Синдром Олбрайта (Albright) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 28.01.2026
наследственная остеодистрофия Олбрайта, по имени американского врача эндокринолога F. Albright, 1900-1969; синоним - псевдогипопаратиреоз) - фиброзная остеодисплазия нескольких костей скелета (порок развития костной ткани с частичным замещением ее хрящевой) и очаговая пигментация кожи в сочетании с эндокринными расстройствами. Основные проявления: симптомы гипопаратиреоза, связанные с невосприимчивостью тканей к паратгормону; изменения скелета. Диагностируют заболевание чаще между 5-м и 10-м годами жизни на основании низкого роста, диспропорции скелета с преобладанием укорочения нижних конечностей, короткой шеи, круглого лица, короткопалости с преимущественным укорочением I, III, IV, V метакарпальных и метатарзальных костей, из-за чего при сжатии руки в кулак отсутствует выпуклость в области IV и V пястно-фаланговых суставов; задержки умственного развития; нарушения минерального обмена (гиперфосфатемия, гипокальциемия, судороги, спазмы мышц, изменения скелета, в тяжелых случаях - патологические переломы, нарушение прорезывания зубов; эктопические кальцификаты - подкожные, в области базальных ганглиев). Выделяют два типа псевдогипопаратиреоза, отличающиеся биохимическими показателями: снижение содержания кальция в сыворотке крови, снижение экскреции фосфатов с мочой и повышение иммунореактивности паратиреоидного гормона плазмы наблюдается при обоих типах; экскреция цАМФ снижена при I и не изменена при II типе; концентрация фосфора в сыворотке повышена при I типе и не изменена или немного повышена при II типе. При цитогенетическом исследовании в ряде случаев выявляют микроделеции: сегмента q37 хромосомы 2 при I типе и сегмента q12 - q13.2 хромосомы 20. Тип наследования - предположительно Х-сцепленный доминантный для I типа; не установлен для II типа. Лечение: препараты кальция и витамина D в больших дозах; в случае выраженной деформации трубчатых костей - хирургическая коррекция.
F. Albright, C. H. Burnett, P. H. Smith, W. Parson. Pseudohypoparathyroidism: an example of «Seabright-Bantam syndrome». Endocrinology, Baltimore, 1942; 30: 922-932.
Найдено научных статей по теме — 4
Синдром Кушинга у новорожденного мальчика с синдромом маккьюна-олбрайта
Маказан Надежда Викторовна, Орлова Елизавета Михайловна, Карева Мария Андреевна, Поддубный Игорь Витальевич, Толстов Кирилл Николаевич, Полякова Галина Александровна, Богданова Полина Сергеевна, Петеркова Валентина Александровна, Дедов Иван Иванович
ВИПАДОК іЗ ПРАКТИКИ. ПОєДНАННЯ СИНДРОМУ МАК-КЮНА — ОЛБРАЙТА З АКРОМЕГАЛієЮ
СИНДРОМ МАК-КЬЮНА - ОЛБРАЙТА - БРАЙЦЕВА (ОБЗОР ЛИТЕРАТУРЫ И КЛИНИЧЕСКИЙ СЛУЧАЙ)
В статье приведены результаты обзора литературы, касающиеся синдрома Мак-Кьюна Олбрайта Брайцева, и описан клинический случай данного синдрома.
Роль молекулярно-генетических методов исследования в диагностике синдрома маккьюна-олбрайта-брайцева
Маказан Надежда Викторовна, Орлова Елизавета Михайловна, Колодкина Анна Александровна, Карева Мария Андреевна, Калинченко Наталья Юрьевна, Васильев Евгений Витальевич, Тюльпаков Анатолий Николаевич, Петеркова Валентина Александровна
Синдром МакКьюна-Олбрайта-Брайцева (МОБ) редкое генетическое заболевание, в основе которого лежат соматические мутации в гене GNAS.
Похожие термины:
Синдром Форбса-Олбрайта
по именам американских врачей A. P. Forbes и F. Albright, 1900-1969) - одно из эпонимических названий синдрома галактореи-аменореи, обусловленного аденомой гипофиза. A. P. Forbes, P. H. Henneman, G. C. Griswold, F. Albright. Syndrome characterize
БАТЛЕРА-ОЛБРАЙТА СИНДРОМ
описан американскими врачами A. Butler и F. Albright, 1900-1969; синоним - почечный тубулярный ацидоз, дистальный тип) - наследственное заболевание из группы тубулопатий, связанное с нарушением секреции ионо
Синдром Олбрайта-Хадорна
Первичное нарушение калиевого обмена с периодическими гипокалиемическими параличами мышц. Проявляется пароксизмами общей слабости, болями в костях, спине, остеопорозом костей, гипо- или арефле
МАРТИНА-ОЛБРАЙТА СИНДРОМ
Martin E., Albright F.]. Проявление нарушения утилизации тканями гормона паращитовидных желез. Характерны: гипокальциемия, гиперфосфатемия, ахондроплазия, умственное недоразвитие, судорожные тетанически
МАККЬЮНА-ОЛБРАЙТА СИНДРОМ
описан американскими педиатром D. J. McCune, 1902-1976, и эндокринологом F. Albright, 1900-1969; синоним - болезнь Олбрайта) - множественная фиброзная остеодисплазия с преждевременным половым развитием и пигмент
Синдром Олбрайта-Мак-Кьюна-Штернберга
Проявляется в детском возрасте. Этиология не известна. Проявляется нарушением функций шишковидной железы и других структур промежуточного мозга, что ведет к избытку продукции гонадотропинов и к
Псевдогипопаратиреоз. Остеодистрофия наследственная. Олбрайта синдром
Своеобразная остеодистрофия, проявляющаяся коротким туловищем, круглым лицом, укороченными метатарзальными, метакарпальными и фаланговыми костями, экзостозами. Олигофрения, эпилепсия, гиперкин
Псевдогипопаратиреоз
Псевдогипопаратиреоз (болезнь Олбрайта) - наследственная остеодистрофия, обусловленная резистентностью периферических тканей к паратгормону, что сопровождается расстройством кальциево-фосфорного обмена, задержкой физического и умственного развития. Псевдогипопаратиреоз протекает с явлениями диффузного остеопороза, тоническими судорогами, переломами и деформацией костей, отложением кальция в мышцах и сосудах, образованием камней в мочевыводящих путях, задержкой роста и отставанием умственного развития. С целью диагностики псевдогипопаратиреоза проводятся определение уровня кальция, щелочной фосфатазы, паратгормона в крови; экскреции фосфора и кальция с мочой; функциональная проба с введением паратгормона; рентгенодиагностика. Лечение псевдогипопаратиреоза предполагает медикаментозную коррекцию гипокальциемии.
МКБ-10
Общие сведения
Псевдогипопаратиреоз является редкой наследственной патологией; в эндокринологии известно всего около 300 случаев данного заболевания. Псевдогипопаратиреоз по своим проявлениям напоминает гипопаратиреоз. Однако, если в основе гипопаратиреоза лежит первичный дефицит паратиреоидного гормона, обусловленный снижением функциональной активности паращитовидных желез, то при болезни Олбрайта псевдогипопаратиреоидный синдром вызывается нарушением чувствительности тканей-мишеней к действию паратгормона при его достаточном уровне секреции.
Впервые псевдогипопаратиреоз был описан в 1942 г. Ф.Олбрайтом и по автору получил название «болезни Олбрайта» или «наследственной остеодистрофии Олбрайта». Первый вариант заболевания характеризуется аномалиями развития скелета, резистентностью периферических тканей к паратгормону, гипокальциемией и клинической картиной, сходной с идиопатическим гипопаратиреозом. Второй вариант болезни Олбрайта протекает по нормокальциемическому типу и получил в литературе название псевдопсевдогипопаратиреоза. Описаны случаи развития в одной семье как нормо-, так и гипокальциемических вариантов заболевания, а также переход одной формы в другую.
Причины псевдогипопаратиреоза
При любых вариантах псевдогипопаратиреоза заболевание носит наследственный характер с аутосомно-доминантным типом наследования, сцепленным с X-хромосомой. Изучение родословных показывает, что число женщин с псевдогипопаратиреозом в 2 раза превышает количество больных мужчин; кроме того, болезнь Олбрайта не передается от отца к сыновьям.
Патогенез
Псевдогипопаратиреоз обусловлен генетической резистентностью скелета и почек к действию паратиреоидного гормона. Это связано с дефектом специфических рецепторов плазматических мембран клеток-мишеней и недостаточностью ферментов аденилатциклазы, протеинкиназы, циклического 3, 5- аденозинмонофосфата (АМФ) в дистальных канальцах нефрона, что сопровождается реабсорбцией фосфора. Снижение выделения фосфора с мочой приводит к гиперфосфатемии и вторичной гипокальциемии.
Поскольку при псевдогипопаратиреозе паращитовидные железы остаются интактными, гипокальциемия может вызывать стимуляцию секреции паратгормона и развитие вторичного гиперпаратиреоза. При псевдогипопаратиреозе обычно имеет место компенсаторная гиперплазия паращитовидных желез (развитие аденом не типично), изменения со стороны костной ткани (остеопороз, кисты), отложение кальцинатов в скелетных мышцах, подкожной клетчатке, а также в почках, стенках артерий, миокарде, конъюнктиве и роговицы глаза. Псевдогипопаратиреоз часто сочетается с артериальной гипертензией, сахарным диабетом, артериитом, полиартритом.
Симптомы псевдогипопаратиреоза
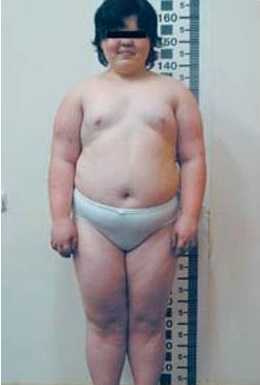
Клинические проявления псевдогипопаратиреоза аналогичны признакам идиопатического гипопаратиреоза. Эндокринные симптомы при псевдогипопаратиреозе включают низкорослость, ожирение и гипергликемию, лунообразное лицо, олигоменорею. Нередко болезнь Олбрайта сочетается с другими эндокринными нарушениями - акромегалией, синдромом Кушинга, гинекомастией, гипотиреозом или гипертиреозом.
Костно-суставные признаки псевдогипопаратиреоза включают брахидактилию с заметным укорочением 1, 4 и 5 пястных и плюсневых костей; экзостозы, дисхондроплазию, изменения в эпифизарных концах костей, резорбцию костей пальцев рук. Характерны задержка прорезывания зубов, гипоплазия зубной эмали, подкожные оссификаты.
Неврологическая симптоматика псевдогипопаратиреоза представлена приступами тонических судорог, которые могут возникать спонтанно или под воздействием любых раздражителей. Для больных с псевдогипопаратиреозом типично отставание в умственном развитии. Среди прочих симптомов псевдогипопаратиреоза отмечается мочекаменная болезнь, гематурия, рвота, развитие лентикулярной катаракты. При псевдопсевдогипопаратиреозе отсутствуют гипокальциемия, гиперфосфатемия, остеомаляция и судорожный синдром.
Диагностика
В типичных случаях псевдогипопаратиреоз диагностируется у детей в возрасте 5-10 лет с учетом характерной симптоматики, лабораторных, рентгенологических данных, генетических тестов. У больных с псевдогипопаратиреозом определяется гипокальциемия, гиперфосфатемия, нормальная или повышенная активность щелочной фосфатазы в крови, увеличенное содержание паратгормона в сыворотке крови; уменьшение экскреции кальция и фосфора с мочой.
Нечувствительность почечных канальцев к паратиреоидному гормону подтверждается с помощью теста, основанного на определении количества выводимых с мочой фосфатов и циклического аденозинмонофосфата. При псевдогипопаратиреозе в ответ на внутривенное введение паратгормона достоверное повышение содержания фосфатов и цАМФ в моче отсутствует. Рентгендиагностика (рентгенография костей и суставов, денситометрия) выявляет специфические изменений в костной ткани.
Для исключения поражения других органов и дифференциальной диагностики псевдогипопаратиреоза от сходных по проявлениям синдромов, проводятся консультации эндокринолога, уролога, невролога, офтальмолога, гинеколога. Дополнительное обследование может включать УЗИ почек и мочевого пузыря, УЗИ щитовидной железы, ЭЭГ, ЭКГ, осмотр глазного дна (офтальмоскопию) и др.
Лечение псевдогипопаратиреоза
При псевдогипопаратиреозе необходима коррекция гипокальциемии, в связи с чем проводится терапия препаратами кальция в дозировках, позволяющих поддерживать нормальную концентрацию кальция в крови. Показан прием витамина D и его активных форм под контролем концентрации кальция в сыворотке крови. Лечение паратиреоидным гормоном не эффективно. Для нормализации концентрации кальция и устранения симптомов вторичного гиперпаратиреоза необходимо соблюдение диеты с ограничением употребления фосфора.
При функциональной недостаточности других желез проводится заместительная терапия соответствующими гормонами. При возникновении судорог показано внутривенное введение растворов кальция.
Прогноз
Своевременная квалификация и рациональная терапия псевдогипопаратиреоза позволяет говорить о положительных прогнозах на жизнь и возможности контроля за течением заболевания. Учитывая генетический характер псевдогипопаратиреоза, целесообразно проведение медико-генетического консультирования для оценки риска появления болезни Олбрайта у потомства.
Синдром Олбрайта

Синдром Мак-Кьюна-Олбрайта является гетерогенным заболеванием, которое независимо друг от друга описали американский педиатр D. J. McCune и эндокринолог F. Albright в 1936-1937 гг. Заболевание редкое, врожденное, но не наследственное, сопровождается метаболическими нарушениями, признаками преждевременного полового развития и деформаций скелета. Первые клинические проявления отмечаются в раннем детстве или подростковом возрасте в виде нарушений развития, полиоссальной фиброзной дисплазии костей, кожной пигментации по типу «кофе с молоком» и эндокринных аномалий, включая тиреотоксикоз, синдром Кушинга, акромегалию.
Приблизительно в 10 раз чаще диагностируют синдром Олбрайта Мак-Кьюна у девочек, чем у мальчиков. Википедия отмечает, что частота встречаемости в человеческой популяции — 1 на 100 тыс. или на миллион особ.
В результате постзиготной мутации гена GNAS1, участвующего в клеточном сигнализировании белков GS-α , обеспечивающих сопряжение рецепторов лютеинизирующих и фоликолостимулирующих гормонов с молекулами аденилатциклазы в цитологических структурах яичников. Постоянное влияние мутантного белка активизирует аденилатциклазу и повышение внутриклеточного цАМФ, а также стимулирует усиление секреции стероидных женских половых гормонов фолликулярными эстрогенпродуцирующими кистами, вызывающими преждевременное половое развитие даже при отсутствии гонадотропинов. Такая же картина наблюдается и «автономное производство» наблюдается с гормонами щитовидной железы, кортизолом и гормонами роста. Прогрессирование патологии вызвано образованием клеток, уже содержащих мутантные белки.
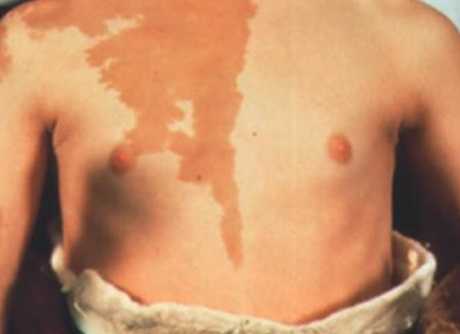
Пигментация при синдроме Олбрайта
Кроме того, рост уровня цАМФ приводит к увеличению продукции меланина и образованию лентигиозных пятен, очаговой гиперпигментации на фоне нормальных значений АКТГ и меланотропина.
Фиброзно-кистозная дисплазия в результате усиленной кальцинации под действием эстрогенов осуществляется путем замещения нормальных костных структур и образованных кальцинатов фиброзными массами, при этом нарушается равновесие процессов резорбции и регенерации костей, пролиферации мезенхимальных стромальных клеток. Наблюдается формирование очагов деструкций и псевдоопухолей.
Классификация
Болезнь Олбрайта бывает разной, в зависимости от тяжести течения различают:
- скрытое заболевание - без каких-либо признаков костной или эндокринной патологии, а также без симптоматики необычной пигментации;
- неполные формы синдрома, например, в процесс патогенеза вовлечения костная ткань, возможны эндокринные изменения и псевдогипопаратиреоз, но нет кожных проявлений;
- псевдогипопаратиреозили наследственная остеодистрофия Олбрайта — заболевание, которое также поражает костные ткани, но механизм и этиология отличаются — патология вызвана нарушением резистентности к паратгормону, метаболизма кальция и фосфора; псевдогипопаратиреоз также проявляется в детском возрасте и выражается в виде низкого роста, круглого лица, умственной отсталости, ожирения, гипотиреоза, гипогонадизма, сахарного диабета и пр.
Причины
Синдром Олбрайта Маккьюна возникает в результате генетических нарушений на ранних этапах эмбриогенеза. До сих пор не установлен тип наследования, все описанные случаи были спорадические.
Симптомы
Клиническая картина заболевания проявляется в виде целого симпатокомплекса, состоящего из:
- преждевременного гонадотропиннезависимого полового развития, развивающегося позже и медленнее нежели при других формах ранней половозрелости;
- пятнистой пигментации кожи - лентиго, пятна с четкими границами светло-коричневого цвета обычно есть от рождения на груди, спине пояснице, бедрах и в местах костных деформаций;
- эндокринных расстройствах, вызывающих тахикардию, необъяснимую утрату веса, глазные симптомы гипертиреоидизма;
- полиоссальной фиброзной остеодисплазии, мышечной слабости и остеопороза, вовлекающей в процесс чаще длинные кости, нарушения проявляются в виде изуродованных черт лица, более вытянутом телосложении, искривленных и асимметричных, укороченных запястьях, что затрудняет движения во время ходьбы, вызывает хромоту, сильные боли и повышает вероятность патологических переломов.

Первые признаки преждевременной половой зрелости начинаются с маточных кровотечений, не имеющих цикличного характера и вызваны кратковременными повышениями концентраций эстрогенов и гонадотропных гормонов в кровяном русле. Такие колебания провоцируют образования кист в яичниках. У детей в возрасте 5-10 лет наблюдается ранний рост груди и гинекомастия. Её нежная этиология не приводит к формированию вторичных половых признаков — Синдром Мак-Кьюна-Олбрайта-Брайцева не вызывает раннего полового оволосения или дифференцировки фигуры. Истинное половое созревание возможно после окончания пубертатного периода.
Анализы и диагностика
При подозрении на синдром Мак Кьюна Олбрайт проводят:
- рентгенологические исследования, которые выявляют псевдокисты, сегментарное поражение любых отделов скелета - чаще нижних конечностей, в более редких случаях — верхнего плечевого пояса и черепа;
- УЗИ брюшной полости позволяет обнаружить кисты яичников;
- лабораторные исследования для выявления повышенного уровня щелочной фосфатазы в кровотоке, сниженного количества гонадотропинов - в анализах мочи и повышенной экскреции лютеинизирующего гормона, 17-гидрокси и 17-оксикортикостероидов.
Лечение
Лечение назначают симптоматическое, оно имеет свои сложности в зависимости от индивидуальных особенностей и клинической картины. Аналоги гонадолиберина не оказывают терапевтического эффекта, так как процесс секреции эстрогенов спровоцирован не избытком гонадотропинов. Для купирования процесса гонадального стероидогенеза у особ мужского пола эффективным может оказываться кетоконазол и андрокур. При лечении медроксипрогестерона ацетатом возможно развитие гипокальциемии, поэтому, если процесс развития ранней половозрелости сопровождается тяжёлыми множественными поражениями костных тканей, то терапию необходимо назначать с осторожностью.
Для угнетения гиперэстрогенизиции и удлинения скелета могут быть назначены препараты тестостерона. Чтобы противодействовать дисплазии костей следует вводить:
- бисфосфонаты;
- ингибиторы антагонистов остеокластов.
Больным необходимо регулярно, каждые 3 мес. посещать эндокринолога для коррекции проблем с щитовидной и другими железами.
Синдром Олбрайта (Albright) - синонимы, авторы, клиника

- Анализы
- Беременность и роды
- Беременность обзор
- Бесплодие и репродуктивный статус
- Бессонница и расстройства сна
- Болезни желудочно-кишечного тракта
- Болезни легких и органов дыхания
- Болезни органов кровообращения
- Болезни щитовидной железы
- Боль в спине
- Восстановительная медицина
- Генитальный герпес
- Гинекология
- Головная боль
- Грипп
- Депрессия
- Детская онкология
- Детские болезни
- Детское развитие
- Заболевания молочных желез
- Здоровое питание. Диеты
- Здоровье и душевное равновесие
- Из истории болезни
- Иммунология
- Импотенция (эректильная дисфункция)
- Инфекции передаваемые половым путем
- Кардиология
- Кровеносная и лимфатическая система
- Лейкемии
- Лечение рака
- Лимфогранулематоз
- Маммология
- Медицина в спорте
- Менопауза
- Микробиология, вирусология
- Педиатрия (детские болезни)
- Проблемная кожа
- Путь к себе
- Рак желудка
- Рак молочной железы (рак груди)
- Рак ободочной, прямой кишки и анального канала
- Рак предстательной железы (рак простаты)
- Рак тела матки
- Рак шейки матки
- Рак яичников
- Сахарный диабет
- Сексология и психотерапия
- Сексуальная жизнь
- Современная контрацепция
- Урология
- Уход за кожей
- Факты о здоровом старении
- Химиoтерапия
- Школа здоровья
- Эндокринология
| Псевдогипопаратиреоз. Болезнь Олбрайта |
| Автор: http://www.eurolab.ua/ |
| 22 Февраля 2011 |
|
Псевдогипопаратиреоз (греч. pseudēs ложный + гипопаратиреоз; синоним: наследственная остеодистрофия Олбрайта, болезнь Олбрайта) - редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития. Полагают, что в основе псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию паратгормона в результате дефекта комплекса специфический циторецептор - паратгормон - аденилатциклаза, что нарушает процесс образования в почках циклического 3', 5'-АМФ, являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип Ia псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип Iб псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз II типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Т.о., теряется чувствительность органов-мишеней, в частности почек, к паратгормону. В результате уменьшается экскреция фосфора с мочой, возникает гиперфосфатемия, вторично развивается гипокальциемия. Так как при псевдогипопаратиреозе паращитовидные железы интактны, то в ответ на гипокальциемию, стимулирующую продукцию паратгормона, может развиться вторичный гиперпаратиреоз. Повышенное образование паратгормона не вызывает увеличения выведения фосфора и цАМФ с мочой из-за генетически обусловленной резистентности почечных канальцев к паратгормону, но сопровождается изменениями в костной ткани, характерными для гиперпаратиреоза, что свидетельствует о сохранении нормальной чувствительности остеокластов к паратгормону. При псевдогипопаратиреозе активность щелочной фосфатазы в сыворотке крови повышена или находится в пределах нормы (0,5-1,3 мкмоль неорганического фосфора на 1 мл сыворотки крови за 1 ч инкубации при 37°; определение по Боданскому). Все варианты псевдогипопаратиреоза представляют собой наследственное заболевание, характер наследования аутосомно-доминантный. Низкая плодовитость мужчин, страдающих Псевдогипопаратиреоз, объясняет редкость его передачи от отца к сыну; женщины болеют в 2 раза чаще, чем мужчины. Обычно при псевдогипопаратиреоз обнаруживают компенсаторную гиперплазию паращитовидных желез (наличие в них аденом не характерно). В костной ткани отмечают изменения, типичные для гиперпаратиреоза - диффузный остеопороз, появление кист (так называемые бурые опухоли, гигантоклеточные опухоли). Высвобождающийся из костей кальций откладывается в виде кальцинатов в подкожной клетчатке, а также в почках, мышцах, миокарде, стенках крупных артерий, конъюнктиве глаза и по периферии роговицы. У больных с псевдогипопаратиреозом обнаруживают низкий уровень 1,25(ОН)2D3 - функционально активной формы витамина Псевдогипопаратиреоз связанный с резистентностью почек к паратгормону. Уменьшение количества активной формы витамина D в организме приводит к снижению всасывания кальция в кишечнике и гипокальциемии, что вызывает еще больший выброс в кровоток паратгормона. Вследствие этого мобилизация костного кальция усиливается, развивается выраженная остеомаляция. Клинические признаки псевдогипопаратиреоза сходны с симптомами идиопатического гипопаратиреоза. Отмечаются приступы тонических судорог, возникающие спонтанно или под влиянием каких-либо раздражителей. Кальцинаты в подкожной клетчатке проявляют тенденцию к изъязвлению. Подкожная оссификация часто выражена до такой степени, что имитирует оссифицирующий миозит. Характерны задержка умственного развития, отставание в росте, лунообразное лицо, ожирение и брахидактилия, особенно укорочение первой, четвертой и пятой пястных и плюсневых костей. Могут наблюдаться множественные экзостозы, дисхондроплазия, проявления вторичного гиперпаратиреоза в виде субпериостальной резорбции костей пальцев рук; изменения в эпифизах костей такие же, как при фиброзной остеодисплазии. Часто отмечают рвоту, а также гематурию вследствие образования оксалатных камней в мочевых путях, выявляют лентикулярную катаракту, гипоплазию зубной эмали. У больных с псевдогипопаратиреозом наряду со снижением чувствительности к паратгормону органов-мишеней может наблюдаться резистентность к другим гормонам, зависимым от аденилатциклазной системы, например половых желез к гонадотропным гормонам , щитовидной железы к тиреотропному гормону, органов-мишеней к глюкагону и антидиуретическому гормону. Отмечается повышенная частота аутоиммунных болезней и диабета сахарного, наблюдаются гипотиреоз и гипертиреоз. Выделяют также псевдопсевдогипопаратиреоз, который характеризуется отсутствием гипокальциемии, гиперфосфатемии, судорог и остеомаляции. Диагноз в типичных случаях заболевания устанавливают у детей в 5-10 лет на основании характерной клинической картины, множественных аномалий развития костного скелета, наличия гипокальциемии, гиперфосфатемии, нормальной или повышенной активности щелочной фосфатазы в сыворотке крови, уменьшенного выделения кальция и фосфора с мочой, повышенного содержания паратгормона в крови. Наличие резистентности почечных канальцев к паратгормону подтверждает тест, основанный на определении количества фосфатов и цАМФ, выводимых с мочой. Отсутствие достоверного повышения содержания фосфатов и цАМФ в моче после введения больному паратгормона свидетельствует о резистентности почек к действию паратгормона. У больных с идиопатическим и послеоперационным гипопаратиреозом, наоборот, после внутривенного введения 200 ЕД паратгормона в моче содержание фосфатов и цАМФ в течение 4 ч увеличивается в 2-10 раз по сравнению с исходным уровнем. Выделение с мочой оксипролина у нелеченых больных с псевдогипопаратиреозом находится в норме или несколько повышено, а при гипопаратиреозе - понижено. Рентгенодиагностика асевдогипопаратиреоза основана на выявлении специфических изменений в костях и мягких тканях. Псевдогипопаратиреоз в сочетании с гипогонадизмом у лиц женского пола необходимо дифференцировать с Шерешевского - Тернера синдромом, с которым псевдопсевдогипопаратиреоз сходен фенотипически. При синдроме Шерешевского - Тернера половой хроматин отсутствует, на месте яичников располагаются соединительнотканные тяжи, не определяемые при ректальном и ультразвуковом исследовании. Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. Начальную дозу рассчитывают из 2000 МЕ/кг массы тела в сутки, но не более 100 000 МЕ в сутки. Во избежание передозировки препаратов витамина D необходим контроль за концентрацией кальция в крови каждые 3-7 дней в течение первых двух недель лечения и каждый месяц в течение последующих 2-3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2-3 месяца. Можно применять кальцитрин, дигидротахистерол, оксидевит, а также другие препараты активных форм витамина D. Диета с ограничением фосфора помогает нормализовать концентрацию кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами. Лечение паратгормоном не эффективно. Для купирования судорожных приступов внутривенно вводят 10% раствор кальция хлорида или кальция глюконата; внутрь - 5-10% раствор кальция хлорида по 1 столовой ложке 3-4 раза в день: кальция глюконат, кальция лактат - до 10 г в день. Прогноз при рациональной терапии благоприятный. Учитывая наследственный характер Псевдогипопаратиреоз, необходимо медико-генетическое консультирование в отношении возможности появления Псевдогипопаратиреоз у потомства. Болезнь ОлбрайтаИз самого названия болезни «псевдогипопаратиреоз» и приставки «псевдо» сразу становится понятно, что она имитирует такое заболевание как гипопаратиреоз. Также данное заболевание носит название болезнь Олбрайта. Выражается псевдогипопаратиреоз в нарушении обмена фосфора и кальция в организме человека. Кроме того, достаточно существенным образом данное заболевание сказывается на темпе умственного, а также физического развития человека. Нечувствительность периферических тканей к паратиреоидному гормону считается главной причиной, по которой может возникнуть псевдогипопаратиреоз. СимптомыЭто заболевание имеет достаточно широкий список симптомов, которые довольно похожи на симптомы, свойственные идиопатическому гипопаратиреозу.
Болезнь Олбрайта может сочетаться с различными эндокринными нарушениями, такими как синдромом Кушинга, акромегалией, поражениями паращитовидных желез, гинекомастией. Кроме того, выделяют также псевдогипопаратиреоз, который характеризуется тем, что при нем отсутствуют гиперфосфатемия, гипокальциемия, остеомаляция и судороги. Группа рискаСуществует определенная группа риска, в которую входят родственники (дети) болевшего псевдогипопаратиреозом. Это связано с тем, что заболевание носит наследственный аутосомно-доминантный характер. Как правило, подобная наследственность передается женщинам, которые в этом случае болеют чаще. Это объясняется тем, что мужчины с заболеванием Олбрайта имеют существенно сниженную плодовитость, а кроме того редко подобная наследственность может быть передана от отца к сыну. Немного успокаивающим фактором является тот факт, что псевдогипопаратиреоз является довольно редким заболеванием костной системы. ДиагностикаВыявление вышеупомянутых синдромов должно послужить поводом для немедленного обращения к квалифицированному специалисту в надежном медицинском учреждении. При своевременной постановке правильного диагноза и рациональной терапии прогнозы довольно положительные. При этом следует также провести соответствующие консультации на медико-генетическом уровне в отношении вероятности появления псевдогипопаратиреоза у потомства пациента. Читайте также:
|
