 1968), «острая мозжечковая энцефалопатия» (Bray и др., 1969), «офтальмо-мозжечково-миоклонический синдром» (Lemerle и др., 1969), «RIMEL-синдром» Pampiglione and Maia, 1972), «атаксия-опсоклонус-миоклонус-синдром» (Pinsard и др., 1980), «энцефалопатия Кинсбурна» (Brandt и др., 1974). Последнее название, наряду с ОМС, встречается в литературе наиболее часто.
Заболевание наблюдается преимущественно в детском возрасте, хотя имеются данные о возникновении ОМС и у взрослых: как правило, заболевание развивается как проявление паранеопластического процесса при различных онкологических заболеваниях. Распространенность заболевания неизвестна. Считается, что ОМС — довольно редкое состояние: так, в США его распространенность составляет 1 случай на 10 000 000 человек. Данных о распространенности ОМС среди детей нет. Частота возникновения ОМС среди мальчиков и девочек примерно одинакова.
Возраст появления первых симптомов при ОМС варьирует от 4 мес до 6 лет и составляет в среднем 17-19 мес (Ferrandez-Alvarez и др., 1978; Boltshauser и др., 1979; Hammer и др., 1995). Основной симптомокомплекс представлен выраженными изменениями поведения, нарушением координации, тремором, миоклонусом и специфическими движениями глазных яблок. Дебют неврологической симптоматики, как правило, связывают с перенесенным инфекционным заболеванием или проведенной иммунизацией.
Опсоклонус — один из кардинальных симптомов ОМС — представляет собой миоклонический гиперкинез глазодвигательных мышц и проявляется быстрыми толчкообразными хаотичными, преимущественно горизонтальными движениями глазных яблок. Может наблюдаться беспорядочная смена горизонтальных, вертикальных, диагональных, круговых и маятникообразных движений различной частоты и амплитуды. Наиболее соответствует кинематике движений глазных яблок при ОМС слово «танец», что и определило одно из первых названий заболевания — синдром «танцующих глаз». Для описания этого характерного симптомокомплекса, отличающегося от нистагма, K. Orzechowski еще в 1913 г. предложил термин «опсоклонус» (от греч. оps — глаз, klonos — беспорядочные движения). Опсоклонус часто сочетается с молниеносными движениями век, напоминающими трепетание крыльев насекомых, что определяет своеобразный клинический «рисунок» патологических движений, практически не встречающийся при других заболеваниях. Характерным является также сохранение опсоклонуса во время сна.
Нарушения поведения отмечаются у всех детей с ОМС. Наиболее распространенными симптомами являются выраженная возбудимость, бессонница, необходимость в укачивании, ночные кошмары, агрессия по отношению к окружающим, аутоагрессия.
Миоклонические подергивания мимических мышц, мышц туловища, конечностей также достаточно специфичны для ОМС и проявляются быстрыми, внезапными мышечными сокращениями, которые иногда имеют настолько небольшую амплитуду, что их можно спутать с генерализованным тремором. Нередко миоклонус затрагивает веки, губы. В конечностях миоклонии выражены преимущественно в проксимальных отделах, чаще проявляются, когда ребенок пытается встать. Интересно заметить, что тремор и миоклонии сохраняются во время сна. Попытки повернуться в кровати сопровождаются такими массивными миоклониями, что дети просыпаются. Манипуляции игрушками нарушены из-за миоклонуса и интенционного тремора. Переход из горизонтального положения в вертикальное, а также попытки ходьбы приводят к некоординированным излишним движениям, особенно в нижних конечностях (синдром «танцующих ног«).
Роль мозжечка и его связей с другими отделами нервной системы в формировании клинической картины ОМС несомненна. Концепция «дисметрии или атаксии мыслей» вводится многими авторами при изучении роли мозжечка и его связей, особенно с корой лобной доли. Дисфункцией корково-мозжечковых связей можно объяснить нарушение поведения и настроения при ОМС, нарушение других мозжечковых связей может приводить к двигательным нарушениям.
В настоящее время принято выделять две группы ОМС: параинфекционный ОМС и паранеопластический ОМС.
Патогенез ОМС до сих пор остается неизвестным, однако очевидно, что в его развитии участвуют аутоиммунные механизмы, причем это относится как к параинфекционному, так и к паранеопластическому ОМС.
Pranzatelli M. R. и др. (2004) исследовали активность В-лимфоцитов в спинномозговой жидкости (СМЖ). В группе 56 детей с ОМС был обнаружен высокий процент В-лимфоцитов СД5(+)- и СД5(-)-субпопуляций, в контрольной группе субпопуляции В-лимфоцитов отсутствовали. Наличие аутореактивных СД5(+)-клеток коррелировало с тяжестью неврологической симптоматики и длительностью ОМС. Этими же авторами в ходе исследования 36 детей с ОМС было установлено, что при сохранении нормального количества лимфоцитов в СМЖ, имеется увеличение субпопуляции CD19(+) и γ-δ-T-клеток, снижение соотношения СD4/СD8. Найденные изменения сохранялись длительное время от начала заболевания и коррелировали с выраженностью неврологической симптоматики.
Сообщается о возможной роли IgG- и IgM-аутоантител: анти-Yo-антитела к цитоплазме и аксонам клеток Пуркинье, анти-Ri- и анти-Hu-антитела к ядрам нейронов, к нейрофиламентам, а также антимитохондриальных антител в патогенезе ОМС.
Предполагается и участие дофаминергической системы в возникновении клинических симптомов ОМС. Так, в ходе исследования, проводившегося в 1995 г. с участием 27 детей с ОМС и 47 детей контрольной группы, в СМЖ определялись метаболиты серотонина — 5-гидроксииндолуксусная кислота и метаболит дофамина — гомованилиновая кислота. В группе детей с ОМС эти показатели оказались на 30-40% ниже, чем в контрольной группе.
Имеются единичные патологоанатомические исследования ОМС, главным образом при нейробластомах. Изменения в головном мозге в основном не определяются, однако описаны уменьшение числа клеток Пуркинье и демиелинизация; изменения определяются главным образом в зубчатом ядре мозжечка.
Дополнительные обследования у детей с ОМС применяют при дифференциальной диагностике (миоклонус при дегенеративных заболеваниях, постаноксический миоклонус, церебеллит, острый рассеянный энцефаломиелит, рассеянный склероз, опухоли задней черепной ямки и др.) и с целью исключения нейробластомы. При исследовании СМЖ, как правило, не выявляется отклонений от нормы, возможны невысокий лимфоцитарный плеоцитоз, повышение уровня иммуноглобулинов. Магнитно-резонансная томография (МРТ) головного мозга неспецифична, иногда отмечаются очаговые изменения плотности в стволе и мозжечке. Необходимо исследование онкомаркеров, в частности α-фетопротеина крови, определение показателей катехоламинового обмена в крови и моче, ультразвукового исследования (УЗИ) органов брюшной полости и забрюшинного пространства для исключения нейробластомы. При отсутствии изменений на УЗИ используются методы компьютерной томографии (КТ) и МРТ органов грудной полости, малого таза и забрюшинного пространства. На электроэнцефалограмме изменения неспецифичны, полиграфическая запись свидетельствует о неэпилептическом генезе миоклоний.
ОМС является достаточно редким заболеванием с неустановленной этиологией и патогенезом, поэтому до сих пор в отношении него не разработано стандартизированных схем терапии. У небольшого числа пациентов ОМС может регрессировать спонтанно, без медикаментозного лечения или при назначении неспецифической симптоматической терапии.
Существует две наиболее распространенные схемы терапии ОМС. Одна из них основана на применении АКТГ, другая — кортикостероидов (преднизолон). Эффективность АКТГ у детей при ОМС, по данным американских исследователей, отмечается в 80-90% случаев. При назначении АКТГ или кортикостероидов, многие пациенты довольно быстро — в течение первых дней или месяца — демонстрируют положительный эффект: значительно уменьшается атаксия, исчезает опсоклонус, улучшается поведение. Однако в большинстве случаев терапию приходится продлевать на срок не менее 6 мес, а у ряда пациентов она должна проводиться в течение нескольких лет. Исследователи подчеркивают, что заболевание может носить волнообразный характер, неврологическая симптоматика способна рецидивировать на фоне интеркуррентных инфекций, поэтому продолжительность терапии и сроки ее отмены должны определяться с учетом клинического состояния пациента.
В ряде исследований не было выявлено статистически достоверных доказательств преимущества назначения АКТГ перед кортикостероидами. Описаны случаи, когда пациенты, не ответившие на терапию преднизолоном в суточной дозе 2 мг/кг, хорошо реагировали на терапию АКТГ, и наоборот.
Даже при нейробластомах ряд исследователей отдают предпочтение преднизолону и АКТГ в сравнении с полихимиотерапией. Наиболее распространенные схемы терапии включают преднизолон в суточной дозе 1 мг/кг или АКТГ от 10 до 40 ЕД в сутки. Все пациенты, которым проводится гормональная терапия, должны находиться под контролем врача, так как риск побочных эффектов гормональной терапии часто превышает ожидаемый результат. В литературе обсуждаются преимущества короткого и длительного курсов кортикостероидной терапии, а также высоких и низких доз АКТГ и кортикостероидов.
В последние годы особое внимание уделяется применению нормального человеческого IgG. Назначение его показано в случае параинфекционного ОМС, хотя и при терапии ОМС, вызванного нейробластомой, также выявлялся отчетливый положительный эффект. Курсовая доза иммуноглобулина составляет 2 г/кг в течение 3-5 дней, описана схема 2 г/кг в течение 6-8 ч в первые сутки, затем 1 г/кг в течение 2 сут. Многие авторы подчеркивают, что решение о повторном введении иммуноглобулина определяется степенью выраженности клинических проявлений.
Считается, что прогноз параинфекционного ОМС более благоприятный, чем паранеопластического. Многие исследователи (Bataller L. и соавт., 2001; и др.) подчеркивают, что при параинфекционном ОМС скорость и степень улучшения зависят от возраста: чем старше пациент, тем медленнее восстанавливается неврологический дефицит, тем больше остаточных явлений при достижении ремиссии. Некоторые авторы считают, что параинфекционный ОМС регрессирует самостоятельно и не требует лечения, другие склоняются к тому, что пациенты с параинфекционным ОМС быстрее выздоравливают на фоне терапии путем внутривенного введения иммуноглобулина или кортикостероидов.
Таким образом, энцефалопатия Кинсбурна, или синдром опсоклонуса-миоклонуса, представляет собой достаточно уникальный клинический синдром. Необходимы дальнейшие исследования с целью изучения этиологии и патогенеза этого заболевания, поиск новых специфических методов лечения.
Е. С. Ильина, кандидат медицинских наук
М. Ю. Бобылова
Российская ДКБ Росздрава, РГМУ, Москва
Миоклоническая эпилепсия
Миоклоническая эпилепсия — заболевание, основу которого оставляют миоклонические эпилептические пароксизмы. Эпизоды миоклонических судорог у больных сочетаются с генерализованными клонико-тоническими эпиприступами, абсансами. Сопутствующая неврологическая симптоматика зависит от формы эпилепсии. Диагностика включает сбор анамнеза, оценку неврологического и психического статуса, электроэнцефалографию, генеалогический анализ, биохимические исследования, нейровизуализацию. Лечение проводится антиконвульсантами, при резистентности — комбинацией противоэпилептических препаратов.
Миоклонические судороги (миоклонии) представляют собой непроизвольные сокращения отдельной мышцы/мышечной группы. Соответственно, эпилепсия с преобладанием в клинической картине миоклоний получила название миоклоническая. Понятие «миоклоническая эпилепсия» (МЭ) включает ряд заболеваний, разнородных по этиопатогенезу, возрасту дебюта, особенностям симптоматики. В подавляющем большинстве случаев они характеризуется сочетанием миоклоний и генерализованных тонико-клонических судорожных приступов, имеют генетическую обусловленность. Встречаемость МЭ различна, некоторые нозологические формы являются настолько редкими, что в литературе по неврологии описано не более 100 клинических случаев.
Причины миоклонической эпилепсии
Обычно ведущим является генетический фактор. Чёткое аутосомно-доминантное наследование прослеживается при синдроме Драве, аутосомно-рецессивное — в отдельных случаях ранней миоклонической энцефалопатии. Некоторые заболевания имеют полигенное наследование. Локализация генетических дефектов установлена не для всех наследственных форм, исследования в этом направлении продолжаются. К генетически детерминированным патологиям относится и симптоматическая МЭ, возникающая вследствие дисметаболических процессов, обусловленных наличием дефектных генов. Образованию спонтанных мутаций в геноме способствуют:
- Внутриутробные инфекции. Инфекционный процесс неблагоприятно отражается на развитии плода. Особенно опасны вирусные инфекции, поскольку вирусы способны провоцировать аномальную перестройку отдельных генов.
- Хронические заболевания беременной. Сахарный диабет, сердечная недостаточность, хронические заболевания лёгких, эндокринная патология матери приводят к гипоксии, метаболическим расстройствам на ранних стадиях развития зародыша. В результате происходят сбои формирования ЦНС, отдельных механизмов обмена веществ.
- Повышенный радиоактивный фон. Радиация оказывает мутагенное влияние на живые организмы. Развивающийся плод наиболее подвержен подобному воздействию. Следствием является возникновение структурных, дисметаболических, функциональных аномалий, влекущих за собой повышенную эпилептическую активность.
- Прием тератогенных медикаментов. Самолечение, незнание о своей беременности в раннем периоде, медицинская необходимость фармакотерапии приводят к приёму опасных для плода медикаментов. Химические вещества оказывают повреждающее воздействие на отдельные гены, вносят изменения в существующие метаболические механизмы.
- Токсические воздействия на плод.Алкоголизм, наркомания, курение женщины в период беременности сопровождаются проникновением токсических веществ в организм плода. Подобно тератогенным фармпрепаратам они способны повредить отдельный локус генома, в результате возникает миоклоническая эпилепсия.
Патогенез
Идиопатические варианты МЭ развиваются вследствие генетически обусловленной повышенной возбудимости церебральных нейронов, приводящей к эпилептогенной активности мозга. Симптоматическая миоклоническая эпилепсия формируется в результате обменных нарушений, накопления в нервных клетках патологических соединений (полисахаридных включений, прионных белков).
При болезни Лафоры, миоклонической энцефалопатии младенцев повышенная эпиактивность обусловлена дисметаболизмом нейронов в условиях разрастания глиальных элементов (при гибели нейронов, нарушении апоптоза астроцитов). Нейрональная гипервозбудимость вызывает возникновение патологической нервной импульсации, идущей к мышечным волокнам. Результатом являются отдельные мышечные сокращения (миоклонии), тонические, клонические судороги. Различная локализация миоклоний отражает локальное возбуждение разных зон мозговой коры. При диффузном распространении гипервозбуждения возникает клонико-тонический пароксизм с тотальным вовлечением мышечных групп.
Классификация
В основе группировки отдельных видов МЭ лежит этиологический принцип. Согласно Международной классификации эпилепсии 1989 года выделяют 3 основные группы:
- Идиопатические — наследственно обусловленные формы. Характерна манифестация симптоматики в детском/подростковом возрасте. Идиопатическими являются доброкачественная миоклоническая эпилепсия младенчества (ДМЭМ), юношеская миоклоническая эпилепсия (ЮМЭ), болезнь Унферрихта-Лундборга, синдром Драве.
- Криптогенные — не имеющие установленной этиологии. Отличаются выраженной резистентностью к фармакотерапии, наличием сопутствующей очаговой симптоматики, интеллектуального дефицита. К криптогенным относятся эпилепсия с миоклонически-астатическими приступами, эпилепсия с миоклоническими абсансами.
- Симптоматические — возникающие на фоне происходящих в организме патологических процессов. В большинстве случаев обусловлены метаболическими нарушениями. Симптоматическими считаются ранняя миоклоническая энцефалопатия, болезнь Лафоры, миоклонические пароксизмы при подостром склерозирующем панэнцефалите, болезни Крейтцфельдта-Якоба.
Впоследствии были выявлены генетические аспекты возникновения криптогенных форм МЭ. Учитывая результаты исследований, Международное общество неврологов предложило относить ранее считавшиеся криптогенными виды МЭ к идиопатическим.
Симптомы миоклонической эпилепсии
Базовым симптомом выступают пароксизмы миоклоний, затрагивающие различные мышечные группы конечностей, реже — лица, еще реже — туловища. Миоклонии выглядят как мышечные подёргивания, при вовлечении мышц одной группы сокращения приводят к непроизвольным двигательным актам, напоминающим гиперкинезы. Миоклонический эпилептический пароксизм происходит при сохранённом сознании, может протекать с перемещением сокращений по различным мышцам. Миоклоническая эпилепсия характеризуется комбинацией миоклоний с клонико-тоническими приступами и/или абсансами. В зависимости от нозологической формы наблюдаются задержка психического развития, атаксия, пирамидная недостаточность, расстройства мышечного тонуса, зрительные нарушения.
ДМЭМ дебютирует в возрастном периоде от 6 месяцев до 3 лет. Приступы захватывают верхние конечности, лицо, шею, могут имитировать наклон головы, моргание, кивки головой. Заболевание редко сопровождается интеллектуальным снижением. Миоклоническая эпилепсия юношеского возраста (манифестация в возрасте 12-18 лет) отличается присоединением тонико-клонических эпизодов, отсутствием неврологического дефицита. Синдром Драве клинически проявляется на первом году жизни, сопровождается олигофренией, нарушениями поведения, пирамидным дефицитом. Семейная миоклония Унферрихта-Лундборга начинается в 5-16 лет, сочетается с тремором, атаксией, дизартрией, психическими расстройствами.
Миоклонически-астатические пароксизмы отличаются возникающей на фоне миоклоний потерей устойчивости. Пациенты описывают приступ как «удар под коленки», «подкашивание ног», вынуждающее становиться на колени, падать. Миоклонические абсансы представляют собой эпизоды кратковременного отключения сознания с миоклоническими сокращениями плечевого пояса, мышц конечностей, периорбитальной области. Заболевание возникает у детей 2-12 лет.
Миоклоническая эпилепсия симптоматического характера отличается прогрессированием симптоматики, выраженным когнитивным дефицитом, прочими неврологическими нарушениями, наличием проявлений основного заболевания, клинико-лабораторных признаков метаболических расстройств.
Осложнения
Клонико-тонические, астатические приступы осложняются травмированием пациента вследствие падения. Генерализованные судороги с утратой сознания опасны западением языка, перекрытием дыхательных путей и асфиксией. Аспирация слюны, рвотных масс приводит к последующему развитию пневмонии. Длительный миоклонический пароксизм, непрерывно следующие кластерные сокращения перерастают в миоклонический эпистатус. В эпилептическом статусе возможны серьёзные дыхательные расстройства, остановка сердца, развитие отёка головного мозга.
Диагностика
Миоклоническая симптоматика входит в клинику многих болезней, эпилептических синдромов. Диагноз «миоклоническая эпилепсия» устанавливается только при превалировании миоклонических приступов над другими клиническими проявлениями. Диагностика направлена на верификацию нозологической формы эпилепсии, при выявлении вторичного характера миоклоний — на поиск основной патологии. Основными диагностическими этапами являются:
- Сбор анамнестических данных. Большое значение имеет возраст дебюта, характер начала, порядок развития симптоматики.
- Неврологический осмотр. Проводится неврологом, направлен на выявление миоклонических сокращений, очагового дефицита, определение уровня психического развития, степени когнитивных расстройств, оценку психического статуса.
- Электроэнцефалография. У большинства пациентов регистрируются диффузные интериктальные симметричные эпилептогенные разряды, иктальные высокоамплитудные спайки. В ряде случаев для выявления эпиактивности требуется суточный ЭЭГ-видеомониторинг, проведение провокационных проб (ЭЭГ при вспышках света, гипервентиляции, резких звуковых сигналах). Результаты исследований оцениваются нейрофизиологом, эпилептологом.
- Нейровизуализация. До закрытия родничков осуществляется путём нейросонографии, у детей старше года — при помощи МРТ головного мозга. Взрослым может проводиться МСКТ. Морфологические изменения церебральных тканей характерны для симптоматических МЭ.
- Лабораторные исследования. Производятся при подозрении на наличие обменных расстройств. Включают биохимический анализ крови и мочи, специфические анализы.
- Консультация генетика. Сбор семейного анамнеза, составление генеалогического древа позволяют определить наследственный характер эпилепсии, установить тип наследования.
Дифференциальная диагностика осуществляется с неэпилептическим миоклонусом, отличительной особенностью которого выступает фокальный характер миоклоний, отсутствие реакции на провокацию, нормальная ЭЭГ-картина. Дифференцировка МЭ необходима также с судорожным синдромом инфекционной этиологии, фебрильными судорогами, синдромом Леннокса-Гасто, мозжечковой миоклонической диссинергией Ханта.
Лечение миоклонической эпилепсии
Терапия базируется на антиконвульсантах. Подбор фармпрепарата и дозировки осуществляется индивидуально. Препаратами выбора выступают производные вальпроевой кислоты, обладающие противоэпилептическим эффектом в равной степени в отношении миоклонических, клонико-тонических, абсансных пароксизмов. В фармакорезистентных случаях показано комбинированное лечение вальпроатами, бензодиазепинами, этосуксимидом, барбитуратами, антиконвульсантами нового поколения (топираматом, леветирацетамом). Важным моментом является исключение провоцирующих приступы факторов: резких звуков, вспышек света, эмоциональных всплесков, физических перегрузок, перегреваний.
Прогноз и профилактика
Наиболее прогностически неблагоприятна ранняя миоклоническая энцефалопатия, смертность составляет половину случаев заболевания, остальные дети являются глубокими инвалидами. Миоклоническая эпилепсия при болезни Лафоры, Крейтцфельдта-Якоба плохо поддаётся противоэпилептической терапии, сопровождается прогрессирующим интеллектуальным распадом. ДМЭМ и ЮМЭ отличаются доброкачественным течением, редко приводят к когнитивному дефициту. Более 50% случаев ДМЭ заканчиваются спонтанным выздоровлением.
МЭ не имеет специфических мер профилактики. К мероприятиям, способным предупредить рождение больного ребёнка, относятся планирование беременности, ранняя постановка на учёт, исключение неблагоприятных воздействий на плод. Ведение беременности должно включать разъяснительные беседы с женщиной по поводу необходимости охранительного режима, тератогенной опасности лекарственных средств, пагубного воздействия на будущего ребёнка вредных привычек.
Читайте также: