Синдром Пелицеуса-Мерцбахера (Pelizaeus-Merzbacher) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
Неврология:
Популярные разделы сайта:
Болезнь Пелицеуса - Мерцбахера (БПМ): причины, диагностика, лечение
Болезнь Пелицеуса - Мерцбахера (БПМ):
1. Сцепленное с Х-хромосомой, наследуемое по рецессивному типу заболевание, поражающее преимущественно мальчиков. Отдельные случаи возникновения болезни Пелицеуса - Мерцбахера у девочек подтверждают возможность аутосомно - рецессивного типа наследования.
2. Медленно прогрессирующее нарушение миелинизации ЦНС В зависимости от характера нарушения миелинизации и от особенностей начала заболевания было выделено три типа болезни, тип I (классический), тип II (врожденный) и тип III (переходный). Все вместе эти типы известны также как тигроидная лейкодистрофия: вследствие наличия периваскулярных островков нормального миелина ткани приобретают "тигровый" (полосатый) вид. Эта группа заболеваний характеризуется относительной сохранностью аксонов.
3. Начало в младенчестве; врожденные формы проявляются с первых дней жизни.
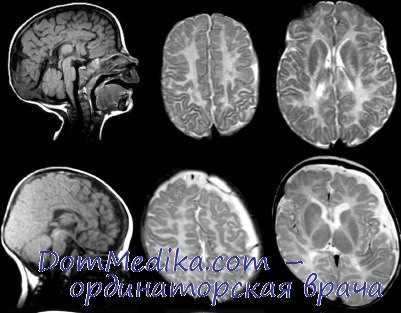
4. Признаки болезни Пелицеуса - Мерцбахера:
а. Крупноразмашистый нистагм, появляющийся обычно до достижения ребенком 3-х месячного возраста.
б. Осцилляторные движения головы в сочетании с плохим её удерживанием.
в. Мозжечковые симптомы: атаксия, интенционный тремор, скандированная речь.
г. Врожденный ларингеальный стридор.
д. Недостижение нормальных показателей развития (врожденный тип) либо прогрессирующее ухудшение психомоторных функций (классический тип)
е. По мере прогрессирования болезни могут развиваться спастика, атрофия зрительных нервов, припадки и непроизвольные движения.
ж. Окончательный диагноз может быть установлен только на аутопсии. Врожденный стридор и нистагм у мальчиков должны настораживать врачей в отношении возможного наличия данного заболевания. Инструментальные методы не могут отличить данное заболевание от других форм поражения белого вещества. На КТ выявляется значительное снижение плотности белого вещества и его прогрессирующая атрофия. МРТ обнаруживает нарушение миединизации; мозг постоянно напоминает мозг новорожденного.
5. Лечения болезни Пелицеуса - Мерцбахера не существует.
6. Болезнь Пелицеуса - Мерцбахера имеет прогрессирующее течение с периодами стабилизации. При врожденных формах смерть обычно наступает в течение первого года жизни. При классической форме болезни Пелицеуса - Мерцбахера больные умирают во второй или третьей декаде жизни.
Азбука Genetics-info: Лейкодистрофия Пелицеуса-Мерцбахера
Лейкодистрофия Пелицеуса-Мерцбахера — Х-сцепленное наследственное заболевание, характеризующееся поражением центральной нервной системы. Заболевание относится к группе лейкодистрофий.

Это Х-сцепленный синдром, связанный с аномалиями белого вещества головного и спинного мозга. Эта форма лейкодистрофии приводит к ненормальному развитию компонентов (преимущественно жиров или белков), которые составляют белое вещество (миелиновую оболочку) мозга. Миелиновая оболочка - это защитное покрытие нервов, без которого они не могут нормально функционировать. Причина аномалии - мутация в гене PLP1 .
Болезнь Пелицеуса-Мерцбахера подразделяют на 2 типа: классический и внутриутробный. Хотя они отличаются по тяжести, клиническая картина может совпадать.
Классический тип является более распространенным. В течение первого года жизни люди, страдающие классической болезнью Пелицеуса-Мерцбахера, обычно испытывают слабый мышечный тонус (гипотония), непроизвольные движения глаз (нистагм) и замедленное развитие двигательных навыков, таких как ползание или ходьба. По мере того, как ребенок становится старше, проявления нистагма могут уменьшаться, но развиваются другие двигательные расстройства, включая мышечную ригидность (спастичность), проблемы с движением и равновесием (атаксия) и непроизвольные подергивания. Когнитивные способности могут быть нарушены, но речь и язык обычно присутствуют.
Внутриутрбный тип болезни Пелицеуса-Мерцбахера является наиболее тяжелым из двух типов. Симптомы обычно присутствуют при рождении или развиваются в первые несколько недель жизни. Особенности включают нистагм, проблемы с питанием, свистящий звук при дыхании, прогрессирующую спастичность, приводящую к деформациям суставов (контрактурам), которые ограничивают движение, наблюдаются затруднения речи, атаксия и судороги. Дети с этим типом синдрома часто имеют низкий рост и плохо набирают вес. Те, кто страдает от внутриутробной болезни Пелицеуса-Мерцбахера, не ходят и не развивают эффективное использование своих верхних конечностей.
Диагноз может быть заподозрен на основании тщательной клинической оценки, подробной истории болезни пациента и различных специализированных обследований, таких как магнитно-резонансная томография (МРТ). Распознавание ранних дефектов миелинизации, таких как отсутствие миелинизации в мозжечке и стволе мозга, может помочь в ранней диагностике тяжелых форм синдрома. Доступно молекулярно-генетическое тестирование на ген PLP1 для подтверждения диагноза. Разработана и пренатальная диагностика и преимплантационная генетическая диагностика.
На сегодняшний день не существует лекарства от синдрома Пелицеуса-Мерцбахера, как и не существует стандартного курса лечения. Ведение пациента обычно осуществляет многопрофильная команда, состоящая из специалистов в области неврологии, физиотерапии, ортопедии, пульмонологии и гастроэнтерологии .
Прогноз для пациентов с болезнью Пелицеуса-Мерцбахера плохой, с прогрессирующим ухудшением клинических проявлений вплоть до смерти. Пациенты, страдающие тяжелым (внутриутробным) типом, могут умереть в младенчестве или детстве, обычно от аспирации. При хорошем уходе пациенты могут жить до 30 лет и дольше. Выживаемость до шестого, седьмого десятков может наблюдаться для людей с классическим типом.
Лейкодистрофия
Лейкодистрофия — нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
МКБ-10

Общие сведения
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр.), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.

Причины возникновения лейкодистрофии
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину — метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток и т. п.
Симптомы лейкодистрофии
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Виды лейкодистрофии
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно-клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван-Богарта-Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Лейкодистрофия Галлервордена-Шпатца чаще всего стартует в 10-летнем возрасте. Проявляется дисфункцией стриопаллидарной системы, затем на фоне гиперкинезов прогрессирует тетрапарез, развивается гемералопия и пигментный ретинит, наблюдается снижение интеллекта, возникают эпиприступы.
Диагностика лейкодистрофии
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха — отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) — возможность пренатального диагностирования.
Лечение лейкодистрофии
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение — в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес.), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.
Невральная амиотрофия Шарко-Мари-Тута
Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.

Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» - чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте.
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание - приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Диагностика
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Медикаментозная терапия
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру - баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Немедикаментозная терапия
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа - ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Хирургическое лечение
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства - метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Экспериментальное лечение
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 - ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута - тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
1. Оценка качества жизни больных с наследственной невропатией Шарко—Мари—Тута в Красноярском крае/ Шнайдер Н.А., Глущенко Е.В., Козулина Е.А.// Бюллетень Сибирской медицины. - 2011.
2. Клинико-генетическая характеристика наследственной невропатии Шарко—Мари—Тута (на примере Красноярского края): Автореферат диссертации/ Глущенко Е.В. - 2011.
3. Ведение и реабилитация пациентов с наследственной невропатией Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В.// Комплексная реабилитация: наука и практика. - 2010.
4. Наследственная невропатия Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В., Кантимирова Е.А. и др. - 2010.
Азбука


Синдром Ларсена - это остеохондродисплазия, характеризующаяся вывихами крупных суставов и характерными черепно-лицевыми аномалиями.

Болезнь Крона — воспалительное заболевание пищеварительного тракта неизвестной этиологии с возможностью поражения любого его отдела от ротовой полости до перианальной области.

Болезнь Краббе — это наследуемая энцефалопатия детского возраста с быстро прогрессирующей церебральной дегенерацией, демиелинизацией, проявляющаяся повышением мышечного тонуса, приступами гиперпирексии и нарушениями интеллекта .
Болезнь Краббе - аутосомно-рецессивное, поражающее белое вещество центральной и периферической нервной системы .

Синдром Коффина-Сириса - редкое врожденное мультисистемное генетическое заболевание, характеризующееся аплазией или гипоплазией дистальной фаланги или ногтя на пятых пальцах стоп, задержкой в развитии, умственной отсталостью, грубыми чертами лица и другими клиническими проявлениями.

Синдром Корнелии де Ланге — врожденное заболевание, характеризующееся множественными пороками развития и умственной отсталостью.
Болезнь Книста (дисплазия Книста, метатропная дисплазия, тип II) - нарушение роста костей, характеризующееся карликовостью и другими нарушениями скелета и проблемами со зрением и слухом. Синдром Книста получил свое название по имени австрийского педиатра W. Kniest. В 1952 г. он сообщил о пятидесятилетнем пациенте, который имел низкий рост и тугоподвижность суставов. Больной был слеп, но с нормальным интеллектом.
Болезнь кленового сиропа (лейцино́з, разветвлённоцепочечная кетонурия, болезнь мочи с запахом кленового сиропа) - наследственное заболевание из группы органических ацидемий, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма аминокислот лейцина, изолейцина, валина.
Синдром Клайнфельтера — самая частая и тяжелая форма первичного мужского гипергонадотропного гипо-гонадизма, сопровождающаяся необструктивной азооспермией.
Синдром каудальной регрессии (сакральная или люмбосакральная агенезия, синдром каудальной дисплазии, каудальная дисгенезия, сиреномелия) — редкий врожденный порок развития дистального отдела позвоночника и спинного мозга. Впервые врожденный дефект в виде агенезии дистальной части позвоночника описан в 1852 году.
Синдром Картагенера (Kartagener syndrome, синдром неподвижных ресничек, двигательная цилиопатия, триада Картагенера) - разновидность генетически обусловленного аутосомно-рецессивного расстройства - первичной цилиарной (реснитчатой) дискинезии, характеризующейся цилиарной дисфункцией и нарушением мукоцилиарного клиренса.

Ихтиоз - группа наследственных заболеваний кожи, которая характеризуется нарушением процессов кератинизации. Существует множество форм ихтиоза, а также ихтиоз может быть симптомом других заболеваний. В целом, определяют по меньшей мере 20 различных типов ихтиоза.
Актуальное






Главное
Партнеры








ООО «Национальное медико-фармацевтическое агентство», 2017-2022
Любые нарушения будут рассматриваться и преследоваться согласно статье 146 Уголовного Кодекса РФ «Нарушение авторских и смежных прав»
Информация, представленная на сайте, несет исключительно ознакомительный характер и не может быть использована для постановки диагноза, назначения лечения и не заменяет прием врача.
Читайте также:
- Железистая гиперплазия эндометрия
- Факторы роста яичников. Инсулиноподобные и эпидермальный факторы роста
- Благоприятные сочетания пороков сердца. Миокардит при ревматизме
- Родители и больные шизофренией дети. Нарушения психики детей и родителей
- Что предпринять, если ребенка обижают сверстники? Воспитание личности