Синдром Рубинштейна (Rubinstein) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 28.01.2026
Синдром Рубинштейна-Тейби - аутосомно-доминантное заболевание, вызванное микроделецией хромосомы 16p13.3, или мутациями в генах CREBBP (локализован в локусе 16p13.3) или EP300 (локализован в локусе 22q13.2). Оба гомологичных гена кодируют гистоновые ацетилтрансферазы, которые являются транскрипционными кофакторами, участвующими в регуляции роста, дифференцировки, репарации ДНК, апоптоза, экспрессии многих генов. Мутации в этих генах обнаруживают у 55-60% больных. Доля мутаций в гене EP300 составляет не более 3%. Мутации весьма разнообразны, от точковых замен, до крупных делеций, захватывающих весь ген. Многие точковые замены приводят к образованию преждевременного стоп-кодона или вызывают преждевременный обрыв цепи мРНК при транскрипции (мутации сайта-сплайсинга). На долю крупных делеций приходится около 10-20%, с помощью прямого автоматического секвенирования обнаруживают еще 30-50% мутаций. Частых мутаций в гене CREBBP нет, однако отмечается кластеризация мутаций, с наибольшей частотой в НАТ-домене. Частота заболевания 1:100000-125000 новорожденных. В основном это - спорадические случаи.
Клиническими признаками синдрома являются микроцефалия, черепно-лицевые дисморфизмы, широкий первый палец на руках и ногах (возможно раздвоение первой фаланги большого пальца), задержка умственного развития, низкий рост, высокий риск возникновения новообразований. Среди черепно-лицевых дисморфизмов выделяют густые дугообразные брови, антимонголоидный разрез глаз, эпикант, длинные ресницы, широкую спинку носа, «клювовидный нос» - загнутый книзу кончик носа с выступающей носовой перегородкой, деформированные низкопосаженные уши, микрогнатию, высокое арковидное небо, аномалии зубов. Среди других патологий у больных часто наблюдается обструкция носослезного канала, птоз, врожденная или ювенильная глаукома, различные врожденные пороки сердца, гипермобильность суставов, аномалии кожи (гирсутизм, невусы, келлоиды). У пациентов отмечают характерное выражение лица - гримасу, напоминающую улыбку, с почти закрытыми глазами. Средний IQ между 35 и 50. Хотя познавательные способности весьма ограничены, больные могут налаживать хороший социальный контакт. Их поведение характеризуется короткой продолжительностью концентрации внимания, плохой координацией, частыми сменами настроения. В анамнезе часто встречаются проблемы с кормлением и дыханием в младенчестве, хронические запоры. У больных существует повышенный риск развития опухолей, в основном менингиомы и других опухолей мозга, а также лейкемии. Новообразования, как правило, развиваются до 15 лет, хотя менингиомы встречаются и в зрелом возрасте.
В Центре Молекулярной Генетики методом количественной MLPA проводится поиск крупных делеций / дупликаций хромосомного локуса 16p13.3, включающего гены CREBBP и EP300.
Синдром Корнелии де ланге и Рубинштейна - Тейби
Синдром Корнелии де Ланге (синдром Брахмана-Ланге) — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития.
Синдром Корнелии де Ланге относится к доминантно-наследуемым заболеваниям, большинство случаев являются спорадическими, возникшими de novo. Синдром является генетически гетерогенным.
· Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной нормы);
· Брахицефалия (укорочение черепа в сагиттальном направлении, в результате чего поперечный размер головы увеличивается, а продольный уменьшается);
· Тонкие сросшиеся брови; длинные загнутые ресницы; деформированные ушные раковины; маленький нос, открытые вперед ноздри, атрезия хоан; тонкая верхняя губа;
· Микрогения; высокое нёбо или расщелина нёба; нарушение прорезывания зубов;
· Миопия, косоглазие, астигматизм, атрофия зрительных нервов, колобома зрительного нерва;
· Маленькие кисти и стопы, отсутствие или значительное недоразвитие проксимальных отделов конечностей, вследствие чего кисти и стопы кажутся прикрепленными непосредственно к туловищу, уменьшение количества пальцев;
· Врожденные пороки внутренних органов (сердца, почек, пилоростеноз, крипторхизм) и др.
У всех больных отмечаются отставание в росте, глубокая умственная отсталость; типичны рецидивирующие респираторные инфекции. Выделяют два варианта синдрома: первый (классический) с выраженной пренатальной гипоплазией, значительной задержкой физического и интеллектуального развития, грубыми пороками развития; второй — с аналогичными лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков развития
Диагностика. Современная медицина пока со 100% уверенностью не может выявить этот синдром у плода. Однако есть данные о том, что во время беременности у женщины в крови не обнаруживается плацентарный белок РАРРА. Это может указывать на наличие заболевания у плода, однако достоверно поставить диагноз только по этому анализу невозможно.
Синдром Рубинштейна-Тейби (синоним: синдром широкого I пальца кистей и стоп, специфического лица и умственной отсталости)
Тип наследования неизвестен.
Клиника. Основными признаками синдрома являются отставание психофизического развития, дисплазия лица, аномалии развития пальцев. У 94 % больных рост ниже среднего. Костный возраст значительно отстает от паспортного (94 %). Отмечается умственная отсталость (100 %) (в большинстве случаев — глубокая олигофрения, задержка моторного и речевого развития).
Комплекс черепно-лицевых дисморфий включает брахицефалию, микроцефалию, большой и поздно закрывающийся родничок, выступающий лоб с низким ростом волос, приподнятые дугообразные брови, антимонголоидный разрез глаз (93 %), широкую переносицу, эпикант (62 %), длинные ресницы, птоз, широкую спинку носа (71 %), загнутый книзу кончик носа (клювовидный нос) (90 %), гипоплазию крыльев носа (72 %), умеренную ретрогнатию, гримасу, напоминающую улыбку, высокое арковидное небо (93 %).
Со стороны глаз отмечаются косоглазие (79 %) и аномалии рефракции (58 %). Ушные раковины деформированы, уменьшены или увеличены (74 %).
Аномалии пальцев заключаются в расширении, укорочении и уплощении ногтевых фаланг первых пальцев кистей и стоп (100 %), иногда терминальных фаланг других пальцев кисти (60 %), вальгусной деформации межфаланговых суставов, удвоении ногтевой фаланги первых пальцев стоп (30 %), реже — проксимальной фаланги, в ряде случаев — полидактилии стоп, частичной синдактилии кистей и стоп.
Отмечаются также лордоз, кифоз, сколиоз, аномалии грудины и рёбер, уплощение крыльев тазовых костей.
В половине случаев встречаются гирсутизм, ярко-красный невус на коже лба, затылка, боковой поверхности шеи.
Пороки развития внутренних органов включают незаращение артериального протока и дефекты перегородок сердца, одностороннюю аплазию почек, удвоение почек,гидронефроз, расширение мочеточников или их стеноз, дивертикул мочевого пузыря,крипторхизм (в 79 % случаев), нарушение лобуляции легких, агенезию или аплазиюмозолистого тела (обнаруживаемую при пневмоэнцефалографии или на вскрытии).
При цитогенетическом исследовании в ряде случаев выявляют микроделецию сегмента р13.3 хромосомы 16.
124.Витамин Д-резистентный рахит, клиническая характеристика, методы диагностики, генетический риск.
Семейный гипофосфатемический рахит, или фосфат-диабет, или витамин-D-резистентный рахит .Х-сцепленный гипофосфатемический рахит - наиболее распространенная форма гипофосфатемии, с частотой встречаемости около1 : 20000-25000. Заболевание имеет Х-сцепленный доминантный тип наследования. Характеризуется полной пенетрантностью для гипофосфатемии и неполной — для костных изменений. Женщины передают патологический признак дочерям и сыновьям c вероятностью 50%, мужчины — только дочерям с вероятностью 100%. У мальчиков болезнь протекает тяжелее, чем у девочек. мутации в гене PHEX через гипотетический фосфатурический гормон приводят к почечной потере фосфата и нарушениям обмена витамина D
Синдром Рубинштейна-Тейби
Синдром Рубинштейна-Тейби - генетически гетерогенное (по последним данным) наследственное заболевание, характеризующееся поражением центральной нервной системы, деформациями костей скелета и рядом других пороков развития. Симптомами этого состояния являются прогрессирующая умственная отсталость, низкий рост, расширение фаланг пальцев, полидактилия на ногах, разнообразные нарушения со стороны внутренних органов. Диагностика синдрома Рубинштейна-Тейби производится на основании данных настоящего статуса пациента, молекулярно-генетических анализов и других исследований. Специфического лечения данной патологии не существует, применяют симптоматическую терапию в зависимости от типа пороков и нарушений.
Общие сведения
Синдром Рубинштейна-Тейби - генетическое заболевание, которое сопровождается нарушениями интеллектуального и физического развития, а также разнообразными пороками скелета и внутренних органов. Впервые данная патология была выявлена в 1963 году и описана американскими педиатрами Дж. Рубинштейном и Г. Тейби - сами исследователи назвали заболевание «синдромом широкого первого пальца кистей и стоп, специфического лица и умственной отсталости». Дальнейшие исследования в области генетики подтвердили, что синдром Рубинштейна-Тейби является аутосомно-доминантной патологией, но подавляющее большинство случаев возникает вследствие спонтанных мутаций различного типа. Встречаемость составляет порядка 1:100 000-125 000, мальчики и девочки поражаются примерно с одинаковой частотой. Особенностью синдрома Рубинштейна-Тейби является повышенный риск развития различных онкологических заболеваний - главным образом, некоторых типов лейкозов, опухолей мозговых оболочек и нервных тканей.
Причины синдрома Рубинштейна-Тейби
Синдром Рубинштейна-Тейби обладает выраженной генетической гетерогенностью, однако различные типы этого заболевания не имеют каких-либо особенностей клинического течения. Наиболее часто причиной этой патологии выступают повреждения гена CREBBP, который расположен на 16-й хромосоме. Он кодирует так называемый CREB-связывающий белок, являющийся важным транскрипционным фактором, контролирующим экспрессию огромного количества генов. Появление в гене CREBBP различных изменений (точечных мутаций, транслокаций, микроделеций в 16-й хромосоме) приводит либо к образованию дефектного белка, неспособного выполнять свои функции, либо к полной блокировке его выделения. Это становится причиной развития синдрома Рубинштейна-Тейби.
В последние годы молекулярно-генетические исследования данной патологии подтвердили, что в ее развитии играют роль и мутации гена EP300, который располагается на 22-й хромосоме. Продуктом экспрессии данного гена является протеин p300, который, как и CREB-связывающий белок, относится к группе транскрипционных факторов. По данным исследований, идентифицировать характер генетического дефекта при синдроме Рубинштейна-Тейби удается в 55-60% случаев, примерно у половины пациентов обнаруживаются мутации гена CREBBP, еще у 40-45% выявляются делеции и другие хромосомные перестройки, затрагивающую 16-ю хромосому, и лишь у 3% больных наблюдаются мутации гена EP300. Все это указывает на возможность участия других генов в развитии этого заболевания.
Дефект белка-фактора транскрипции или его малое выделение (по причине гаплонедостаточности) приводит к многочисленным нарушениям, обусловленным недостаточной стимуляцией ряда других генов. Это можно заметить по характерной клинической картине синдрома Рубинштейна-Тейби. Оба транскрипционных фактора влияют на активность образования новых связей между нейронами, формирование долговременной памяти, стимуляцию иммунного ответа, развитие репродуктивной системы. Недостаточность этих белков ведет к целому каскаду патологических процессов в организме, что и составляет клинику синдрома Рубинштейна-Тейби. Кроме того, вышеуказанные транскрипционные факторы также влияют на активность антионкогенов, поэтому при их дефекте значительно возрастает риск развития злокачественных новообразований.
Симптомы синдрома Рубинштейна-Тейби
Симптоматика синдрома Рубинштейна-Тейби характеризуется значительным разнообразием у разных больных, что отражается на тяжести течения заболевания и его прогнозе. Обычно уже при рождении можно обнаружить некоторые признаки этой патологии - деформации лица и черепа (микро- или брахицефалия, расширение переносицы, эпикант, клювовидный нос), высокое арковидное нёбо, изменение формы и положения ушных раковин, расширенные фаланги пальцев, особенно первых. На ногах у больных синдромом Рубинштейна-Тейби нередко выявляется полидактилия. Иногда уже при рождении определяются признаки пороков развития внутренних органов - бледность или цианоз (при поражении легких или сердца), длительная желтуха новорожденных, крипторхизм.
При дальнейшем течении синдрома Рубинштейна-Тейби отмечается прогрессирующее отставание ребенка в физическом и умственном развитии от здоровых сверстников, затрудненное развитие речи и моторных навыков. Также наблюдается косоглазие, миопия. Выражение лица больных характеризуется гримасой, напоминающей улыбку, возникает гирсутизм, примерно у половины пациентов определяется наличие красного невуса в области лба, затылка или шеи. В старшем возрасте у больных синдромом Рубинштейна-Тейби выявляются искривления позвоночного столба различного характера и низкий рост (не более 150-160 сантиметров). Могут определяться деформации грудной клетки и (реже) другие скелетные аномалии, у мальчиков в большинстве случаев отмечается крипторхизм.
Постоянным признаком синдрома Рубинштейна-Тейби является умственная отсталость. Как правило, диагностируется ЗПР, олигофрения, значительная задержка речевого развития по сравнению со здоровыми сверстниками. Отмечаются нарушения концентрации внимания, больные легко отвлекаются на посторонние раздражители при выполнении какого-либо задания, возможны резкие перепады настроения. При этом лица с синдромом Рубинштейна-Тейби хорошо идут на контакт, легко социализируются. Среди других неврологических симптомов заболевания нередко обнаруживается плохая координация движений, изредка наблюдаются судорожные приступы.
Синдром Рубинштейна-Тейби может также проявляться различными нарушениями со стороны внутренних органов - головного мозга, сердца, почек и мочевыделительных путей, терминальных отделов пищеварительной системы. Кроме того, у таких больных значительно повышается риск возникновения онкологических заболеваний, в основном развивающихся в раннем возрасте. К ним относят различные формы лейкозов, меланому, некоторые типы лимфом. Поэтому до пубертатного периода больные синдромом Рубинштейна-Тейби должны периодически проходить обследования у онколога для ранней диагностики злокачественных новообразований.
Диагностика и лечение синдрома Рубинштейна-Тейби
Для выявления синдрома Рубинштейна-Тейби используют данные общего осмотра больного, рентгенологических исследований и молекулярно-генетических анализов, вспомогательную роль могут играть УЗИ и МРТ. При осмотре определяются характерные для этого заболевания аномалии развития лица (изменение формы и размеров черепа и носа, антимонголоидный разрез глаз), укорочение и расширение фаланг больших пальцев на руках и ногах, иногда - полидактилия. У взрослых больных синдромом Рубинштейна-Тейби также выявляют уменьшение роста, глубокую умственную отсталость, искривления позвоночника. На рентгенограммах можно увидеть изменения костей фаланг, позвоночника и грудной клетки, костный возраст несколько отстает от фактического.
Молекулярно-генетическая диагностика синдрома Рубинштейна-Тейби выполняется врачом-генетиком, который может использовать множество методов для определения этого заболевания. Точечные мутации в генах CREBBP и ЕР300 выявляются посредством прямого автоматического секвенирования кодирующей последовательности. Методика FISH используется в том случае, когда подозревается наличие делеций или транслокаций на 16-й хромосоме, также приводящих к развитию синдрома Рубинштейна-Тейби. Так как на основании фенотипических данных крайне сложно определить возможный тип генетического дефекта, в рамках диагностики этого заболевания нередко приходится применять сразу несколько техник современной молекулярной генетики.
Вспомогательные методы диагностики позволяют выявить сопутствующие нарушения внутренних органов при синдроме Рубинштейна-Тейби. К таким методам относят ультразвуковые исследования (УЗИ почек и мочевыделительных путей, ЭхоКГ), электрокардиографию, магнитно-резонансную томографию и другие методики. Нередко у больных синдромом Рубинштейна-Тейби диагностируют врожденные пороки сердца (открытый Боталлов проток, дефект межжелудочковой перегородки), аритмии, аномалии почек (удвоение, гипоплазия) и мочевыделительных путей (атрезии на различных участках). Также при этом заболевании нарушается формирование мозолистого тела головного мозга, что определяется при помощи магнитно-резонансной томографии. У части больных синдромом Рубинштейна-Тейби отмечаются пороки развития легких и пищеварительной системы.
Специфического лечения данной патологии на сегодняшний момент не существует, все терапевтические мероприятия сводятся к облегчению симптомов, коррекции аномалий и пороков развития, угрожающих жизни больных. Назначаются ноотропные средства, препараты кальция и витамина Д, для уменьшения выраженности умственной отсталости рекомендуют специализированную психологическую помощь. В ряде случаев при синдроме Рубинштейна-Тейби применяют хирургические методы лечения - для устранения пороков сердца, аномалий развития прямой кишки и мочевыделительных путей, крипторхизма. Также может потребоваться лечение у офтальмолога, а при признаках развития злокачественного новообразования - консультация и лечение у онколога.
Прогноз и профилактика синдрома Рубинштейна-Тейби
Согласно мнению большинства специалистов, прогноз синдрома Рубинштейна-Тейби чаще всего неопределенный или неблагоприятный. Это связано с тем, что данное заболевание характеризуется значительным спектром разнообразных нарушений - от относительно безопасных для жизни (легкие скелетные аномалии, умственная неполноценность) до ярко выраженных, способных привести к смерти в раннем возрасте (тяжелые пороки развития сердца, легких, почек). Кроме того, значительно повышен риск развития онкологических патологий. Поэтому прогноз синдрома Рубинштейна-Тейби составляется строго индивидуально, исходя из конкретной клинической картины и общего состояния больного. Каких-либо специфических методов профилактики этого заболевания из-за его частого спонтанного развития на сегодняшний день не разработано.
Рубинштейна Тейби

синдром Рубинштейна - Тейби
Как поставить диагноз, какие обследования провести, какие бывают частые ошибки при ведении пациентов с наследственной патологией?
Начнём с разбора клинического примера.
Информация на сайте размещена по согласованию с родителями ребенка.
К неврологу - эпилептологу обратилась мама с ребенком в возрасте 11 лет 7 месяцев с просьбой дать разрешение на посещение бассейна. Семья прибыла из другого региона несколько месяцев назад.
При подробном опросе выяснились жалобы и анамнез.
- С рождения специфический фенотип , свойственный генетической патологии: антимонголоидный разрез глаз, гипертелоризм, макростомия, широкий I палец кистей, булавовидные широкие терминальные и проксимальные фаланги пальцев кистей и стоп.
- Задержка психоречевого развития . Говорить первые слова начал после 1 года, словарный запас к 5 годам составлял 20 слов; фразовой речи не было; после 5 лет отмечалось регресс в речи. К настоящему времени говорит 5 слов, общается жестами, речь понимает на бытовом уровне частично. Посещал детский сад. В настоящее время учится в школе по программе 8 вида, читать и считать не умеет, буквы знает не все. Посещает реабилитационный центр, где занимается с логопедом и психологом.
- Задержка моторного развития : голову начал удерживать с 2 месяцев, сидеть самостоятельно с 9 месяцев, самостоятельно начал ходить с 1 года 4 месяцев.
- Стереотипии - открывает-закрывает рот, раскачивается, кружится вокруг себя, крутит волосы.
- Особенности поведения - в глаза почти не смотрит, отстранен, в контакт вступает избирательно.
- Шаткая, неустойчивая походка ; ходит медленно, широко расставив ноги.
- Ожирение 2-3 степени.
- Отставание в физическом развитии - низкий рост .
- Частые дневные мочеиспускания. Энурез каждую ночь.
- В 8 месяцев во время нахождения в стационаре по поводу острого пиелонефрита с подъёмом температуры до 37,8 С, отмечался единичный тонико-клонический судорожный приступ в течение 3 минут. Был поставлен диагноз: Эписиндром. Назначен депакин-хроно, который пациент получал в течение 4 лет. Приступов больше никогда не было. По ЭЭГ эпиактивности не было.
- Были случаи падений, запинания при ходьбе в детском саду, которые были расценены как приступы.
Напомним, что у ребенка атаксия, проявляется шаткой походкой, медлительностью, неуклюжестью; а также ожирение, а значит «грузность». Эти приступы не были эпилептическими, а это результат моторной неловкости.
- Наблюдается у психиатра и невролога . Регулярно 2-3 раза в год проводят курсы лечения ноотропами (используют пантогам, кортексин, актовегин, церебрализин, глиатилин, глицин), без явного эффекта от лечения. Оформлена инвалидность у психиатра. С целью реабилитации планировали посещать бассейн, но из бассейна запросили справку от эпилептолога для разрешения посещения бассейна, ссылаясь на диагноз: Эписиндром.
Ребенок рожден от молодых здоровых родителей. Наследственность не отягощена. Беременность протекала с легким гестозом, отёчным вариантом; с угрозой самопроизвольного выкидыша. Роды срочные, самопроизвольные. Масса при рождении 3050 мг, рост 50 см, 8/9 баллов по Апгар.
У родителей есть 4-летний второй здоровый ребенок.
В контакт вступает избирательно. Интеллект снижен. В речи - отдельные звуки, слоги. Команды понимает частично, выполняет избирательно - даёт себя осмотреть, открывает рот в ответ на просьбу, подаёт ручку, ходит.
Частые, каждые 5 минут, стереотипии в виде открывания-закрывания рта, отведения глаз и лица в сторону, усиливающиеся при волнении.
Дисморфии. Череп микроцефальный, окружность головы 51 см. В двигательной сфере: силовых парезов нет. Мышечный тонус диффузно умеренно снижен. Сухожильные рефлексы живые, равные. Координация: легкая атаксия в виде моторной неловкости, неустойчивой походки. Ожирение 2-3 степени.
Консультация генетика (август 2013 года). Заключение: Моногенная патология. Синдром Рубинштейна - Тейби. Аутосомно-доминантный тип наследования. Прогноз по заболеванию - без выздоровления. Рекомендовано: наблюдение психиатра, невролога, генетика; симптоматическая терапия. Направление на МСЭК.
ЭЭГ (02.2013 год) - Диффузные изменения биоэлектрической активности головного мозга. МПА и эпиактивности нет.
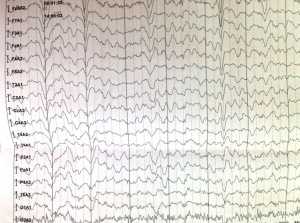
ЭЭГ при синдроме Рубинштейна - Тейби
Электромиография (08.2013 год) - Данных за поражение мотонейронов спинного мозга не получено. Нейропатия n. tibialis с двух сторон в форме миелинопатии.
Заключение
Нами были выявлены следующие симптомы:
врожденный специфический фенотип: антимонголоидный разрез глаз, гипертелоризм, микростомия, широкий I палец кистей, булавовидные широкие терминальные и проксимальные фаланги пальцев кистей и стоп;
умственная отсталость с регрессом речевых навыков;
В анамнезе в 8 месяцев единичного приступа судорог на фоне течения острого пиелонефрита с субфебрильной температурой; при отсутствии эпиактивности на ЭЭГ.
Тип течение заболевания - медленно прогрессирующее; первые проявления с рождения .
Подтверждена генетиками моногенная патология, синдром Рубинштейна - Тейби.
Можно поставить неврологический диагноз:
Энцефалопатия как проявление генетической патологии - синдрома Рубинштейна - Тейби в форме когнитивных нарушений, аутистического симптомокомплекса, атактического синдрома, миатонического синдрома, энуреза.
Фебрильный приступ в анамнезе. Данных за активную эпилепсию в настоящее время нет.
Сопутствующий диагноз: Ожирение 2-3 степени. Отставание в росте.
Даны рекомендации:
Контроль ЭЭГ 1 раз в год.
Симптоматическая терапия. В настоящее время в лечении не нуждается.
Рациональное питание с учетом рекомендаций эндокринолога.
Занятия с ребенком. Логопедические занятия. Занятия с психологом.
ЛФК. Адаптивная физкультура (принципы занятий адаптивной физкультурой и спортом описаны в статье Паралимпиада в Сочи 2014).
Осмотр через 1 год с результатами ЭЭГ.
Бассейн посещать не рационально в связи с выраженными стереотипиями (открывает-закрывает рот), поведенческими (аутистическими) расстройствами из-за риска несчастного случая. Рекомендованы занятия в «сухом» бассейне.
Была проведена беседа с мамой о прогнозе, режиме, тактике лечения (нет оснований назначать ноотропы, лечение только симптоматическое) и наблюдения. Задача: улучшить качество полноценной жизни в сложившихся обстоятельствах; заниматься адаптивной физической культурой, социальная реабилитация.
Сведения из литературы.
«Наследственные синдромы по Дэвиду Смиту» и другие.
Описывают характерные черты синдрома Рубинштейна - Тейби:
синдром широкого I пальца конечностей и терминальных фаланг пальцев кистей и стоп; специфическое лицо в виде антимонголоидного разреза глаз и макростомии; умственная отсталость, отставание в росте.
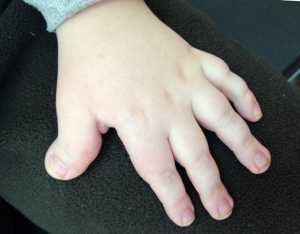
кисть при синдроме Рубинштейна - Тейби
Этиология
Большинство случаев синдрома Рубинштейна - Тейби спорадические, вызванные спонтанной точечной мутацией или делецией в локусе 16p13.3 хромосомы. Ген CCREBBP этой хромосомы кодирует CREB- связывающий белок. Этот белок ядра клетки регулирует проявляемость гена через цАМФ.
Клиническая картина Синдрома Рубинштейна - Тейби
Физическое развитие
Описаны задержка роста с первых лет жизни, отставание костного возраста (75-94%). В среднем мужчины достигают роста и веса 153 см, а женщины 147 см.
Психомоторное развитие
Все пациенты (100%) с умственной отсталостью, чаще в тяжелой степени; 90% случаев у детей задержано моторное и речевое развитие.
Неврологические синдромы
У 85% - нарушение координации в виде атаксической походки, у 67% - мышечная гипотония, у 40% - гиперрефлексия.
У 57% - есть изменения по ЭЭГ, и у 23% развивается симптоматическая эпилепсия.
Дисморфии черепно-лицевой области
Микроцефалия (35%), брахиоцефалия, позднее закрытие большого родничка (24%), выпуклый лоб (33%).
Антимонголоидный разрез глаз (93 %), длинные ресницы (87%), дакриоцистит новорожденных (43%), гипертеллоризм (71 %), эпикант (62 %), птоз (36%), густые (76%) и приподнятые изогнутые брови (73%), косоглазие (75%), аномалии рефракции (58%), энофтальм (22%) или экзофтальм, широкую спинку носа, большой нос с опущенным кончиком (клювовидный) (90 %), гипоплазия крыльев носа (72 %), макростомия (56%), микрогнания (49%), гримасу, напоминающую улыбку, «готическое» (узкое и высокое) нёбо (93 %), низкий рост волос на лобной (24%) и затылочной области (42%), низкопосаженные деформированные уши (81%), аномалия зубов в виде скученности.
Аномалии кистей и стоп
Расширение, укорочение и уплощение проксимальных и дистальных фаланг первых пальцев кистей и стоп (100 %), чаще всех пальцев кисти (87 %); X-образная деформация межфаланговых суставов; удвоение конечной фаланги первых или пятых пальцев стоп (30 %), полидактилия или синдактилия; высокие подушечки пальцев (31%); плоскостопие (72%).
Аномалии скелета
Варианты нарушений осанки: усиление кифоза (37%), гиперлордоз, сколиоз(42%), аномалии ребер и грудины, воронкообразная грудная клетка, гипоплазия и разворот наружу тазовых костей (26%).
Аномалии со стороны мочеполовой системы
Крипторхизм (78%); различные пороки развития почек (52%): односторонняя гипоплазия почки, удвоение почек, расширение мочеточников, гидронефроз.
Врожденные пороки сердца
Различные врожденные пороки сердца (30%), чаще открытый аортальный проток, дефект межжелудочковой перегородки.
Аномалии на коже
Гипертрихоз (75%), гемангиомы (25%); ярко-красный невус в области лба, затылка, шеи; склонность к образованию келоидных рубцов (22%), «кофейные» пятна на коже.
Среди прочих симптомов встречаются:
Патологическая синкинезия, стереотипии; агенезия мозолистого тела; катаракта, глаукома, калабома; вывих надколенника, вывих головки лучевой кости, остеохондропатия головки бедренной кости; болезнь Гиршспрунга; двурогая матка, раннее телархе; искривление полового члена, гипоспадия; нарушение строения легких.
Течение и прогноз
В раннем возрасте затруднения при кормлении, склонность к запорам (40-70%), частые инфекции дыхательных путей; возможен желудочно-пищеводный рефлюкс, иногда требующий оперативного лечения. При аномалии дыхательных путей возможны аспирации. Частые отиты приводят к тугоухости. Часты вросшие ногти и паранихии.
Задержка психомоторного развития.
Средние временные показатели: сидеть с 11 месяцев, ползать с 1г 3 месяцев, ходить с 2,5 лет, говорить первые слова с 2 лет 1 месяца. К 7 годам могут научиться читать (67%), словарный запас чаще остается ограничен и соответствует 7 годам. Походка неустойчивая, неловкость при движениях.
Детям показаны логопедические занятия, ортопедическая коррекция, лечебная физкультура и массаж.
Изучив особенности течения болезни, разбирая клинический пример Синдрома Рубинштейна-Тейби, мы можем сделать несколько выводов:
1. Описаны характерные черты синдрома Рубинштейна - Тейби : синдром широкого I пальца конечностей и терминальных фаланг пальцев кистей и стоп; специфическое лицо в виде антимонголоидного разреза глаз и макростомии; умственная отсталость; отставание в росте.
2. Для подтверждения диагноза: синдрома Рубинштейна - Тейби требуется генетическое обследование. Учеными в ыявлена мутации в локусе 16p13.3 хромосомы.
3. Синдром был описан Рубинштейном и Тейби в 1963 году. Встречается в популяции 1 на 25-30 тысяч, с равной вероятностью среди мужчин и женщин. Аутосомно-доминантный тип наследования.
4. Данный клинический случай отражает самые частые заблуждения и ошибки при лечении пациентов с синдромом Рубинштейна - Тейби.
4.1. Учитывая тот факт, что механизмы биохимического осуществления синдрома Рубинштейна-Тейби в настоящее время не изучены, представляется неоправданной интенсивная терапия случайно выбранными ноотропами . Это может привести к усилению стереотипий и спровоцировать эпилептические припадки (симптоматическая эпилепсия встречается у 23% детей с синдромом Рубинштейна - Тейби).

стереотипии при синдроме Рубинштейна - Тейби
4.2. Единичный фебрильный приступ был расценен как «эписиндром». Диагноз эписиндром некорректный.
Варианты диагноза могут быть:
А. Судорожный синдром при каких-либо острых состояниях (кровоизлияние в головной мозг, нейроинфекция и прочее).
В. Симптоматическая эпилепсия.
В данном случае не было оснований для постановки диагноза эпилепсия , так как единичный приступ был спровоцирован течением острого пиелонефрита, сопровождающегося субфебрильной температурой. По ЭЭГ не выявлено наличия эпилептиформной активности.
В длительном приеме , в течение 4 лет, противоэпилептичекого препарата (вальпроевой кислоты) показаний не было.
5. Основные направления в лечении пациентов с синдромом Рубинштейна - Тейби:
5.1. Профилактика возможных осложнений (запоров, аспираций, инфекций, травм).
5.2. Симптоматическая терапия (например, при гипертермии - жаропонижающие).
5.3. Рациональное питание как у здорового человека с учетом возраста или с учетом рекомендаций эндокринолога.
5.4. Достаточная двигательная активность. Занятия адаптивной физической культурой.
5.5. Логопедические занятия. Обучение по коррекционной программе.
5.6. Посещение центра реабилитации. Участие в программах для людей с ограниченными возможностями.
Это позволит немного расширить эти ограниченные возможности пациентов с наследственной патологией.
В написании статьи принял участие эксперт по наследственной неврологической патологии к.м.н. Корень Олег Леонидович.
Синдромы врожденных аномалий, вовлекающих преимущественно конечности
Термин «синдром» используется для обозначения устойчивых сочетаний патологических симптомов, как уточненной этиологии, так и этиологически неясных. В клинической генетике и тератологии термин «синдром» имеет несколько значений. Он часто применяется как синоним «болезни», когда речь идет о единой нозологической форме с определенной этиологией. При этом синдромом чаще называют врожденные нарушения.
Протокол "Синдромы врожденных аномалий, вовлекающих преимущественно конечности"
Код по МКБ:
- Отсуствия (недорозвития) ногтей, надколенника
- Сиреномиелии (сращение нижних конечностей)
- Тромбоцитопения с отсутствием лучевой кости (TAR-синдром)
Q87.4 синдром Марфана

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
1. Умственная отсталость.
2. Легкая умственная отсталость.
3. Умеренная умственная отсталость.
4. Тяжелая умственная отсталость.
5. Глубокая умственная отсталость.
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, расторможенность, врожденные пороки развития конечностей.
Физикальное обследование
Синдром Холта-Орама (фенотипические проявления) - пороки развития верхних конечностей варьируют от изменений I пальца кисти (гипоплазия, трехфаланговый I палец) и гипоплазия лучевой кости (50%). Левая рука поражается чаще. У 50% больных I палец не противопоставлен остальным пальцам кисти. Из скелетных изменений наблюдаются также гипоплазия лопаток и ключиц, сколиоз, воронкообразная деформация грудины, клинодактилия, синдактилия, гипоплазия или аплазия других пальцев кисти и костей запястья (40%). В 85% случаев синдрома обнаруживаются врожденные пороки сердца: дефекты межпредсердной перегородки (наиболее часто), дефект межжелудочковой перегородки (50%), открытый артериальный проток, коарктация аорты, стеноз легочной артерии, пролапс митрального клапана. В редких случаях описывают гипертелоризм, отсутствие большой грудной мышцы, расщелину неба.
Синдром Клиппеля-Треноне-Вебера (синонимы: гемангиэктазия гипертрофическая; невус остеогипертрофический варикозный) - фенотипические проявления: типичным признаком синдрома является гипертрофия одной или нескольких конечностей, что связано с врожденным пороком развития артерио-венозной системы и лимфатических сосудов. Отмечается гиперплазия мягких тканей и кости пораженной конечности, трофические язвы и отеки. Гемангиомы бывают единичные или множественные, капиллярные, кавернозные, кистозные, различающиеся по размерам и локализации (любая часть тела, но чаще всего на голени, ягодицах, животе, нижней части туловища), преобладает одностороннее поражение. Описаны гиперпигментация кожи, синдактилия, полидактилия. Челюстно-лицевые аномалии включают асимметричную гипертрофию лица, микроцефалию, макроцефалию. Наблюдаются глаукома, катаракта, колобома.
Синдром Рубинштейна-Тейби (фенотипические проявления), синонимы: синдром широкого I пальца кисти и стопы с лицевыми аномалиями. Клиническая картина характеризуются умственной отсталостью, задержкой роста и специфическими особенностями лица и тела, из которых основные - короткий и широкий I палец кисти и стопы, своеобразное лицо с длинным загнутым носом, антимонголоидным разрезом глаз, гипертелоризмом, недоразвитием верхней челюсти, брахимикроцефальной структурой черепа. Отмечается низкий рост волос на лбу, иногда линия роста волос спускается в центре углом. У больных, как правило, высокое небо, встречается его расщелина. Часто имеются синдактилия и полидактилия стоп, реже кистей, косолапость, врожденный вывих бедра. Задержка в росте в основном постнатальная.
Тромбоцитопения с отсутствием лучевой кости (TAR-синдром). Тромбоцитопения отмечается в 100% случаев и появляется в первые 4 мес. жизни, но с возрастом число тромбоцитов повышается до нормального уровня. Тромбоцитопенические кризы могут провоцироваться стрессом, инфекциями и хирургическими вмешательствами. Нарушена агрегация тромбоцитов и уменьшено время их жизни. Мегакарициты отсутствуют в 66% случаев, в 12% случаев их количество снижено. В 60-70% случаев заболевания отмечается лейкемоидная реакция на 1-м году жизни, особенно в период кровотечений; в это время нарастает тромбоцитопения и появляется гепатоспленомегалия. Основной скелетной аномалией является двустороннее отсутствие или гипоплазия лучевой кисти (100%), часто в сочетании с аномалиями кисти (ограничения разгибания, радиальная девиация, гипоплазия карпальных костей и фаланг при обязательной сохранности I пальца), укорочением и деформацией локтевой кости, отсутствием ее (в 20% случаев - двусторонним, 8% - односторонним), аномалией плеча (50%).
Синдром Марфана - для больных типичны высокий рост и длинные тонкие конечности, особенно дистальные отделы. Часто отмечаются сколиоз (60%), кифоз, воронкообразная или килевидная грудная клетка, долихоцефалия, узкое лицо, высокое дугообразное небо, плоскостопие. Поражения глаз включают двусторонний подвывих хрусталика (75%), часто сочетающийся с иридодонезом (дрожание радужки). Отмечаются высокая степень миопии, отслойка сетчатки, мегалокорнеа и голубые склеры.
Ассоциация VATER - основные проявления включают аномалии позвоночника (полупозвонки, кифосколиоз, менингоцеле) (82%), дефекты перегородки и другие пороки сердца (50%), атрезию ануса (70%), трахеопищеводный свищ с атрезией пищевода (78%), дисплазию лучевой кости, преаксиальную полидактилию и синдактилию (65%), аномалию почек.
Лабораторные исследования: общий анализ крови и мочи.
Инструментальные исследования:
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза.
Читайте также: