Синдром Риддока (Riddoch) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 22.01.2026
Синдром Прадера-Вилли- врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 25000-10000 новорожденных. Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание. Заболевание характеризуется выраженной мышечной гипотонией при рождении, сохраняющейся в течение первого года жизни ребенка. Сосательный и глотательный рефлексы снижены, что затрудняет кормление ребенка. Из-за гипотонии у таких детей задерживается развитие двигательных функций: они с трудом учатся держать голову, сидеть и т. д. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает.
Позднее, к второму-четвертому году жизни появляются постоянное чувство голода и отсутствие насыщения, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей. Из-за тяжелого ожирения грозным осложнением является обструктивное апноэ (остановка дыхания) во сне.
Рост больных нередко снижен. Часто отмечается долихоцефалия (удлиненная форма головы), миндалевидный разрез глаз, низко расположенные ушные раковины, широкая переносица, маленький рот с тонкой верхней губой. Стопы и кисти больных диспропорционально маленькие (акромикрия). У 75% детей наблюдается слабая пигментация кожи, волос и радужки.
У мальчиков при рождении отмечается недоразвитие полового члена, мошонки,крипторхизм, а у девочек недоразвитие половых губ, иногда и матки. В дальнейшем заболевание проявляется задержкой или отсутствием полового созревания, бесплодием.
Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед.). Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарем, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, косоглазие.
Продолжительность жизни больных может достигать 60 лет и более. Нередко у таких детей развивается сахарный диабет.
Лечение
Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным.
Младенцы со сниженным мышечным тонусом должны получать массаж и другие виды специальной терапии. Комплекс лечебных мероприятий включает также диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). Рекомендуется терапия гормоном роста.
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Медико-генетическое консультирование
Родителям ребенка с синдромом Прадера-Вилли рекомендуется пройти генетическое обследование, прежде чем планировать дальнейшую беременность, поскольку существует риск того, что следующий ребенок у тех же родителей родится также с синдромом Прадера-Вилли, что зависит от механизма, вызвавшего генетический сбой.
Синдром Риддока - Riddoch syndrome
Синдром Риддока (также известный как Феномен Риддока) является формой нарушение зрения часто вызвано поражения в затылочная доля которые ограничивают способность больного различать предметы. Только движущиеся объекты в скотома видны, статические - невидимы для пациента. [1] Движущиеся объекты не имеют цвета или деталей. Субъект может осознавать только движение без его визуального восприятия (гносанопсия ), [2] или общая форма движущегося объекта может восприниматься как тень, подобная контуру. [3] Синдром назван в честь Джорджа Риддока, который был временным офицером в Медицинском корпусе Королевской армии и обследовал солдат, которые были ослеплены огнестрельными ранениями в мозг. [2]
По крайней мере, одна пациентка смогла использовать кресло-качалку, приводя неподвижное окружение в движение относительно ее головы, чтобы улучшить свое восприятие движения. В конце концов, она смогла сделать то же самое, просто произвольно двигая головой. [3]
Синдром Прадера-Вилли ( Синдром гипотонии-ожирения )
Синдром Прадера-Вилли - это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
МКБ-10

Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 - 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.
Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% - наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом - полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек - недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Диагностика
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения - ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза - показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации - основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития - синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция - увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии - бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия - прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина - карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике - предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
1. Синдром Прадера-Вилли у детей: новое в этиологии, патогенезе и лечении/ Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В.// Российский вестник перинатологии и педиатрии - 1999 - №4.
2. Синдром Прадера-Вилли в Беларуси: генетическая структура и фенотипическая характеристика/ Хурс О.И., Политыко А.Д., Румянцева Н.В. и др.//Известия Национальной Академии наук Беларуси - 2010 - № 1.
3. Clinical report-health supervision for Children with Prader-Willi Syndrome, the Committee on Genetics/ McCandless S.E.// Pediatrics - 2011 - №1.
4. Prader-Willi syndrome: clinical and genetic fi ndings/ Butler M.G., Thompson T.// Endocrinologist - 2000 - №10.
Синдром Прадера-Вилли: как распознать патологию развития?

Синдром Прадера-Вилли — генетическая патология, диагностируемая с частотой от 1:10 000 до 1:25 000 случаев. Причем количество людей с данным нарушением различается не только в зависимости от распространенности заболевания в различных регионах, но и напрямую коррелирует с развитостью медицинского обслуживания. При отсутствии генетика в клинике, стертых симптомах родители и окружающие нередко считают, что ребёнок просто «хорошо кушает», не любит активные игры и немного запаздывает в развитии, возможно, ему нужно больше внимания и дополнительных занятий. А к моменту поступления в школу выясняется, что к лишнему весу и отставанию в умственном развитии привели не социальные факторы, а генетическая патология, коррекцией которой надо было начинать заниматься с самого рождения.
Синдром Прадера-Вилли

Заболевание названо по именам исследователей, впервые описавших симптомокомплекс. Швейцарские педиатры Андреа Прадер и Генрих Вилли выделили из массы маленьких пациентов группу детей со схожими нарушениями в развитии и описали ее в середине прошлого века как отдельное заболевание.
Позднее было выявлено, что к возникновению данного синдрома приводит отсутствие или нарушение работы 7 генов (сегмент 11.2-q13) на хромосоме №15, причем из-за различия в симптомах при отсутствии или частичном нарушении выраженность заболевания может значительно варьироваться — от тяжелых случаев, диагностируемых при рождении, до стертых форм.
Изначальная причина нарушений в геноме не ясна, а вот наследуется эта патология всегда по мужской линии при наличии соответствующей генетической идеосомии у материи, и передается от отца к детям, хотя здоровые копии генов могут приводить к скрытому наследованию, и малыши с таким заболеванием рождаются и у людей без наличия синдрома.
Генетическая предрасположенность — дополнительный фактор, повышающий возраст постановки диагноза у малышей до предшкольного (и даже более позднего) обследования: внешние проявления синдрома Прадера-Вилли, достаточно нехарактерные для любой другой семьи, при явном генетическом наследовании выглядят очень логично: ребёнок просто похож на папу, и внешностью, и аппетитом, и характером.

Синдром Прадера-Вилли — достаточно редкая патология развития, однако опасна она не только своими проявлениями, но и неизвестностью. Характерная внешность детей, поведенческие нарушения, отставания в развитии могут сопутствовать не только множеству иных заболеваний, но и наследоваться от родителей. При такой картине понятно, что обращение к специалистам может затянуться на весь предшкольный период, а порой дети остаются без правильного диагноза и необходимого лечения, просто потому, что очень похожи на папу, а это считается нормой.
В моей практике был мальчик Володя, которого привели в развивающий центр в 8 лет с установкой «подготовить к школе». Предшкольное тестирование весной ребёнок не прошел из-за несоответствия уровня развития (память, внимание, мыслительные процессы) требованиям общеобразовательного учреждения, но родители считали, что всему виной то, что Вова не посещал детский садик, а проводил время в основном со старенькой бабушкой в деревне. Она же его и «раскормила» на козьем молоке, лишнего веса у мальчика было достаточно.
Володя действительно «проваливал» тесты на развитие по возрасту, отличался хорошим аппетитом, отличной зрительной памятью и склонностью к играм в лего-конструкторы, иногда упрямился и «взрывался». В целом он соответствовал уровню пятилетнего ребёнка. Довольно долго, месяца три после начала занятий, наши специалисты также считали причиной проблем мальчика социально-педагогическую запущенность. Но время шло, буквы не запоминались, а потом забирать Володю пришел папа с еще более выраженной внешней симптоматикой: невысокий рост, полнота, выраженная переносица, миндалевидные глаза, акромикрия и т. п. Вот с этого момента картина начала проясняться.
После длительных переговоров с мамой и уговоров папы Вова получил наконец консультацию у генетика и направление в школу специального вида. Стало ясно, что общеобразовательная школа пока не готова к обучению ребёнка, отсутствует класс коррекции, и выбрасывать деньги на изучение букваря в частном центре в данный момент смысла не имеет, нужны логопеды-дефектологи, эндокринолог, специальная диета и много работы, чтобы наверстать упущенное время и предупредить сопутствующие заболевания.
Интеллект, страдающий у больных синдромом Прадера-Вилли, в случае семейного наследования заболевания осложняет коррекцию. Нередко отец не хочет принять, что и сам он болен, и ребёнок нуждается в лечении. Частая смена настроения, повышенная ригидность, агрессивность, которые также являются симптомами болезни, приводят к необходимости длительной предварительной работы с членами семьи перед началом непосредственных занятий с детьми. Парам также необходима консультация генетика перед зачатием следующих детей.
Проявления генетической патологии у детей

Вероятность развития патологии можно заподозрить еще в процессе вынашивания. На необходимость консультации и обследования у генетика могут указывать пониженная активность плода, его неправильное положение в полости матки, многоводие, а также несоответствующий срокам уровень хорионического гонадотропина человека у беременной женщины.
Дети появляются на свет в срок, с симптомами незначительной внутриутробной асфиксии, в среднем каждый третий ребёнок рождается с ягодичным предлежанием. У мальчиков часто отмечают недоразвитие полового члена, мошонки, явления крипторхизма. У девочек — недоразвитие внешних половых органов или всей репродуктивной системы.
Первые годы жизни характеризуются отставанием в психомоторном развитии. Мышечная дистония различной степени тяжести приводит как к запаздыванию появления навыков держать головку, сидеть, ползать, так и может быть выражена нарушениями сосательного, глотательного рефлексов, в тяжелых случаях детей приходится кормить через зонд и подключать к аппарату искусственной вентиляции легких.
Явления дистонии проходят в среднем к 7-8 годам. Но высокой двигательной активностью дети не отличаются, чему способствует еще один фактор: ожирение.
В период 2-4 лет у малышей развивается постоянное чувство голода и отсутствует сигнал о насыщении, что приводит к быстрому образованию лишнего веса, откладывающемуся в основном на туловище и проксимальных областях конечностей. Ожирение может доходить до тяжелых стадий и грозить остановкой дыхания во сне (ночным апноэ).
К частым внешним проявлениям относят также низкий рост, долихоцефалию (удлиненную форму черепа), миндалевидную форму глаз, близорукость, миопию, низкое расположение ушей по сравнению с нормой, выраженную переносицу, тонкую верхнюю губу, акромикрию (непропорциональное развитие ступней и кистей рук, выглядящих слишком маленькими по сравнению с туловищем).
Ранее считалось, что синдром Прадера-Вилли обязательно вызывает значительную умственную отсталость и коэффициент интеллекта у взрослых составляет от 20 до 85, сопровождаясь идиотией.
В соответствии с исследованиями специалистов, установлено, что, в зависимости от выраженности симптоматики и коррекционной работы, только 40% больных отличаются интеллектом ниже среднего.
Ученые, работавшие с подростками, выявили еще более утешительные цифры: IQ может быть выше 85 баллов (нормой считается 90-115) у 5% больных, у 27% контрольной группы IQ составляет от 70 до 85 баллов, что соответствует невыраженной умственной отсталости, 39% детей отличались коэффициентом в 50-70 баллов (незначительная умственная отсталость), 27% — выраженная форма, и только 1% соответствовал критериям глубокой умственной отсталости.
Дети с данным синдромом характеризуются нестандартным развитием когнитивных способностей: имея развитые навыки визуального восприятия, чтения, они редко могут полностью выразить мысли или пересказать смысл прочитанного из-за неразвитости речи, то есть понимание превышает экспрессию.
Страдает восприятие информации на слух, логическое мышление, мелкая и крупная моторика, зрительная память. Однако специалисты утверждают, что, используя сильные стороны и корректируя слабые, можно добиться значительного улучшения всех мыслительных процессов.
Лечение, коррекция и осложнения заболевания

Специфической терапии, позволяющей изменить геном или устранить последствия хромосомных нарушений, не существует. Поэтому при работе с маленькими пациентами специалисты выбирают симптоматическое, коррекционное направление и профилактику осложнений.
Для снижения выраженности эндокринных нарушений необходима строгая диета (для детей-дошкольников менее ограниченная, но хорошо сбалансированная, для детей старше с калорийностью не более 1000 ккал в день), режим питания, контроль вплоть до запирания продуктов в отдельных шкафах. По назначению проводят гормонотерапию, инъекции рекомбинантного гормона роста и стимуляцию полового развития в случае нарушений полового развития.
Эндокринолог необходим детям с симптомом Прадера-Вилли и по причине вероятности развития сахарного диабета из-за снижения толерантности к глюкозе.
К подростковому периоду может возникнуть необходимость в наблюдении у психиатра: дети с синдромом Прадера-Вилли нередко страдают обсессивно-компульсивным расстройством, могут проявлять самоагрессию, отличаться высокой тревожностью.
Узкие специалисты, о которых нельзя забывать:
- стоматолог, из-за вязкости слюны наблюдается раннее возникновение кариеса;
- окулист, часто синдром сопровождается миопией и косоглазием;
- ортопед: гипотония мышц вызывает ортопедические нарушения, сколиоз;
- андролог и гинеколог: синдром Прадера-Вилли может сопровождаться нарушениями развития органов половой сферы.
Необходимо помнить, что из-за своих особенностей и дети, и взрослые с синдромом Прадера-Вилли могут испытывать затруднения в определении своего самочувствия, не уметь выразить вербально свои мысли, чувства, не обращаться вовремя к родителям или за медицинской помощью, из-за чего процент хронических соматический заболеваний в данной группе катастрофически высок.
Следить за здоровьем ребёнка с данным диагнозом надо с раннего младенчества, при этом обучая обращать внимание на признаки заболеваний, сообщать о них, понимать, когда и какая помощь требуется. При неполном нарушении работы генов и курсе коррекции дети с синдромом Прадера-Вилли вырастают во взрослых, способных к самостоятельной жизни, работе, образованию семьи и воспитанию собственных здоровых детей при условии своевременной консультации у генетика.
Синдром Рейно
Синдром Рейно является заболеванием, при котором нарушается кровоснабжение сосудов кистей или стоп. Данное заболевание постепенно вызывает трофические изменения тканей. Болезнь и синдром Рейно встречается у 3-5% людей, преимущественно его развитию подвержены женщины. В 85% случаев синдром Рейно является признаком другого заболевания.
Врачи-ревматологи Юсуповской больницы имеют опыт лечения синдрома Рейно, поэтому они готовы предложить пациентам с признаками данной патологии комплексное обследование и последующие лечение. При проведении диагностики специалисты выясняют причины, провоцирующие расстройство артериального кровообращения в сосудах конечностей.
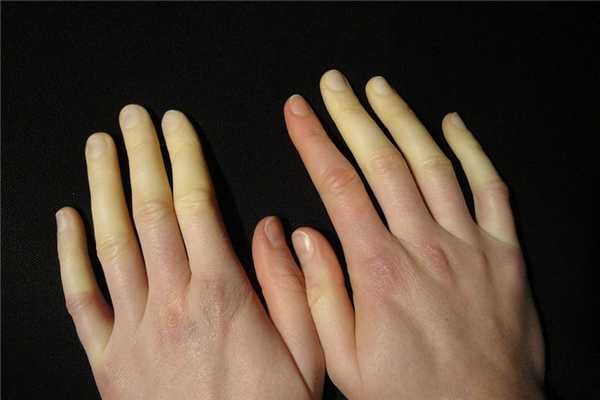
Синдром Рейно: МКБ 10
Синдром Рейно в классификации МКБ 10 отнесен к болезням периферических сосудов, кодирующихся под шифром I73. Для данного заболевания присвоен код I73.0. Синдром Рейно зачастую развивается на фоне патологий соединительной ткани, так, при диагностике артрита или склеродермии в некоторых случаях выявляется данное заболевание. Данная взаимосвязь объясняется тем, что стенки сосудов состоят из соединительной ткани, при заболеваниях которой их структура нарушается.
Синдром Рейно, симптомы и лечение при котором взаимосвязаны, характеризуется поражением капилляров и артериол, их стенки воспаляются и заметно сужаются. Резкое сужение сосудов вызывают также провоцирующие факторы, такие как стресс и холод. Проявления синдрома Рейно возникают у больных чаще всего в отделенных частях тела: пальцах ног и рук, подбородке, кончике носа, где кровь медленнее циркулирует.
Синдром Рейно, код в МКБ 10 для которого I73.0, имеет синонимичные названия: феномен Рейно, синдром Рейно-Лериша и Рейно болезнь. При постановке диагноза врачами-ревматологами Юсуповской больницы используется Международная классификация болезней 10 пересмотра.
Синдром Рейно у женщин
Синдром Рейно симптомы и лечение у женщин начинается в более раннем возрасте. Среди людей, страдающих данным заболеванием в возрасте до 50 лет, отмечается преобладание в пять раз болезни у женщин. Общими проявлениями синдрома Рейно являются: бледность, синюшность кожных покровов и потеря чувствительности определенных участков конечностей или лица.
Некоторые женщины игнорируют признаки заболевания на ранней стадии в результате чего развиваются другие серьезные заболевания, успешное лечение которых определяет своевременное выявление нарушения. Синдром Рейно развивается в несколько фаз:
- вазоконстрикторная фаза характеризуется появлением бледности кожных покровов, которая может сохраняться до 15 минут;
- цианотическая фаза длится несколько минут, бледность при данной фазе сменяется синюшностью;
- фаза реактивной гиперемии отличается покраснением кожи.
Данная последовательность наблюдается не у всех пациентов во время приступа. Продолжительность и порядок фаз зависит от течения болезни и общего состояния организма.
Синдром Рейно у мужчин
При игнорировании признаков и отсутствии терапии синдром Рейно прогрессирует. Во время приступов мужчины и женщины испытывают общие симптомы:
- болевой синдром, развитию которого предшествует недостаточное кровоснабжение тканей и нарушение обмена веществ;
- побледнение кожных покровов отмечается в первые минуты после действия провоцирующего фактора. Бледность кожи связана с резким спазмом сосудов и нарушением циркуляции крови;
- чувство онемения и покалывание, которые исчезают после нормализации кровообращения;
- синюшность кожных покровов появляется после бледной окраски, что объясняется застойным явлением и кровенаполнением вен;
- покраснение кожных покровов обусловлено притоком крови к сосудам, испытавшим спазм.
Синдром Рейно, симптомы и лечение у женщин и мужчин схожие, может развиваться при аутоиммунных ревматических заболеваниях. Врачи-ревматологи клиники терапии Юсуповской больницы занимаются научной деятельностью и изучают мировой опыт лечения синдрома Рейно.
Диагностика синдрома Рейно
Пациенту с признаками синдрома Рейно следует обратиться к врачу-ревматологу на консультацию, также в Юсуповской больнице при признаках данного нарушения обязательна консультация сосудистого хирурга. В ходе комплексной диагностики специалисты проводят исследования для выявления сопутствующих патологий, которые могли вызвать болезнь Рейно.
Диагноз синдром Рейно определяется специалистами после сбора анамнеза, изучения симптоматики и комплексного обследования пациента. При диагностике синдрома Рейно используются следующие лабораторные исследования:
- биохимический анализ крови;
- общий анализ крови для определения скорости оседания эритроцитов;
- общий анализ мочи позволяет выявить органические и функциональные поражения почек;
- иммунологические анализы;
- коагулограмма, при которой изучается свертываемость крови. При синдроме Рейно увеличена способность тромбоцитов и эритроцитов, а также отмечается повышенная вязкость крови.
В диагностическом центре Юсуповской больнице имеется европейское оборудование для исследования полученных материалов. Высокая точность результатов диагностики обеспечивается не только современными устройствами, но и профессионализмом сотрудников Юсуповской больницы. Синдром Рейно, лечение которого врачами-ревматологами Юсуповской больницы проводится по индивидуальному плану, может контролироваться различными методами в зависимости от течения болезни.
Синдром Рейно: лечение
Комплексный подход к устранению причин синдрома Рейно позволяет вылечить болезнь без хирургического вмешательства. При развитии вторичного синдрома Рейно, связанного с патологиями соединительной ткани, пациенту назначаются лекарственные средства для устранения симптоматики и причин болезни.
При самостоятельном синдроме Рейно медикаментозное и хирургическое лечение проводится в исключительных случаях. Синдром Рейно, симптомы и лечение которого взаимосвязаны, требует исключения провоцирующих факторов, таких как вибрации, стресс, курение, охлаждение. Врачами-ревматологами Юсуповской больницы в план лечения включаются также физиотерапевтические процедуры. Профессиональные психологи с многолетним опытом работы обучают пациентам приемам снятия стресса и напряжения, релаксации.
Лечение синдрома Рейно в Москве
Пациенты Юсуповской больницы получают медицинские услуги высокого качества, так как врачи и персонал ответственно подходят к выполнению своих обязанностей. Синдром Рейно, симптомы которого появляются у пациентов неожиданно, вызывает множество вопросов и опасений. Врачи-ревматологи Юсуповской больницы доступно объясняют пациентам особенности данной болезни, наиболее оптимальные методы ее лечения.
В клиники терапии Юсуповской больницы, где оказывают помощь пациентам с синдромом Рейно, имеется современное оборудование для выявления нарушений кровообращения и сопутствующих патологий, которые могут ухудшать качество жизни человека. При лечении болезни специалисты используют современные подходы и средства, обладающие минимальным количеством побочных эффектов.
Для комфортного пребывания пациентов в Юсуповской больнице действует система предварительной записи по телефону, поэтому для посещения специалиста не требуется длительного ожидания.
Читайте также: