Синдром Робена (Robin) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Синдром Пьера Робена, также называемый секвенция (последовательность) Пьера Робена (сокр. СПР) — это врожденный порок развития, характеризующийся тремя основными признаками: небольшой нижней челюстью (микрогнатия), смещением языка к задней части ротовой полости (глоссоптоз) и аномальным отверстием в небе (хейлосхизис или расщелина нёба).
Считается, что секвенция Пьера Робена вызывается множеством факторов, которые приводят к ряду физических изменений в полости рта. В настоящее время считается, что нижняя челюсть недостаточно растет, что приводит к смещению языка к задней части горла. Учитывая ограниченный размер полости рта, язык также выталкивается вверх, что мешает естественному закрытию развивающегося неба. Эти изменения происходят во время беременности и приводят к черепно-лицевым аномалиям, которые обычно обнаруживаются при рождении. Кроме того, измененная анатомия полости рта затрудняет дыхание, которые могут варьироваться по степени тяжести от легкого нарушения до опасного для жизни респираторного расстройства. Поскольку пища должна проходить через измененную полость рта, чтобы попасть в желудочно-кишечный тракт, также часто возникают трудности с кормлением. Признаки СПР могут присутствовать в виде изолированной последовательности или как часть генетического синдрома. Люди с изолированной СПР чаще всего имеют мутации вблизи гена SOX9.
Синдром Пьера Робена был назван в честь доктора Пьера Робена, французского хирурга-стоматолога, который впервые заметил его особенности в начале 20 века. Хотя точная причина не совсем ясна, в настоящее время считается, что множественные факторы приводят к последовательным физическим изменениям в полости рта, что в конечном итоге приводит к обструкции дыхательных путей. По этой причине проблемы с дыханием — частые проявления СПР. Проблемы с кормлением также распространены, поскольку полость рта также является проводником в желудочно-кишечный тракт.
Признаки и симптомы
Синдром Пьера Робена (СПР) включает в себя физические изменения в процессе развития, которые приводят к изменению анатомии полости рта. Поскольку воздух и пища проходят через рот и в глотку, проблемы с дыханием и кормлением являются частым явлением.
При СПР характерно изменение формы и положения нижней челюсти. Как правило, он имеет уменьшенную длину и расположен в направлении спины (микроретрогнатия). В свою очередь, эти изменения нижней челюсти могут повлиять на положение языка по направлению к задней части рта («западение» язык). Анатомические аномалии синдрома Пьера Робена также часто включают U-образную расщелину неба, которая влияет на динамику дыхания и развитие речи.
В частности, смещение языка к задней части рта предрасполагает его к падению в сторону горла. Это может заблокировать дыхательные пути и вызвать затруднение дыхания. Это отклонение может вызвать разной степени тяжести осложнения, от легкого нарушения дыхания до опасного для жизни респираторного расстройства. Обструкция дыхательных путей также может возникать ночью в случае связанного с ней состояния, называемого «обструктивным апноэ во сне». Это нарушение сна, при котором дыхание временно останавливается и возобновляется из-за периодической закупорки дыхательных путей.
Поскольку пища, поступающая в желудочно-кишечный тракт, также проходит через рот и горло, трудности с кормлением также могут возникать из-за аномальной анатомии полости рта. В зависимости от степени тяжести это может привести к таким проблемам, как удушье (аспирация) или меньшая прибавка в весе, чем ожидалось (что врачи называют «неспособностью к развитию»). Также чаще встречается кислотный (гастроэзофагеальный) рефлюкс у детей с СПР.
Другие возможные проявления СПР включают сердечно-сосудистые и легочные заболевания, такие как шумы в сердце, высокое кровяное давление в легочных артериях (легочная гипертензия) и сужение отверстия между легочной артерией и правым желудочком сердца (легочный стеноз). Также распространены аномалии опорно-двигательного аппарата, в том числе в руках, ногах, стопах и позвоночнике. Воспаление среднего уха (средний отит), обычно сопровождающееся повторными инфекциями уха, встречается примерно у 80% пациентов, а глазные дефекты отмечаются примерно у 10-30% пациентов. Еще одна частая особенность — присутствуют зубы при рождении (натальные зубы).
Причины и факторы риска
В настоящее время точная причина синдрома Пьера Робена неизвестна. Наиболее широко распространено мнение о том, что множественные факторы приводят к последовательности физических изменений в полости рта. Считается, что эти изменения происходят в несколько этапов, а не в виде отдельных событий. В частности, считается, что неспособность нижней челюсти полностью развиться на ранних сроках беременности заставляет язык располагаться по направлению к спине и высоко в ротовой полости, что, в свою очередь, препятствует закрытию неба.
Синдром Пьера Робена (СПР) как состояние может возникать само по себе («изолированная СПР») или как признак множественных аномальных расстройств («синдромальная СПР»). Когда СПР возникает сам по себе, наиболее часто поражается ДНК рядом с геном SOX9. Ген SOX9 позволяет производить белок SOX9, который играет важную роль в развитии скелета. У пораженных людей часто наблюдаются мутации в областях ДНК, которые положительно модулируют активность SOX9 (энхансеры). Когда эти области повреждены, активность гена SOX9 снижается, что приводит к выработке меньшего количества нормального белка SOX9. Считается, что это играет роль в черепно-лицевых аномалиях, характерных для СПР.
В настоящее время считается, что большинство случаев изолированного синдрома Пьера Робена возникает спорадически или в результате новых генетических изменений, а не передается по наследству от одного поколения к другому. В более редких семейных случаях изолированного СПР исследования отдавали предпочтение аутосомно-доминантному типу наследования. Синдроматический СПР наследуется по тому же генетическому шаблону, что и состояние, с которым оно связано, что означает, что это может варьироваться в зависимости от расстройств.
Доминирующие генетические нарушения возникают, когда для того, чтобы вызвать определенное заболевание, необходима только одна копия аномального (патологического) гена. Аномальный ген может быть унаследован от любого из родителей или может быть результатом мутировавшего (измененного) гена у пораженного человека. Риск передачи патологического гена от пораженного родителя к потомству составляет 50% при каждой беременности. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Синдром Пьера Робена поражает мужчин и женщин в равных количествах, с оценочной распространенностью около 1 на 8 500-14 000 человек.
Связанные расстройства
Симптомы следующих расстройств могут быть похожи на Синдром Пьера Робена (СПР) или могут быть связаны с ними. Сравнения могут быть полезны для дифференциальной диагностики.
- Синдром Стиклера — редкое заболевание соединительной ткани, которое чаще всего поражает глаза, уши, скелет и суставы. Признаки и симптомы могут включать: близорукость (миопия), отслоение сетчатки (отделение сетчатки глаза от поддерживающих ее слоев глазного яблока), потерю слуха, характерный внешний вид лица с плоскостностью средней части лица и боль в суставах. У пораженных людей также могут быть признаки последовательности Пьера Робена, в частности, необычно маленькая нижняя челюсть (микрогнатия), смещение языка к задней части ротовой полости (глоссоптоз) и аномальное отверстие в нёбе (расщелина неба).
- Синдром делеции хромосомы 22q11.2 (велокардиофациальный синдром) — заболевание, вызванное отсутствием небольшого фрагмента 22-й хромосомы. Делеция хромосомы 22q11.2 связана с рядом проблем, включая врожденные пороки сердца, патология нёба, дисфункцию иммунной системы, включая аутоиммунные заболевания, низкий уровень кальция (гипокальциемия) и другие эндокринные аномалии, такие как проблемы с щитовидной железой и дефицит гормона роста, желудочно-кишечные проблемы, трудности с кормлением,заболевания почек, потеря слуха, судороги, патология скелета, незначительные лицевые различия, а также различия в обучении и поведении. Симптомы делеции хромосомы 22q11.2 чрезвычайно разнообразны, даже среди членов одной семьи. — редкое генетическое заболевание, характеризующееся характерными аномалиями головы и лица, особенно тяжелой микрогнатией. Сопутствующие проявления включают пороки развития глаз, аномалия уха, которые могут привести к потере слуха, и многие друге состояния и отклонения.
Другие родственные синдромы и состояния СПР включают: хромосома 11, частичная трисомия 11q; синдром трисомии 18 (синдром Эдвардса); цереброкостомандибулярный синдром; синдром Катела Манцке; кампомелическая дисплазия; синдром Мебиуса; и CHARGE синдром.
Диагностика
Синдром Пьера Робена можно обнаружить, пока плод еще находится в утробе матери. Обученный медицинский персонал может визуализировать характерные особенности секвенции Пьера Робена с помощью ультразвуковой визуализации. Если диагноз не был диагностирован ранее, черепно-лицевые аномалии обычно обнаруживаются после рождения во время физического осмотра. Младенцы с тяжелой обструкцией дыхательных путей могут иметь респираторный дистресс при рождении и могут потребовать медицинского вмешательства.
Не существует единого стандартного теста, который обычно используется для диагностики изолированной секвенции Пьера Робена, хотя молекулярно-генетическое тестирование может использоваться для выявления изменений ДНК, связанных с геном SOX9.
При подозрении на синдромальную секвенцию Пьера Робена настоятельно рекомендуется проконсультироваться с генетиком. Этот медицинский работник может провести лабораторное обследование для подтверждения подозреваемого состояния.
Стандартные методы лечения
Лечение синдрома Пьера Робена является многогранным и индивидуальным процессом, при этом операция проводится только для решения функциональных проблем, которые могут возникнуть у пациента. Хирургическое лечение может быть показано пациентам с СПР с более тяжелыми клиническими состояниями, часто связанными с нарушением дыхательных путей.
Младенцев с синдромом Пьера Робена следует внимательно наблюдать на предмет затрудненного дыхания. Размещение ребенка на животе (положение лежа), а не на спине, может помочь предотвратить падение языка обратно к горлу. Если это не решит проблему обструкции дыхательных путей, можно ввести в нос небольшие трубчатые инструменты, такие как «носоглоточные дыхательные пути», чтобы дыхательные пути оставались открытыми. Если обструкция дыхательных путей еще более серьезна, в горло младенца в больнице может быть вставлена трубка (интубация) или, в редких случаях, может быть сделано хирургическое отверстие в трахее через шею (трахеостома), чтобы помочь ребенку дышать.
Чтобы закрыть волчью пасть (расщелина нёба), операция обычно проводится в возрасте от 12 до 18 месяцев. Однако врачи могут отложить корректирующую операцию, чтобы позволить отверстию во нёбе закрыться самостоятельно по мере естественного роста.
Для решения проблем, связанных с кормлением, можно использовать различные специально адаптированные бутылочки и соски. Если проблемы с кормлением не решены и являются серьезными, может временно потребоваться зонд для кормления, чтобы помочь с правильным набором веса.
Симптоматическое и поддерживающее лечение может быть обеспечено с использованием подхода мультидисциплинарной команды, чтобы наилучшим образом удовлетворить потребности пострадавшего. При нарушении речи ребенок должен участвовать в логопедических занятиях или находиться под наблюдением логопеда. Врачи уха, горла и носа (отоларингологи) и аудиологи могут обеспечить последующее наблюдение по вопросам, связанным с ухом и слухом. При рецидиве ушных инфекций могут быть рекомендованы хирургические дренажные трубки. Комбинация ортодонтов, челюстно-лицевых хирургов и стоматологов может работать вместе, чтобы контролировать полость рта, например, стараясь избежать скученности зубов и обеспечить правильное выравнивание зубов. Для выявления глазных аномалий можно обратиться к офтальмологу.
Также для пациентов и их семей полезно генетическое консультирование.
Прогноз
Все новорожденные со значительным СПР подвержены риску внезапной смерти. Имеющееся данные о синдроме внезапной детской смерти (СВДС) показывают, что риск СВДС увеличивается, когда младенцы спят в положении лежа. Новорожденные с секвенцией Пьера Робена уже имеют нарушенные дыхательные пути, и им также обычно требуется лежать. Соответственно, следует внимательно следить за этими новорожденными.
Младенцы с СПР заслуживают того, чтобы к ним применялся междисциплинарный подход, предполагающий участие знающей и опытной команды, способной предоставить всестороннюю оценку, реалистичный план лечения и соответствующее последующее наблюдение. Вовлечение семьи на ранних этапах оценки, текущие медицинские исследования, вопросы, касающиеся ухода за ребенком и будущего планирования, обычно приводят к удовлетворению даже в самых сложных медицинских сценариях.
В исследовании 103 пациентов, наблюдаемых в среднем в течение 8,6 лет (диапазон 0,1-21,9 года), Logjes et al задокументировали 10%-ную смертность (n = 10) при среднем возрасте пациентов 0,8 года (диапазон 0,1-5,9 года). Из 10 умерших младенцев у 9 была синдромальная СПР; семь из девяти умерли от дыхательной недостаточности по разным причинам, а двое других умерли от аритмии из-за гипернатриемии и синдрома Веста с эпилептическим статусом. Младенец с несиндромным СПР умер от ишемии головного мозга после дистракционного остеогенеза нижней челюсти.

Высшее образование (Кардиология). Врач-кардиолог, терапевт, врач функциональной диагностики. Хорошо разбираюсь в диагностике и терапии заболеваний дыхательной системы, желудочно-кишечного тракта и сердечно-сосудистой системы. Закончила академию (очно), за плечами большой опыт работ.
Синдром Робена
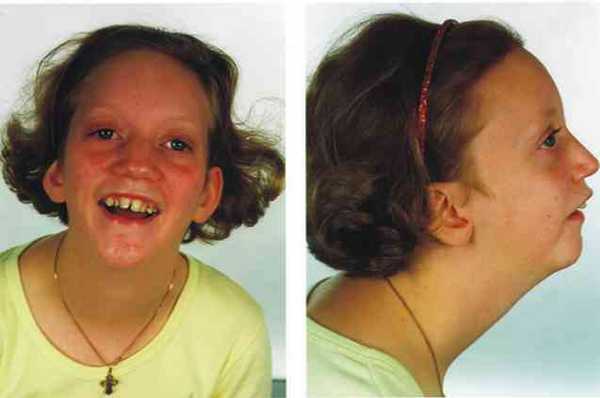
Синдром Ро́бена — врождённый порок челюстно-лицевой области, характеризующийся тремя основными клиническими признаками: недоразвитием нижней челюсти (нижней микрогнатией), глоссоптозом (недоразвитием и западанием языка) и наличием расщелины нёба. Робена синдром встречается на каждые 10—30 тыс. новорожденных.
Этиология и патогенез
Факторы возникновения и механизм развития не выяснены. Имеются указания на то, что в основе Робена синдрома лежат нарушения эмбрионального развития, вызванные разнообразной патологией дородового периода.
Клиническая картина и диагностика
Сразу же после рождения появляется резкое нарушение дыхания, связанное с западением языка. Ребёнок беспокоен, выражена синюшность. Из-за нарушения акта глотания во время кормления ребёнка может наступить удушье. Характерен внешний вид новорожденного — так называемое «птичье лицо», возникающее в связи с недоразвитием нижней челюсти (от слабо выраженного недоразвития до полного её отсутствия). Наряду с типичной триадой (нижняя микрогнатия, глоссоптоз и расщелина нёба) у больных с синдромом Робена имеются и другие пороки развития (врождённая катаракта, миопия, пороки сердца, мочеполовой системы, аномалии развития грудины и позвоночника, а также полидактилия (присутствие лишних пальцев) и врождённое отсутствие конечностей). Умственное недоразвитие отмечается примерно у 20 % больных. Диагноз основывается на клинических проявлениях этого синдрома.
Лечение и профилактика
Лечение должно проводиться практически сразу после рождения. В лёгких случаях для профилактики нарушения дыхания достаточно держать новорожденного в положении лёжа на животе или вертикально, наклонив голову к груди. При кормлении нельзя держать ребёнка горизонтально из-за опасности попадания пищи в дыхательные пути и развития аспирационной пневмонии с последующим смертельным исходом.
При резко выраженной нижней микрогнатии (недоразвитие нижней челюсти и значительном смещении языка кзади) требуется выведение языка вперёд в физиологическое положение оперативным путём. Рекомендуется пришивать язык к нижней губе, щекам или нижней челюсти в нескольких точках сроком на 1—2 месяца. В тяжёлых случаях накладывается гастростома.
В более старшем возрасте устраняют расщелину нёба, проводят ортопедическое и оперативное лечение нижней микрогнатии. Показаны занятия с логопедом для установления правильной речи. Профилактика заключается в исключении всех неблагоприятных факторов в дородовом периоде развития ребёнка.
Существует альтернатива дистракционного метода лечения. Суть метода - это установка индивидуально изготовленной из пластмассы платы, которая крепится на протезный клей на верхнюю челюсть. Эта плата давит на корень языка, тем самым выдвигая челюсть вперед, а также закрывает расщелину, решая проблемы с питанием, дыханием и всеми остальными трудностями.
Синдром Робинова
Синдром Робинова — это орфанное наследственное заболевание, которое характеризуется задержкой роста с множественными фенотипическими аномалиями. Патология возникает при аутосомно-рецессивном и аутосомно-доминантном наследовании мутаций. Болезнь характеризуется мезомелической карликовостью, патогномоничными врожденными пороками строения лица, отставанием в развитии. С диагностической целью проводятся специальные молекулярно-генетические исследования, применяются различные методы визуализации. При синдроме Робинова возможно только симптоматическое лечение: хирургическая коррекция аномалий развития, нейропсихологическая программа реабилитации больных.
МКБ-10

Общие сведения
Мезомелическая карликовость была описана в 1969 г. американским генетиком М. Робинов в сотрудничестве с врачами Ф. Сильверманом и Х. Смитом, поэтому в некоторых медицинских источниках болезнь называют синдромом Робинова-Сильвермана-Смита. Патология встречается крайне редко — 1 случай на 500 тыс. новорожденных. Наибольшая база данных о таких пациентах собрана в США благодаря Фонду синдрома Робинова, который занимается статистической работой, оказывает поддержку людям, столкнувшимся с этим заболеванием.

Причины
Благодаря достижениям современной генетики в 2000 г. были установлены точные генетические предпосылки синдрома Робинова. Заболевание может наследоваться по аутосомно-доминантному типу (когда болен один из родителей), по аутосомно-рецессивному типу (если оба родителя — носители мутантного гена). Развитие мезомелической карликовости вызвано следующими генными дефектами:
- МутацииWNT5A, DVL1, DVL3. Все три варианта генетических дефектов передаются аутосомно-доминантным путем. Такой тип синдрома Робинова отличается менее выраженными аномалиями развития, имеет благоприятный прогноз для больных.
- МутацииROR2. Патология этого гена наследуется аутосомно-рецессивным путем, вызывает грубые внутриутробные пороки и, кроме того, обычно сочетается с интеллектуальными расстройствами.
Главным предрасполагающим фактором развития синдрома Робинова являются близкородственные браки, при которых в популяции накапливается большое число мутаций, резко возрастает риск неблагоприятной комбинации генетического материала родителей. Поэтому максимальное распространение болезни наблюдается в Турции и Омане, где браки между родственниками до сих пор встречаются довольно часто. Влияние тератогенных факторов в эмбриональном периоде беременности не исключается, но точных научных данных по этому вопросу пока нет.
Патогенез
Характерные клинические проявления синдрома Робинова обусловлены нарушениями процессов созревания и дифференцировка тканей, которые возникают во время эмбрионального развития ребенка. При аутосомно-доминантном типе синдрома в период эмбриогенеза наблюдается недостаточность протеинов из группы WNT, которые необходимы для правильного формирования соединительной ткани и образования скелета.
Для аутосомно-рецессивного варианта болезни характерны мутации гена, отвечающего за работу тирозинкиназного рецептора, который расположен на поверхности всех типов клеток организма эмбриона. При недостаточности ROR2 происходят грубые нарушения роста и размножения клеток, изменяются биохимические реакции клеточного метаболизма. Вследствие таких мутаций страдает костная и хрящевая ткань, а также ткани внутренних органов.
Симптомы
Признаки патологии четко видны сразу после родов. Особенности черепно-лицевого строения включают большой размер головы (макроцефалию), крупные выпуклые глаза, короткий вздернутый нос с расширенными ноздрями и вдавленной переносицей. Также у ребенка заметны большой рот (макростомия), обнаженные передние зубы в спокойном состоянии, недоразвитая нижняя челюсть (микрогнатия). Зачастую наблюдается выпуклый высокий лоб, низко расположенные уши.
Типичные нарушения строения тела заключаются в мезомелии — недоразвитии средних участков верхних и нижних конечностей (предплечья, голени). Аномалии развития скелета дополняются брахидактилией (короткопалостью), врожденным сколиозом, который вызван пороками строения грудных и шейных позвонков. По мере взросления ребенка становится заменой задержка роста, избыточный вес.
Синдром Робинова, как правило, сопровождается нарушениями полового развития: у девочек выявляется недоразвитие клитора и больших половых губ, у мальчиков — тяжелая гипоплазия полового члена. Если заболевание связано с аутосомно-доминантным типом мутации, клинические симптомы выражены не так интенсивно, отсутствует патологическая полнота, а половое развитие находится в пределах нормы.
Осложнения
Синдрому Робинова нередко сопутствуют соматические болезни, вызванные нарушениями внутриутробного развития. К типичным осложнениям относят гидронефроз с последующей атрофией органа, аномалии формы, размера или положения почек. Нередко диагностируются врожденные паховые или пупочные грыжи. У 10% больных развиваются врожденные пороки сердца и крупных сосудов.
Около 20% пациентов с синдромом Робинова имеют нарушения психического развития по типу умственной отсталости легкой или среднетяжелой степени. В сочетании с выраженными аномалиями скелета это нарушает социализацию детей, провоцирует серьезные психологические проблемы. Вследствие скелетных аномалий такие больные неспособны к большинству видов трудовой деятельности, потому они нуждаются в медико-социальной помощи.
Зачастую фенотипические признаки синдрома Робинова определяются еще в периоде внутриутробного развития при плановом ультразвуковом скрининге. Однако исследование не дает 100% вероятности диагноза, поэтому обследование продолжается после рождения ребенка. Чтобы установить мезомелическую карликовость, врачу требуется полное физикальное обследование пациента и следующие инструментально-лабораторные методы:
- Рентгенография скелета. Рентгенологическое исследование демонстрирует характерное мезомелическое укорочение конечностей, разнообразные аномалии лицевого черепа и другие отклонения от физиологической нормы.
- УЗИ сердца. Учитывая высокую частоту сердечных пороков у страдающих синдромом Робинова, ультразвуковое сканирование и консультация кардиолога (если это необходимо) являются важным этапом диагностики.
- УЗИ органов живота. Инструментальная визуализация используется для оценки структурно-функциональных особенностей почек, мочевыводящих путей, подтверждения диагноза при обнаружении грыж передней брюшной стенки.
- Генетическое тестирование.Молекулярно-генетический анализ методом автоматического секвенирования проводится для диагностики болезни Робинова, выяснения ее клинического варианта, что важно для оценки прогноза заболевания.
Лечение синдрома Робинова
Специфические методы терапии отсутствуют. Медикаментозная терапия при этом заболевании неэффективна, однако она может применяться при сопутствующих соматических аномалиях для стабилизации состояния пациента. Основную роль в лечении таких больных играет помощь пластических и челюстно-лицевых хирургов для максимально возможного устранения скелетных аномалий. Целесообразно проведение следующих типов операций:
- экстракраниальная или интракраниальная коррекция гипертелоризма;
- коррекция положения нижней челюсти с ее фиксацией пластинами и установкой имплантатов для добавления недостающего объема;
- стоматологические операции для восстановления нормального прикуса;
- коррекция длины конечностей с помощью хирургической реконструкции.
Операции выполняются, начиная с раннего возраста ребенка, причем у каждого типа вмешательства есть собственные временные рамки и показания. Для максимально полного устранения недостатков внешности пациентам требуются сложные многоэтапные вмешательства. При наличии жизнеугрожающих пороков хирургическое лечение рекомендовано начать в ранние сроки, чтобы восстановить функционирование организма.
Если синдром Робинова сопровождается интеллектуальными нарушениями, больным требуется помощь педагогов-дефектологов. При легкой степени расстройств дети успешно обучаются по специальным программам, сохраняют работоспособность. При среднетяжелой выраженности нарушений речевое и психическое развитие значительно снижены, поэтому суть реабилитации заключается в привитии пациенту навыков самообслуживания.
Прогноз и профилактика
Синдром Робинова существенно не влияет на продолжительность жизни, но значительно ухудшает ее качество, особенно при несвоевременном проведении пластических операций по коррекции скелетных аномалий. При многоэтапной хирургической помощи удается достичь естественной внешности, улучшить функциональные возможности скелета. У пациентов, страдающих мезомелической карликовостью и сопутствующими соматическими пороками, прогноз неблагоприятен.
Если в семье были случаи диагностированного синдрома Робинова или карликовости, вероятно связанной с генетическими патологиями, перед планированием ребенка необходима консультация генетика. При обследовании обоих партнеров проводится анализ на типичные мутации, на основании результатов которых устанавливается риск рождения больного ребенка. В дальнейшем такие пары подлежат генетическому консультированию на протяжении беременности.
2. WNT5A mutations in patients with autosomal dominant Robinow syndrome/ Person A. D., Beiraghi S., Sieben C. M., Hermanson S., Neumann A. N., Robu M. E., Schleiffarth J. R., Billington C. J. et al// Dev Dyn. — 2010.
3. Клинический случая синдрома Робинова в сочетании с множественным гамартозным ростом/ Ю.Б. Гречанина, Л.В. Молодан. — 2003.
Синдром Пьера Робена Симптомы, причины, лечение
Синдром Пьера Робина (SPR), также известная как последовательность Пьера Робина, представляет собой расстройство генетического происхождения, классифицируемое как черепно-лицевые синдромы или патологии (Arancibia, 2006).
Клинически синдром Пьера Робена характеризуется тремя основными клиническими признаками: микрогнатия, глоссоптоз и обструкция верхних дыхательных путей и переменное наличие расщелины неба (Sridhar Reddy, 2016).
Что касается этиологического происхождения этой патологии, синдром Пьера-Робена обусловлен наличием специфических мутаций в гене SOX9, большинство из которых диагностированы (Genetics Home Reference, 2016).
В целом, этот синдром вызывает важные медицинские осложнения, в том числе дыхательную недостаточность, проблемы с пищеварением животных или развитие других черепно-лицевых пороков развития (Ассоциация аномалий и порочно-лицевых пороков развития, 2016).
С другой стороны, диагноз синдрома Пьера-Робина обычно не подтверждается до момента рождения, в дополнение к клиническим данным важно выполнять различные рентгенологические тесты для выявления изменений кости (Pierre Robin Australia, 2016).
В настоящее время не существует никакого лечения синдрома Пьера Робина, однако, хирургические подходы часто используются для коррекции опорно-двигательного расстройства. Кроме того, респираторная и желудочно-кишечная адаптации важны для предотвращения опасных для жизни медицинских осложнений (Rethe, Rayyan, Shoenaers, Dormaar, Breuls, Verdonck, Devriednt, Vander Poorten and Hens, 2015)..
Характеристика синдрома Пьера Робина
Синдром Пьера Робена - это врожденная патология, клинические проявления которой присутствуют с момента рождения и, кроме того, все его характеристики связаны с наличием черепно-лицевой аномалии (Genetics Home Reference, 2016).
Кроме того, в медицинской литературе мы можем выделить различные термины, используемые в контексте синдрома Пьера Робина: болезнь Пьера Робина, порок развития Пьера Робина или последовательность Пьера Робина (Национальная черепно-лицевая ассоциация, 2016).
На определенном уровне этот синдром был описан в 1891 году Менерадом и Ланнелонгом. В клинических отчетах они описали двух пациентов, клиническое течение которых характеризовалось наличием недоразвития структуры нижней челюсти, расщелины неба и смещения или ретракции языка (Arancibia, 2006).
Однако только в 1923 году Пьер Робин описал клинический спектр этой патологии, сосредоточив свои исследования на случае ребенка, страдающего аномалией нижней челюсти, аномально большим языком и значительными респираторными проблемами (Детская черепно-лицевая ассоциация, 2016 ).
Хотя эта патология в основном отличается черепно-лицевыми рентгенологическими данными, она характеризуется высокой подвижностью, связанной с медицинскими осложнениями, в основном связанными с сердечной недостаточностью и проблемами с питанием..
В частности, синдром Пьера Робина характеризуется высокой смертностью, связанной с обструкцией дыхательных путей, неврологическими нарушениями или изменениями в сердце (Sridhar Reddy, 2016)..
С другой стороны, многие авторы предпочитают называть эту патологию только последовательностью Пьера, поскольку именно аномалии нижней челюсти приводят к появлению остальных типичных признаков и симптомов (Pierre Robin Australia, 2016).
частота
Распространенность синдрома Пьера Робина оценивается примерно в одном случае на 8500 детей, родившихся живыми, из которых более 80% диагностированных случаев связаны с другими медицинскими осложнениями и специфическими синдромами (Arancibia, 2006).
С другой стороны, в случае США заболеваемость синдромом Пьера Робина составляет 1 случай на 3120 рождений в год (Lee, Thottam, Ford and Jabbour, 2015).
В настоящее время не выявлено различий в распространенности синдрома Пьера Робина, связанного с полом, происхождением, географией или конкретными этническими и расовыми группами..
Кроме того, как мы указывали ранее, синдром Пьера Робена представляет собой одну из черепно-лицевых патологий с высокой вероятностью смертности. В Соединенных Штатах приблизительно 16,6% пострадавших умирают из-за развития медицинских осложнений (Lee, Thottam, Ford and Jabbour, 2015).
По порядку возникновения наиболее частыми вторичными медицинскими патологиями являются: сердечные аномалии (39%), изменения в центральной нервной системе (33%) и аномалии в других органах (24%) (Lee, Thottam, Ford and Jabbour, 2015).
Последовательность Пьера Робена отличается от других типов черепно-лицевой патологии наличием трех основных клинических признаков: микрогнатия, глоссоптоз и волчья пасть (Детская черепно-лицевая ассоциация, 2016, Genetics Home reference, 2016, Rehté et al., 2015):
micrognatia
Под термином микрогнатия мы подразумеваем наличие патологического изменения развития структуры нижней челюсти, в частности, окончательная форма имеет уменьшенный размер по сравнению с ожидаемой для уровня развития пораженного человека..
Как следствие, неполное развитие этой черепно-лицевой структуры вызовет широкий спектр изменений, связанных с наличием пороков развития, которые влияют на рот и лицо.
Микрогнатия является медицинским признаком, присутствующим примерно у 91% людей, страдающих синдромом Пьера Робина.
glossoptosis
Под термином глоссоптоз мы подразумеваем наличие аномальной ретракции положения языка в структуре полости рта, в частности, языки должны быть размещены за нормальным продуктом микрофотографии и уменьшением объема полости рта..
Аномалии, связанные с положением и структурой языка, могут вызвать серьезные проблемы с питанием, которые могут привести к серьезным заболеваниям..
Кроме того, в других случаях также можно идентифицировать аномально большой язык (макроглоссия), который, среди прочего, затрудняет дыхание, жевание или выработку функционального языка..
Кроме того, глоссоптоз является одним из наиболее частых клинических признаков синдрома Пьера Робина, наблюдаемых примерно в 70-85% диагностированных случаев. В то время как макроглоссия может наблюдаться в меньшем количестве, примерно у 10-15% пострадавших.
Волчья пасть
Этот термин относится к наличию уродства в небных или ротовых областях крыши, то есть может наблюдаться наличие трещин или отверстий, связанных с неполным развитием нижней челюсти.
Как и другие клинические данные, расщелина неба вызовет значительные изменения в диете.
В дополнение к этим признакам и симптомам, также можно определить другие типы изменений, среди которых включены (Arancibia, 2006, Rehté et al., 2015):
- Пороки развития носа.
- Изменения и скелетно-мышечные дефекты, в основном связанные с развитием oligodactyly (сокращение числа пальцев, менее чем 5 или пальцев ноги) клинодактилией (поперечное отклонение положения пальцев), полидактильный (увеличение числа пальцев), hiperlaxity суставы (аномально преувеличенное увеличение подвижности суставов), дисплазия в фалангах (фаланги с недостаточным или неполным развитием костей) или синдактилия (слияние нескольких пальцев)
- Другие изменения: также возможно выявить пороки развития в структуре конечностей или в позвоночнике.
Наиболее частые медицинские осложнения
В дополнение к медицинским функциям, перечисленным выше, могут появляться и другие, связанные с несколькими системами (Arancibia, 2006, Детская черепно-лицевая ассоциация, 2016, Genetics Home reference, 2016, Rehté et al., 2015):
Сердечные изменения
Сердечные изменения представляют собой одно из медицинских осложнений, оказывающих большее влияние на здоровье человека и представляющих важные риски для его выживания. Однако признаки и симптомы, связанные с сердечно-сосудистой системой, обычно поддаются лечению с помощью фармакологического и / или хирургического подхода..
Некоторые из наиболее частых сердечных аномалий включают стеноз сердца, постоянное овальное отверстие, изменение артериальной перегородки или гипертонию..
Неврологические изменения
Генетическое происхождение синдрома Пьера Робена может также включать развитие различных неврологических расстройств, в основном связанных с наличием аномалий центральной нервной системы (ЦНС)..
Таким образом, некоторые из неврологических расстройств, связанных с синдромом Пьера Робина, могут включать гидроцефалию, порок развития Киари, эпилептические эпизоды или задержку в приобретении психомоторных навыков..
Респираторные расстройства
Респираторные расстройства являются одной из наиболее важных особенностей, поскольку они могут вызвать как смерть пациента из-за дыхательной недостаточности, так и развитие повреждения головного мозга из-за недостатка кислорода в нервных зонах..
Таким образом, во многих случаях требуется хирургическая коррекция, чтобы освободить дыхательные пути, главным образом коррекция дисплазии нижней челюсти или положение языка.
Силовые аномалии
Как и в случае респираторных расстройств, проблемы с питанием возникают в основном из-за пороков развития нижней челюсти.
Синдром или последовательность Пьера Робина, имеет генетическое этиологическое происхождение, связанное с изменениями в гене SOX9. Хотя эта аномалия была выявлена в большинстве единичных случаев синдрома Пьера Робина, некоторые из ее клинических характеристик могут быть связаны с другим типом генетических мутаций (Genetics Home Reference, 2016)..
В частности, ген SOX9 играет фундаментальную роль пропорциональных биохимических инструкций, необходимых для производства белка, участвующего в развитии и формировании различных тканей и органов во время развития плода (Genetics Home Reference, 2016).
Кроме того, текущие исследования показывают, что белок SOX9 может регулировать активность других типов генов, особенно тех, которые участвуют в развитии структуры скелета и, следовательно, нижней челюсти (Genetics Home Reference, 2016).
В результате генетические изменения препятствуют адекватному морфологическому развитию определенных структур и, следовательно, появляются кардинальные клинические признаки: микогнатия, глоссоптоз и волчья пасть.
диагностика
Во многих случаях черепно-лицевые структурные пороки развития могут быть выявлены во время беременности с помощью ультразвука, хотя случаи редки.
В этом смысле чаще всего подозрение на синдром Пьера Робина возникает в постнатальном или детском периоде. У многих из тех, кто пострадал, структуры признаков значительно очевидны, поэтому диагноз подтверждается рентгенологическими исследованиями и физическим осмотром..
Тем не менее, в другом случае необходимо предварительно провести респираторное исследование, а затем рентгенологическое исследование, чтобы определить наличие этого синдрома..
Кроме того, другим фундаментальным аспектом в диагностике этой патологии является исследование других областей, особенно сердечной и нервной системы, поскольку могут появиться другие виды опасных для жизни аномалий..
Наконец, диагностическое вмешательство может включать индивидуальное и семейное генетическое исследование для выявления возможных генетических ассоциаций.
лечение
Типичное лечение синдрома Пьера Робина основано на хирургических вмешательствах для коррекции черепно-лицевых пороков развития (Arancibia, 2006):
- Закрытие небных расщелин.
Кроме того, другие фармакологические подходы также используются для лечения патологий сердца, эпилептических эпизодов и других неврологических явлений..
Кроме того, у затронутых людей обычно возникают трудности, связанные с производством языка, поэтому во многих случаях ранний логопедический подход имеет важное значение.
Основная цель - создать эффективный метод общения с помощью остаточных возможностей и, в свою очередь, стимулирование приобретения новых навыков..
Синдром Пьера Робена ( Аномалия Робена , Последовательность Робена )
Синдром Пьера Робена - это генетически обусловленная аномалия, характеризующаяся гипоплазией нижней челюсти, небной расщелиной и глоссоптозом. Комбинация пороков ЧЛО вызывает затруднения при вскармливании, обструктивное апноэ, диффузный цианоз, риск аспирации пищи и асфиксии. Патология диагностируется по данным рентгенографии и КТ челюсти, фиброларингоскопии, полисомнографии, генодиагностики. Консервативная тактика включает позиционную терапию, СРАР, ортодонтическое лечение. Методы хирургической коррекции порока представлены глоссопексией, пластикой неба, компрессионно-дистракционным остеосинтезом.



Синдром (последовательность, аномалия) Робена включает триаду признаков: микрогнатию, глоссоптоз, расщепление твердого и мягкого неба. Врожденная анатомическая деформация описана в 1923 г. французским зубным врачом Пьером Робеном. По разным оценкам, дети с данной аномалией рождаются с популяционной частотой 1:8500-30000, независимо от пола. Ведущей проблемой младенцев с синдромом Робена является обструкция верхних дыхательных путей и критическое нарушение дыхания вследствие недоразвития нижней челюсти и смещения мягких тканей вглубь полости рта.

Последовательность Пьера Робена может встречаться как изолированный синдром (20-40%) или как часть других генетических патологий. В первом случае челюстно-лицевая аномалия формируется под влиянием неблагоприятных условий внутриутробного развития, в числе которых может быть:
- механическое сдавление плода в полости матки, ограничивающее рост челюсти (рубцами, амниотическими тяжами, опухолями или другим плодом при многоплодной беременности);
- внутриутробные инфекции;
- маловодие;
- дефицит фолиевой кислоты в организме беременной;
- тератогенные воздействия на плод, вызванные вредными экологическими факторами, радиацией, химическими веществами, приемом ЛС.
Кроме этого, аномалия Пьера Робена встречается в структуре генетических синдромов с множественными пороками развития. Всего насчитывается порядка 300 таких заболеваний, среди которых синдромы Стиклера, Хангарта, Эдвардса, кампомелическая дисплазия и др. Более чем в 30% случаев синдром Пьера Робена сочетается с ВПС, скелетными аномалиями, дефектами развития глаз и ушных раковин.
Спорадические случаи возникают в результате генетических мутаций de novo. Синдромальная последовательность наследуется по тому же принципу, что и патология, с которой она ассоциирована (сообщается об аутосомно-рецессивном, аутосомно-доминантном, Х-сцепленном варианте). Согласно исследованиям, генетические мутации затрагивают области хромосом 2 (2q24.1-33.3), 4 (4q32-qter), 11 (11q21-q23.1) и 17 (17q21-q24.3).
Синдром Пьера Робена принято называть «последовательностью», поскольку его признаки представляют собой цепочку взаимовлияющих друг на друга событий. Нарушение развития нижней челюсти у плода происходит в период между 7-й и 11-й неделями гестации под воздействием генетических мутаций или экзогенных факторов. Гипоплазия челюсти способствует неправильному положению языка между небными пластинками, которое препятствует их своевременному закрытию и вызывает образование расщелины.
Нижняя ретрогнатия и маленький объем ротовой полости создают условия для смещения языка в сторону задней стенки глотки и возникновения глоссоптоза. Оттянутый назад язык вызывает у новорожденного серьезные затруднения дыхания, вследствие чего часто возникает инспираторная обструкция ВДП.
Классификация
В зависимости от выраженности нарушения глотания и дыхания выделяют три степени тяжести синдрома Пьера Робена:
- легкая - дыхание не нарушено, имеются небольшие затруднения при кормлении младенца, которые устраняются консервативными методами в домашних условиях;
- средняя - трудности дыхания и кормления выражены умеренно, ребенку требуется стационарная помощь, зондовое питание;
- тяжелая - выраженные затруднения дыхания и самостоятельного питания, требующие постановки трахеостомы и желудочного зонда.
Новорожденные с синдромом Пьера Робена имеют характерное «птичье» лицо, обусловленное гипоплазией нижней челюсти (микрогнатия) и ее смещением назад (ретрогнатия). Объем полости рта полости уменьшен, язык оттянут вглубь (глоссоптоз). Как правило, у пациентов обнаруживается U-образная субмукозная расщелина мягкого неба, также встречается готическое небо.
Смещение языка кзади вызывает проблемы с сосанием в раннем возрасте, из-за чего у ребенка отмечается задержка роста и набора веса, отставание в моторном развитии. Тяжесть дыхательных нарушений варьируется от храпа до опасных эпизодов обструктивного апноэ. В легких случаях дыхательные расстройства возникают только в положении на спине и при беспокойстве ребенка, проходят самостоятельно. В более серьезных ситуациях приступы остановки дыхания длительные (до 20 сек. и более), приводящие к развитию гипоксии и гиперкапнии.
Тяжелые эпизоды апноэ сопровождаются акроцианозом или диффузным цианозом. Постоянная задействованность вспомогательных дыхательных мышц способствует формированию воронкообразной деформации грудной клетки.
Синдромальная последовательность Пьера Робена, как правило, сочетается с врожденными аномалиями других органов: тугоухостью и недоразвитием слухового аппарата, патологией зрительного анализатора (близорукость, катаракта), скелетными деформациями (полидактилия, врожденные ампутации), сердечными пороками. Примерно у половины пациентов обнаруживаются дефекты ЦНС: микроцефалия, гидроцефалия, эпилепсия, задержка психоречевого развития, олигофрения.
Практически все дети с синдромом Пьера Робена страдают речевыми дефектами (открытая ринолалия), нарушением прикуса, рецидивирующими ушными инфекциями. На фоне пролонгированных приступов обструктивного апноэ постепенно формируется гипоксическая энцефалопатия, легочное сердце. В некоторых случаях ребенок может погибнуть от асфиксии. Дисфагические расстройства могут повлечь за собой аспирацию слюны, пищи, желудочного содержимого при срыгивании, что чревато развитием аспирационной пневмонии. Смертность детей составляет порядка 16%, а при наличии сочетанных пороков достигает 41%.
В настоящее время синдром Пьера Робена диагностируется пренатально с помощью скринингового УЗИ в 11-12 недель беременности. После рождения диагноз подтверждается данными клинической картины и инструментальных исследований. Необходимая диагностика:
- Рентгеновские методы. Для первичной оценки анатомии челюстно-лицевой зоны выполняется рентгенография нижней челюсти, гортани и трахеи. В рамках предоперационного обследования обязательно проведение КТ челюстей, лицевого черепа, мягких тканей шеи. Рентгеновская визуализация помогает определить степень микро- и ретрогнатии, обструкции дыхательных путей, рассчитать параметры дистракции.
- Эндоскопия гортани. С помощью фиброларингоскопии уточняется состояние носоглотки, гортани и начальных отделов трахеи. Оценивается уровень и степень обструкции ВДП, обнаруживаются другие аномалии ЛОР-органов.
- Полисомнография. Считается «золотым стандартом» определения тяжести синдрома обструктивного апноэ. С ее помощью выясняется частота и длительность эпизодов остановки дыхания, подбирается оптимальная терапия.
- Другие методы. Регулярно проводится исследование газового состава крови, пульсоксиметрия. Перед планируемой операцией выполняется фотофиксация внешнего вида ребенка в фас и профиль, регистрация прикуса. При синдромальных формах аномалии Робена показана генодиагностика.
Лечение синдрома Пьера Робена
Лечебная тактика при аномалии Робена зависит от выраженности респираторных и дисфагических расстройств. Она может заключаться в проведении позиционной терапии, вспомогательной терапии, хирургического лечения. На различных этапах жизни ребенку требуется сопровождение врача-отоларинголога, челюстно-лицевого хирурга, ортодонта, логопеда.
Консервативное лечение
При легкой степени дыхательной обструкции рекомендуется постуральная терапия. Ребенка выкладывают на живот или на бок с приподнятой или зафиксированной головой, обеспечивающей выдвижение вперед языка и нижней челюсти. Персистирующая обструкция может купироваться с помощью назофарингеального воздуховода. Одним из методов с подтвержденной эффективностью является СРАР-терапия, позволяющая осуществлять неинвазивную масочную вентиляцию под постоянным положительным давлением.
При невозможности самостоятельного питания кормление ребенка производится через назогастральный зонд. Тяжелые степени синдрома Пьера Робена требуют установки трахеостомы или интубации трахеи с подключением ребенка к аппарату ИВЛ.
Консервативные методы позволяют купировать симптомы, но ни один из них не устраняет имеющиеся врожденные аномалии. Поэтому такое лечение должно проводиться очень длительно, до тех пор, пока размеры дыхательных путей ребенка не позволят ему дышать самостоятельно без риска обструкции. После нормализации жизненных функций проводится ортодонтическое лечение, логопедическая коррекция.
Хирургическое лечение
Устранение небной расщелины проводится поэтапно: велофарингопластика выполняется в 6-8 мес., уранопластика - в 12-18 мес. В качестве временной меры при подготовке к радикальному лечению может осуществляться глоссопексия - фиксация языка к нижней губе, альвеолярному отростку. Ранее применявшиеся методы вытяжения нижней челюсти, транспозиции жевательных мышц не являются достаточно эффективными.
Радикальным хирургическим вмешательством, позволяющим удлинить нижнюю челюсть, является компрессионно-дистракционный остеосинтез. Операция заключается в проведении двусторонней остеотомии нижней челюсти с фиксацией на костных фрагментах специального аппарата. Активация устройства обеспечивает постепенное перемещение нижней челюсти вместе с комплексом мягких тканей дна полости рта кпереди. Процесс дистракции занимает около 3-х месяцев, в результате чего становится возможным сначала самостоятельное дыхание, а затем - питание.
Изолированный синдром Робена имеет более благоприятный прогноз, чем синдромальные формы, ассоциированные с множественными пороками и умственной отсталостью. Также существенное значение имеет своевременность и радикальность лечения. Вместе с тем, на данный момент смертность от асфиксии, других осложнений и сопутствующих аномалий остается достаточно высокой.
Для снижения риска рождения ребенка с синдром Пьера Робена рекомендуется не допускать воздействия тератогенов, инфицирования, дефицита микронутриентов в период гестации. Беременным необходимо вовремя проходить генетические и ультразвуковые пренатальные скрининги.
1. Синдром Пьера Робена у детей/ Кириллова Л.Г., Ткачук Л.И., Шевченко А.А., Силаева Л.Ю., Лисица В.В., Мироняк Л.А.// Международный неврологический журнал. - 2010. - №3 (33).
3. Хирургическое лечение новорожденных и грудных детей с синдромом Пьера Робена/ Дубин С.А., Комелягин Д.Ю., Злыгарева Н.В., Строгонов И.А.,Р огинский В.В., Полуэктов М.Г.// Российский вестник детской хирургии, анестезиологии и реаниматологии. - 2011.
4. Клинический случай синдрома Пьера Робена у новорожденного ребенка/ Вахитова Л.Ф., Фазлеева Л.К., Булгакова Л.Г., Варламова О.В.// Практическая медицина. - 2013.
Читайте также:
- Эндоскопическое удаление инородных тел. Инородные тела желудка, толстой кишки
- Генодерматозы
- Лечение IV стадии ретинопатии недоношенных новорожденных. Тракционная отслойка сетчатки (ТОС)
- Возбудитель чумы. Чума. Иерсиния пестис. Yersinia pestis. История чумы. Эпидемии чумы. Распространенность чумы. Эпидемиология чумы.
- Сцепленность генов тяжелых цепей антител. Гены тяжелых цепей IgA