Синдром Нонне-Милроя-Мейжа (Nonne-Milroy-Meige) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 02.02.2026
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Смотреть что такое "Милроя болезнь" в других словарях:
Милроя болезнь — (W. F. Milroy, 1855 1942, амер. врач) см. Нонне Милроя Мейжа синдром … Большой медицинский словарь
Нонне-Милроя-Мейжа синдром — (М. Nonne, 1861 1959, нем. невропатолог; W. F. Milroy, 1855 1942, амер. врач; Н. Meige, 1866 1940, франц. врач; син.: Мейжа болезнь, Милроя болезнь) наследственная болезнь, характеризующаяся сочетанием трофедемы лица и конечностей с низким ростом … Большой медицинский словарь
Болезнь Милроя (Afilroy'S Disease) — см. Лимфедемия. Источник: Медицинский словарь … Медицинские термины
Мейжа болезнь — (Н. Meige, 1866 1940, франц. врач и анатом) см. Нонне Милроя Мейжа синдром … Большой медицинский словарь
Но́нне — Ми́лроя — Ме́йжа синдро́м — (М. Nonne, 1861 1959, нем. невропатолог: W.F. Milroy, 1855 1942. американский врач: Н. Meige, 1866 1940, франц. врач; син.: Мейжа болезнь, Милроя болезнь) наследственная болезнь, характеризующаяся сочетанием трофедемы лица и конечностей с низким… … Медицинская энциклопедия
МЭЙЖА СИНДРОМ — (описан французским врачом Н. Meige, 1866-1940; синонимы - идиопатическая лимфедема, лимфедема второго типа) - наследственное заболевание лимфатической системы, проявляющееся в возрасте 20-35 лет отеками ног; иногда в процесс вовлекаются и руки.… … Энциклопедический словарь по психологии и педагогике
Слоновость — I Слоновость (elefantiasis; синоним: элефантиаз, лимфедема) постепенно прогрессирующий диффузный отек части тела, обусловленный нарушением лимфооттока лимфостазом. Поражаются нижние и верхние конечности, реже наружные половые органы, брюшная… … Медицинская энциклопедия
Гемигипертрофия — I Гемигипертрофия (греч. hēmi полу + Гипертрофия) увеличение размеров половины тела, чаще врожденное. Предполагают, что Г. может быть вариантом дизэмбриогенеза. Наблюдается либо как изолированный симптом, либо (у детей) сочетается с гидроцефалией … Медицинская энциклопедия
Синдром Нонне-Милроя-Мейжа (Nonne-Milroy-Meige) - синонимы, авторы, клиника
Кафедра неврологии и реабилитации Казанского государственного медицинского университета
Синдром Мейжа или сегментарная краниоцервикальная дистония: терминология, история изучения и современный взгляд
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2015;115(12): 133‑136
Термин «синдром Мейжа», или «синдром Брейгеля», применяется специалистами для описания блефароспазма с непроизвольными движениями в нижней части лица и/или жевательных мышцах. Эпонимы «синдром Мейжа» и «синдром Брейгеля» вносят некоторую путаницу в терминологию. Термин «сегментарная краниоцервикальная дистония» объединяет различные блефароспазм-плюс фенотипы и отражает современные представления об их генетической и патофизиологической общности. Ботулинотерапия остается практически единственным методом симптоматического лечения краниоцервикальных дистоний.
Термин «синдром Мейжа» применяется специалистами для описания блефароспазма с непроизвольными движениями в нижней части лица и/или жевательных мышцах. Применение этого термина, как и других эпонимов для описания различных форм дистоний, не поддерживается единогласно исследователями [1]. Во-первых, поскольку Г. Мейж не страдал синдромом, носящим его имя, по мнению Редакционного научного совета, притяжательная форма эпонима неприменима [2]; во-вторых, Г. Мейж не был первым, кто описал комбинацию блефароспаза и нижнелицевых гиперкинезов; в-третьих, только в эпонимическом словаре-справочнике [3] можно найти 3 синдрома, относящиеся к различным областям медицины, названные именем Мейжа (синдром Мейжа, синдром Нонне—Мейжа, синдром Бриссо—Мейжа).
Впервые к блефароспазму и к другим краниальным дистониям привлек внимание H. Wood в 1887 г., описав их как «тоническое сокращение, приводящее к закрыванию глаз с последующим ослеплением. Это сопровождается бесчисленными причудливыми гримасами вследствие усилия мышц-антагонистов по преодолению силы, которая закрывает глаза» [4]. Понятно, что второе утверждение было ошибочным.
В 1910 г. Г. Мейж, французский невролог, наблюдал около 10 пациентов с непроизвольным закрыванием глаз. Блефароспазм сочетался с гиперкинезами жевательных мышц только у 1 из этих пациентов [5]. Более чем через 60 лет G. Paulson, описав 3 пациентов с блефароспазмом и оромандибулярными дистониями, подчеркнул их общую патофизиологическую основу [6].
В 1976 г. D. Marsden для обозначения синдрома блефароспазма и оромандибулярных гиперкинезов предложил термин «синдром Брейгеля», обратив внимание на патологическое выражение лица, изображенного на картине Питера Брейгеля-старшего «Зевака» [1, 7, 8]. С тех пор в литературе начали применяться два эпонимических обозначения.
Анализ литературы показывает, что синдромы Брейгеля и Мейжа остаются плохо очерченными и существуют некоторые разночтения в их интерпретации. Некоторые авторы считают, что существенным проявлением синдрома Брейгеля является широкое дистоническое открывание рта [9]. Большинство же исследователей, придерживаясь предложения D. Marsden, относят к синдрому Брейгеля как открывательные, так и закрывательные оромандибулярные гиперкинезы, если они сочетаются с блефароспазмом [10—12].
В то же время открывательные гиперкинезы могут встречаться в составе сегментарных, мультифокальных и генерализованных дистоний [12], а применительно к ним термин «синдром Брейгеля» не используется [2].
Эпоним «синдром Мейжа» применяется для обозначения как первичной, так и вторичной краниоцервикальной дистонии.
Анатомическая классификация дистоний, локализующихся в области головы, требует обсуждения. Фокальными принято называть дистонии, локализующиеся в области одного сегмента [13—15]. К таким дистониям относят, например, блефароспазм, оролингвальные или ларингеальные дистонии. Локальными могут также считаться и дистонии, ограниченные областью лица (как одним сегментом) — краниальные, блефароспазм-оромандибулярные дистонии. В то же время в оролингвальных гиперкинезах нередко участвуют мышцы, имеющие точки прикрепления как в области головы, так и шеи (m. platyzma, digastricus, mylohyoideus, geniohyoideus и т. д.). И наконец, в дистонии краниальной области вовлекаются не только мимические, но и жевательные мышцы, приводя в действие нижнюю челюсть, не являющуюся частью головы [16].
Нередко блефароспазм сочетается с дистониями ларингеальных мышц, относящихся и к краниальному, и к цервикальному регионам [7, 10, 14, 17]. Все упомянутые обстоятельства указывают на то, что синдром Мейжа, или Брейгеля, правильнее относить к сегментарной краниоцервикальной мышечной дистонии.
Этиологическая классификация краниоцервикальной дистонии включает: первичные краниоцервикальные дистонии — синдромы, при которых дистония — единственное клиническое проявление (за исключением сочетания с тремором); вторичные краниоцервикальные дистонии: сосудистые (ишемический или геморрагический инсульт, ДЦП и др.); инфекционные (ВИЧ, арбовирус и др.); токсические (марганец, угарный газ и др.); лекарствнные (нейролептики, антиэметики, антиконвульсанты); аутоиммуные/воспалительные (рассеянный склероз, системная красная волчанка, болезнь Бехчета и др.); краниоцервикальные дистонии-плюс: наследственные дистонии, не относящиеся к первичным или дегенеративным; дофа-чувствительная дистония (DYT5); миоклонус-дистония (DYT11); дегенеративные заболевания с краниоцервикальной дистонией наследственного и спорадического характера: прогрессирующий надъядерный паралич, болезнь Паркинсона, мультисистемная атрофия, кортикобазальная дегенерация, спиноцеребеллярная атаксия, болезнь Гентингтона, Лубаг (DYT3), паркинсонизм с быстрым началом (DYT12), болезнь Вильсона, синдром Леша—Нихана, нейродегенерации, ассоциированные с пантотенат киназой, митохондриальные энцефалопатии и др.; психогенные краниоцервикальные дистонии — дистонии вследствие воздействия психогенного фактора.
Термин «первичная дистония» объединяет варианты как с наследственной этиологией, так и с генетической предрасположенностью.
Блефароспазм и сегментарная краниоцервикальная дистония определенно имеют генетическую детерминированность [18—20]. По данным ряда исследователей [18, 21—23], у 10% пациентов с блефароспазмом и другими формами краниоцервикальной дистонии есть родственники первой или второй линии родства с подобным проявлениями. Клинико-генетические корреляции показали возможность формирования краниоцервикальной локализации дистонии при различных DYT-мутациях. Сегментарная краниоцервикальная дистония чаще всего является фенотипическим проявлением мутации DYT6, реже DYT7, DYT13 [22, 24, 25]. Иногда краноцервикальные проявления носит дистония с DYT1-делецией [26]. Возможна этническая специфичность в формировании риска и модификации клинического фенотипа краниоцервикальной дистонии. В частности, в российской популяции установлена ассоциация фокальной/сегментарной дистонии с микросателлитным полиморфизмом (СТ/GT/GA)n в 5’-области гена DRD5 [27].
Сегментарная краниоцервикальная дистония может быть одним из симптомов дегенеративного заболевания наследственного или спорадического характера, например одной из форм спиноцеребеллярной дегенерации [28], прогрессирующего надъядерного паралича [29].
Нередко сегментарная краниоцервикальная дистония является следствием медикаментозной блокады дофаминовых рецепторов, инфаркта базальных ядер и других причин [30—32].
Определенные результаты в понимании этиологии, патогенеза и патофизиологии сегментарных краниоцервикальных дистоний могут дать исследования молекулярной, клеточной и системной роли торсина А, TAF1 и THAP1 при первичных дистониях DYT1, DYT3, DYT6 соответственно. Так, торсин, А является посредником взаимодействия между оболочкой ядра и цитоскелетом. Мутантный торсин, А может опосредованно нарушать перемещение транскрипционных факторов и копирование в или из ядра соответственно. Этот белок обладает АТФ-связывающей активностью и может участвовать в процессах образования везикул, конформационных изменениях белков, регуляции клеточных сигналов и функционировании митохондрий. ТAF1 (транскрипционный фактор), являясь основным строительным белком РНК-полимеразы II, играет важную роль в коактивации распознания и модификации процессов транскрипции. THAP1 (танатос-ассоциированный протеин) является одним из ДНК-связывающих проапоптотических факторов и наряду с другими протеинами участвует в клеточной пролиферации, клеточном цикле, сегрегации хромосом [24, 33].
В совокупности вышеуказанные данные свидетельствуют о возможной роли этих белков в процессах высвобождения нейромедиаторов в нигростриарной системе мозга и/или поддержании энергетического потенциала дофаминергических нейронов и нейронопротекции.
Спорадическое проявление первичной краниоцервикальной дистонии объясняется неполной пенетрантностью, вариабельной экспрессивностью дистоний под влиянием факторов внешней среды. Врожденная неадекватная сенсомоторная реактивность к периферическим сенсомоторным раздражителям также рассматривается как один из пусковых механизмов возникновения гиперкинезов [34]. Например, блефароспазм нередко начинается с симптомов раздражения глаза, таких как синдром сухого глаза или блефарит [35]. Сначала пациенты испытывают потребность в частом моргании (адаптивный ответ) с последующим развитием полноценного блефароспазма (дезадаптивный ответ).
Аналогично этому у многих пациентов первичным оромандибулярным гиперкинезам предшествовали травмы лица или стоматологические вмешательства. Таким образом, краниоцервикальные дистонии, как и дистонии в целом, вероятно, являются следствием нарушения сенсомоторной интеграции [36].
Физиологические исследования демонстрируют патологическую активность ствола мозга и сенсомоторной коры [37, 38]. Повышение возбудимости интернейронов мозгового ствола было обнаружено при анализе мигательного рефлекса и массетерного ингибиторного рефлекса [39]. Функциональная МРТ показала повышение активности соматосенсорной и дополнительной моторной коры. Укорочение латентного периода свидетельствовало о гипервозбудимости ингибирующих нейронов коры и определялось как при изолированном блефароспазме, так и в сочетании с оромандибулярными дистониями и не выявлялось у здоровых из группы контроля [37].
Основным методом лечения сегментарной краниоцервикальной дистонии является ботулинотерапия. Успех лечения зависит от правильности выбора мышцы-мишени, дозы препарата, точности инъекции, в конечном счете, от профессионализма врача. Инъекции ботулинических токсинов при сегментарной краниоцервикальной дистонии приводят к значительному объективному и субъективному улучшению при блефароспазме и менее выраженному улучшению при блефароспазм-плюс дистониях [7, 11, 17, 39—42].
По данным Van den Bergh и соавт. [42], эффективность такого лечения при блефароспазме достигает 79%, а при синдроме Мейжа — 53%. Однако следует учесть, что выраженность блефароспазма при блефароспазм-плюс дистонии может быть более значительной. Кроме того, блефароспазм может сочетаться с оромандибулярными гиперкинезами как закрывательного, так и открывательного типа [41], а, как известно, открывательный вариант является трудной задачей для врача, выполняющего инъекции ботулинических токсинов [2, 41, 43—45].
Ботулиничесий токсин ксеомин — первый препарат ботулинического токсина, А (БТА), не содержащий комплексообразующих белков и, как следствие, имеющий низкую способность к образованию антител. Еще одним преимуществом ксеомина, отличающим его от всех других БТА, является сохранение стабильности при комнатной температуре, что упрощает условия транспортировки и хранения препарата [39].
Эффективность ксеомина при лечении блефароспазма и цервикальной дистонии исследовалась неоднократно. D. Dressler [40] обобщил мировой опыт применения ботулотоксина ксеомин при краниоцервикальных дистониях за 5 лет. В обзоре указывается, что эффективность ксеомина (Incobotulinumtoxin A) была подтверждена крупными рандомизированными двойными слепыми и плацебо-контролируемыми исследованиями. Наряду с сопоставимыми с ботоксом (OnabotulinumtoxinA) лечебными эффектами, переносимостью и продолжительностью действия, в силу своей малой молекулярной массы (150 кДa) и отсутствия комплекса белков нетоксичной части, ксеомин демонстрирует аналогичный ботоксу уровень диффузии [40].
По дизайну одного из недавних исследований [45] эффективности и переносимости ксеомина при блефароспазме допускался гибкий график частоты инъекций. Наиболее частыми побочными эффектами были птоз века (31,4%) и сухой глаз (17,6%). Повторные инъекции ксеомина, введенного в интервалах от 6 до 20 нед в соответствии с потребностями пациентов, обеспечили устойчивую эффективность в лечении блефароспазма. При укорочении интервала между инъекциями менее чем в 12 нед частота нежелательных явлений и титр нейтрализующих антител в крови не возрастали [45]. Еще одно исследование [46] подтверждает эти же данные и содержит рекомендации по возможности проведения повторной инъекции ранее 12 нед, если лечебный эффект закончился, не опасаясь вторичной резистентности.
Только при неэффективности ботулинотерапии сегментарная краниоцервикальная дистония может являться показанием для хронической стимуляции глубоких структур мозга (DBS). Признанной мишенью для этих целей считается внутренний сегмент бледного шара [47, 48]. Желаемый эффект может развиваться постепенно, через несколько месяцев от начала стимуляции [48]. Описаны наблюдения, когда после длительного применения дальнейшая хроническая стимуляция при дистонии не требуется, что объясняется нормализацией нейрональных связей под действием DBS.
В заключение отметим, что эпонимы «синдром Мейжа» и «синдром Брейгеля», менявшие свою популярность в разные исторические периоды, вносят некоторую путаницу в терминологию. Термин «сегментарная краниоцервикальная дистония» объединяет различные блефароспазм-плюс фенотипы и отражает современные представления об их генетической и патофизиологической общности. Ботулинотерапия остается практически единственным методом симптоматического лечения краниоцервикальных дистоний. Дальнейшее установление генетических и средовых факторов риска, механизмов клеточной и нейрональной дисфункции неизбежно приведет к разработке патогенетических методов лечения.
Милроя болезнь (лимфедема наследственная)
Врожденная наследственная лимфедема тип IА - болезнь Нонне-Милроя (MIM 1531000) - заболевание лимфатической системы, характеризующееся образованием опухолей конечностей. У больных уже при рождении обнаруживаются отеки ног. Степень отеков варьирует от небольшой припухлости в области лодыжек до выраженного увеличения объема стоп, голеней, бедер. Кожа в области отека может быть нормальной или истонченной. При надавливании остается ямка. Болезнь Нонне-Милроя - лимфедема с ранним началом, наиболее тяжелые поражения наблюдаются в нижних конечностях с рождения. Опухоли конечностей возникают из-за дисфункции лимфатических сосудов. Они могут располагаться на одной или на всех конечностях, отличаются по размеру и положению, а также могут озлокачествлятся. На лимфоангиограмме видна гипоплазия или аплазия лимфатических сосудов.
Данный тип заболевания надо дифференцировать с синдромом Мэйджа (MIM 153200) - лимфедемой II типа, при которой клинические признаки появляются в 20-40 лет. Кроме того, лимфедема может встречаться в ассоциации с другими клиническими особенностями, например аутосомно-доминантная лимфедема с distichiasis.
Популяционная частота - 1:10 000. Тип наследования - аутосомно-доминантный с неполной пенетрантностью. Пенетрантность составляет 80% у гетерозиготных носителей мутантного гена и 99% - у гомозиготных носителей.
Обнаружено сцепление фенотипа заболевания с хромосомой 5q34-q35, где картирован ген рецептора васкулярного эндотелиального фактора роста (VEGFR3 - vascular endothelial growth factor receptor 3, или FLT4). Ген необходим для кардиоваскулярного развития в эмбриогенезе, у взрослых он экспрессируется в лимфатических эндотелиальных клетках. Ген состоит из 31 экзона. В нем обнаружены мутации у больных в семьях, для которых обнаружено сцепление с районом хромосомы 5q34-q35. На сегодня описано 14 мутаций в экзонах 18, 23-25 гена. Однако существуют семьи, для которых сцепление с областью 5q34-q35 исключено.
В 2008 году выделен фенотип, похожий на фенотип при лимфедеме типа IА, сцепленный с хромосомой 6q16.2-q22.1 (лимфедема тип IB, MIM 611944). Ген в этом локусе пока идентифицировать не удалось.
В Центре Молекулярной Генетики проводится поиск мутаций в кодирующей области гена FLT4 методом прямого автоматического секвенирования.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Нонне - Милроя - Мейжа болезнь
Наследственное заболевание, характеризующееся развитием трофедемы лица и/или конечностей. В настоящее время принята классификация, согласно которой выделяют два типа наследственной лимфедемы: тип I (болезнь (Нонне-)Милроя, или первичная врождённая лимфедема) и тип II (болезнь Мейжа, или поздняя лимфедема).
Наследственная лимфедема I типа обусловлена мутацией гена FLT4 (локус 5q35.3), кодирующего рецептор фактора роста эндотелия сосудов-3. Большинство случаев характеризуется аутосомно-доминантным типом наследования с неполной пенетрантностью (около 84%), вариабельной экспрессивностью и возрастом дебюта (от рождения до 35 лет). Заболеваемость - 1:6000 новорождённых; соотношение полов 1:2,3. Как правило, дебют заболевания - с рождения или сразу же после рождения. У ребёнка отмечается дистальный плотный тёплый отёк конечностей, безболезненный, без тенденции к изъязвлению и варикозу. В большинстве случаев отёк сохраняется на всю оставшуюся жизнь; изредка возможно спонтанное выздоровление.
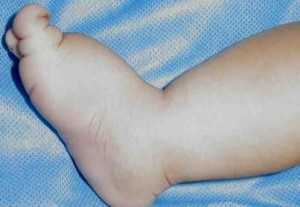

Возможно также развитие хилёзного асцита, гидроторакса, хотя в целом для заболевания не характерны сопутствующие аномалии. Прогноз обычно благоприятный, заболевание в основном характеризуется косметическим дефектом. Описаны сочетания лимфедемы с гидроцеле, дефектами межпредсердной перегородки, аномалиями развития лицевого черепа. Из осложнений описаны лимфангиэктазия кишечника, рецидивирующий септический артирит, (лимф)ангиосаркома.
Впервые наследственная лимфедема нижних конечностей была описана немецким неврологом Максом Нонне в 1891г. (Nonne M. Vier Fälle von Elephantiasis congenita hereditaria // [Virchows] Archiv für pathologische Anatomie und Physiologie und für klinische Medicin, Berlin, 1891. - S.189-196). Американский невролог Уильям Милрой в 1892г. описал 22 случая заболевания (Milroy W.F. An undescribed variety of herditary edema // New York Medical Journal, 1892. - Vol.56. - P.505-508). Эпонимическое название «болезнь Милроя» было предложено английским врачом сэром Уильямом Ослером в его учебнике «The Principles and Practice of Medicine», изданном в 1892г. в Нью-Йорке. Термин «болезнь Нонне - Милроя» получил распространение в немецкоязычной литературе.
Наследственная лимфедема II типа обусловлена мутацией гена FOXC2 (локус 16q24.3), кодирующего один из факторов транскрипции. К аллельным заболеваниям лимфедемы Мейжа относятся сочетание лимфедемы с дистихиазом (двойной ряд ресниц), птозом, жёлтыми ногтями. Наследуется по аутосомно-доминантному типу, дебют после полового созревания. Описаны сочетания поздней лимфедемы с нейросенсорной глухотой, первичной лёгочной гипертензией, мальформациями сосудов головного мозга, экстрадуральными кистами, аномалиями развития позвоночника. Клинически характеризуется развитием трофедемы самой разнообразной локализации, более выраженной ниже пояса.
Довольно часто пациентов беспокоят выраженные боли. Возможно вовлечение в процесс тканей лица и гортани. Несколько выше, чем в популяции, у таких больных риск развития злокачественных новообразований.
Нонне — Милроя — Мейжа синдром
наследственная болезнь, характеризующаяся сочетанием трофедемы лица и конечностей с низким ростом, гипогонадизмом, психической и физической отсталостью.
Смотреть что такое "Нонне — Милроя — Мейжа синдром" в других словарях:
Ме́йжа боле́знь — (Н. Meige, 1866 1940, франц. врач и анатом) см. Нонне Милроя Мейжа синдром … Медицинская энциклопедия
Ми́лроя боле́знь — (W.F. Milroy, 1855 1942, американский врач) см. Нонне Милроя Мейжа синдром … Медицинская энциклопедия
Читайте также: