Синдром Савилла (Savill) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Баршоня — Тешендорфа синдром (Th. Barsony, 1887-1942, венгерский рентгенолог; W. Teschendorf, немецкий рентгенолог, XX в.; синонимы: дивертикулы пищевода множественные ложные, дивертикулы пищевода множественные функциональные, пищевод извитой, пищевод четкообразный, пищевод штопорообразный) — болезнь пищевода неизвестной этиологии, характеризующаяся спастическим сокращением его циркулярных мышц, что придает пищеводу четкообразный вид на рентгенограмме; клинически проявляется непостоянной дисфагией и загрудинными болями.
Бойса симптом (Boys) — появление булькающего звука или урчания при надавливании на боковую поверхность шеи; признак большого дивертикула шейного отдела пищевода.
Бушара болезнь (Ch.J. Bouchard, 1837-1915, французский патолог) — гастрэктазия (расширение полости желудка с растяжением его стенок при стенозе привратниковой части или двенадцатиперстной кишки).
Бэррета язва пищевода (N.R. Barret, род. в 1903 г., английский хирург) — язва пищевода, по клинико-морфологическим признакам напоминающая пептическую язву желудка или двенадцатиперстной кишки.
Кушинга эзофагит (Н.W. Cushing, 1869-1939, американский нейрохирург) — острый эзофагит, иногда развивающийся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кушинга язва (Н.W. Cushing) — язва желудка или двенадцатиперстной кишки, иногда развивающаяся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кэмерона синдром (A.J. Cameron) — язвенные или эрозивные повреждения слизистой оболочки грыжевого выпячивания при грыжах пищеводного отверстия диафрагмы, которые сопровождаются хроническим оккультным или явным кровотечением и железодефицитной анемией.
Ларрея грыжа (D.J. Larrey, 1766-1842, французский хирург). Левосторонняя парастернальная диафрагмальная грыжа, выходящая в средостение через грудино-реберный треугольник.
Лихтенштерна симптом (Lichtenstern, синоним дисфагия парадоксальная, d. paradoxalis) — дисфагия, при которой большие порции пищи легче проходят в желудок, чем малые; возможный признак грыжи пищеводного отверстия диафрагмы.
Лиана — Сигье — Вельти синдром (С.С. Lian, 1882-1969, французский врач; F. Siguier, 1909-1972, французский врач; Н.L. Welti, 1895-1970, французский хирург) — сочетание диафрагмальной грыжи, часто осложненной рефлюкс-эзофагитом, с повторными тромбозами и тромбофлебитами сосудов конечностей и нередко с гипохромной анемией; патогенез неясен.
Монтандона синдром (А. Montandon, швейцарский врач, XX век) — приступы длительной дисфагии вследствие миогенного стеноза шейной части пищевода (области пищеводноглоточного соединения), сопровождающиеся попаданием жидкой пищи в гортань, аспирации и приводящие к нарастающему истощению.
Морганьи грыжа (G.B. Morgagni, 1682-1771, итальянский анатом). Правосторонняя парастернальная грыжа.
Харрингтона грыжа (S. W. Harrington, 1889-1975, американский хирург). Антральная грыжа пищеводного отверстия диафрагмы (типы 1 и 2).
Ценкера дивертикул (F.A. Zenker, 1825-1898, немецкий патолог; синоним ценкеровский дивертикул) — мешковидный дивертикул глоточного конца пищевода, образующийся сначала на его задней стенке, а затем распространяющийся и на боковую. В начале заболевания — глоточные парестезии, перемежающаяся дисфагия, сухой кашель. С увеличением дивертикула в нем происходит задержка пищи, урчание при приеме воды. Мешок сдавливает пищевод, усиливается дисфагия, часты аспирационные пневмонии.
Эпонимные симптомы/синдромы, связанные с патологией кишечника
Алапи симптом (Alapy). Отсутствие или незначительное напряжение брюшной стенки при инвагинации кишечника.
Альвареса синдром (W.C. Alvarez, 1884-1978, американский врач, описан в 1949 г.; синонимы: псевдоилеус; истерическое вздутие живота). Преходящее вздутие живота нейрогенной природы. Чаще наблюдается у истеричных или психопатичных женщин.
Бувре признак (1) (L. Bouveret, 1850-1929, французский врач) — выпячивание в области проекции на переднюю брюшную стенку места перехода подвздошной кишки в слепую, наблюдаемое при непроходимости толстой кишки.
Бувре признак (2) (L. Bouveret; синоним Куссмауля симптом, Adolf Kußmaul, 1822-1902, немецкий терапевт) — периодическое выбухание брюшной стенки в надчревной области и левом подреберье при сужении привратника, обусловленное усиленной перистальтикой желудка.
Вербрайка синдром (J.R. Verbryke, род. в 1885 г., американский врач; синоним синдром печеночного перегиба ободочной кишки) — боль и чувство натяжения в подложечной области в сочетании с диспептическими явлениями при наличии сращений желчного пузыря с печеночным углом ободочной кишки.
Вильмса симптом падающей капли (М. Wilms, 1867-1918, немецкий хирург и онколог) — звук падающей капли жидкости, определяющийся аускультативно на фоне шумов перистальтики при непроходимости кишечника.
Гарднера синдром (Е.J. Gardner, 1909-1989, американский врач-генетик) — наследственная болезнь, характеризующаяся множественным полипозом прямой и ободочной кишок в сочетании с доброкачественными опухолями, чаще костей и кожи (остеомы, фибромы, липомы); наследуется по аутосомно-доминантному типу.
Гейбнер-гертеровская болезнь (O.J.L. Heubner, 1843-1926, немецкий педиатр; С.A. Herter, 1865-1910, американский врач и фармаколог; синоним Ги — Гертера — Гейбнера болезнь) — целиакия.
Данса симптом (J. В. H. Dance, 1797-1832, французский врач) — западение брюшной стенки в правой подвздошной области при илеоцекальной инвагинации.
Жанбона синдром (М. Janbon, современный французский терапевт; синоним syndromus choleriformis, enteritis choleriformis). Описан как гастроинтестинальная симптоматика после лечения окситетрациклином. Патогенез заключается в уничтожении антибиотиками физиологической бактериальной флоры кишечника, что способствует развитию патогенной, устойчивой к антибиотикам.
Ирасека — Цюльцера — Уилсона синдром (A. Jirasek, 1880-1960, чехословацкий хирург; W.W. Zuelzer, 1909-1987, американский педиатр; J.L. Wilson, американский педиатр) — аганглиоз толстой кишки врожденный сегментарный. Патоморфологическая форма болезни Гиршспрунга, при которой в толстой кишке имеются одна или две аганглионарные зоны с нормальным участком кишки между ними; при этом аганглиоз не распространяется на прямую кишку.
Кантора симптом (М.О. Cantor, род. в 1907 г., американский хирург) — нитевидные тени в дефектах наполнения кишки; рентгенологический признак колита и регионарного илеита.
Карно симптом (M.O. Carnot) — боль в эпигастральной области, возникающая при резком разгибании туловища. Бывает при спаечной болезни.
Кенига синдром (F. Konig, 1832-1910, немецкий хирург) — сочетание приступов коликообразных болей в животе, метеоризма, чередования запоров и поносов, урчания при пальпации правой подвздошной ямки, наблюдающееся при хронической непроходимости кишечника в области перехода подвздошной кишки в слепую.
Клойбера симптом (Н. Kloiber, немецкий рентгенолог; синоним Клойбера чаши) — наличие на рентгенограмме живота (при вертикальном положении больного) теней, напоминающих чаши с жидкостью; признак скопления жидкости и газа в кишечнике при его непроходимости.
Кронкхайта — Канада синдром (L.М. Cronkhite, род. в 1919 г., американский врач; W.J. Canada, современный американский рентгенолог) — облысение, атрофия ногтей и гиперпигментация кожи при семейном полипозе органов пищеварительного тракта.
Кюсса синдром (G.Е. Kuss, 1877-1967, французский хирург) — хроническая частичная непроходимость кишечника, обусловленная наличием спаек в области малого таза, например при хроническом сальпингите.
Лобри — Сулля синдром (Ch. Laubry, 1872-1941, французский врач; P.L.J. Soulle, 1903-1960, франц. врач) — сочетание избыточного содержания газа в желудке, метеоризма и высокого стояния правого купола диафрагмы при ишемической болезни сердца, обусловленное рефлекторной гипокинезией пищеварительного тракта.
Мачеллы — Дворкена — Биля симптом (T.E. Machella, F.J. Dworken, H.J. Biel; 1952). Синоним: симптом селезеночного угла. Сильная боль в левой половине живота, вызванная растяжением газами селезеночного угла толстой кишки. Облегчение наступает после опорожнения кишечника и отхождения газов. См. Пайра болезнь, синдром.
Матье симптом (A. Mathieu, 1855-1917, французский врач) — шум плеска в пупочной области при толчкообразной пальпации; признак непроходимости кишечника.
Меккеля дивертикул (J.F. Meckel junior, 1781-1833, немецкий анатом). Врожденный дивертикул подвздошной кишки. Правило двоек: 2 дюйма длиной, в 2 футах от илеоцекального клапана (баугиниевой заслонки), у 2% населения.
Образцова признак (В.П. Образцов, 1849-1920, отечественный терапевт) — шум плеска при пальпации слепой кишки; признак хронического колита.
Обуховской больницы симптом — баллонообразное расширение ампулы прямой кишки, определяемое пальцевым исследованием; признак заворота сигмовидной кишки.
Пайра болезнь, синдром (Erwin Payr, австрогерманский хирург, 1871-1946; синонимы: синдром селезеночного изгиба, flexura lienalis syndrome, splenic flexure syndrome). Первично описана как запоры, обусловленные перегибом и сращением поперечной и нисходящей ободочной кишок («двустволка»). Разнообразная симптоматика (в т. ч. кардиалгии) связана с скоплением газа в области изгиба. Одни считают вариантом СРК (облегчение после опорожнения кишечника и отхождения газов), другие - самостоятельной патологией. Иногда называют также синдромом Мачеллы-Дворкена-Била.
Пиулахса — Хедериха (Пиулакса - Эдерика) болезнь, синдром (испанские врачи P. Piulachs, H. Hederich; синоним: тимпанит при синдроме долихомегаколона, tympanites in dolichomegacolon syndrome). Острое паралитическое расширение толстой кишки (без механической непроходимости). Симптомы: резкий метеоризм и боль в животе. Рентгенологически обнаруживают долихомегаколон.
Протопопова синдром (В.П. Протопопов, 1880-1957, отечественный психиатр; синоним Протопопова триада) — сочетание тахикардии, мидриаза (расширения зрачка) и спастического запора, наблюдающееся при депрессиях.
Рапунцель синдром (описан E.D. Vaughan, J.L. Sawyers и H.W. Scott в 1968 г.). Закупорка кишок, вызванная систематическим проглатыванием волос. В кишках больного образуются трихобезоары. Может потребовать хирургического вмешательства. Наблюдается при психопатиях, шизофрении, эпилепсии, олигофрении, преимущественно в детском возрасте. Рапунцель (Rapunzel) - персонаж одноименной сказки братьев Гримм, девушка с длинными косами.
Сейнта синдром (Ch.F.М. Saint, совр. южноафриканский патолог; синонимы: Сейнта триада, Сена синдром — нрк*, Сента синдром — нрк*) — сочетание грыжи пищеводного отверстия диафрагмы, желчнокаменной болезни и дивертикулеза толстой кишки.
Тавеля болезнь (Е. Tavevl, швейцарский хирург) — периколит после аппендэктомии, характеризующийся образованием спаек и сужением толстой кишки, лихорадкой и расстройствами функции кишечника.
Miserere! (помилосердствуй!) от начала католической молитвы "miserere mei" - помилуй меня (Боже), так по-старому называлась каловая рвота (copremesis) при непроходимости кишечника.
Что такое синдром саванта и почему его не лечат
Синдром саванта The savant syndrome: an extraordinary condition. A synopsis: past, present, future — это состояние, при котором люди с тяжёлыми психическими расстройствами (тем же аутизмом) и серьёзными нарушениями развития демонстрируют ярко выраженную гениальность в какой‑то узкой области. Например, в математике, или просто обладают феноменальной памятью.
Этот талант специалисты называют «островком гениальности». Таким образом подчёркивая, что гениальность в случае саванта окружена тёмным океаном бессознательности, неспособности контролировать себя, разумно мыслить.
Синдром саванта — это очень редкое явление: примерно один случай на миллион Advanced Proficiency and Exceptional Ability in Second Languages человек.
Впервые термин «савант» использовал британский учёный Джон Лэнгдон Даун (именно он описал синдром Дауна) в 1887 году. Тогда же лиц с коэффициентом интеллекта ниже 25 стали называть «идиотами». И Джон Даун сыграл на контрасте.
Он рассказал об удивительных случаях из своей практики — умственно неполноценных детях, один из которых наизусть цитировал труд «Взлёт и падение Римской империи» (причём легко повторял текст от начала к концу и наоборот), а другие великолепно рисовали или считали. Доктор Даун назвал таких детей «идиотскими учёными» (idiot savants; savant по‑французски — «учёный»).
Затем исследователи обнаружили, что термин некорректен. Практически у всех савантов IQ хоть и низок, но всё же выше 40. Определение заменили на «аутичный учёный» (autistic savant) — поскольку у многих савантов проявлялись признаки расстройств аутистического спектра.
Но и от этого варианта отказались, когда выяснилось, что аутизмом страдают The savant syndrome: an extraordinary condition. A synopsis: past, present, future лишь около 50% савантов (по некоторым данным Savant Syndrome FAQs — до 75%). У других же присутствуют иные формы психических нарушений или повреждения центральной нервной системы (ЦНС).
В итоге учёные ограничились термином без эпитетов — синдром саванта, или савантизм.
Как проявляется синдром саванта
Саванты могут демонстрировать таланты в самых разных областях. Единственный общий признак — в большинстве случаев гениальность развивается на фоне глубокой умственной неполноценности.
- Искусство, редкий художественный талант.
- Память, то есть способность с одного взгляда в мельчайших подробностях запоминать огромные массивы данных — цифры, пейзажи, сложные карты, тексты.
- Арифметические вычисления, в том числе самый распространённый Incidence of Savant Syndrome in Finland среди савантов талант — навык календарного счёта. Люди‑календари способны назвать, на какой день недели выпадает та или иная дата в пределах десятилетий и даже столетий. Многие из них могут точно сказать, чем занимались в любой день и час своей жизни.
- Музыка — например, способность до последней ноты запомнить и воспроизвести любое впервые услышанное произведение.
- Ориентация в пространстве. Саванты с этим даром точно запоминают, где что лежит. Они не могут заблудиться даже в незнакомом городе: им достаточно бросить один взгляд на карту, чтобы понять, где они находятся и куда идти.
Самый знаменитый савант в мире — Рэймонд Бэббит из фильма «Человек дождя». У героя Дастина Хоффмана был вполне реальный прототип — мужчина, который помнил наизусть более 6 000 книг, обладал энциклопедическими познаниями в области географии, музыки, литературы, истории, спорта, знал почтовые коды всех городов США и цитировал телефонные справочники.
Откуда берётся синдром саванта
Точных данных нет. Учёные склоняются к тому, что это врождённое генетическое нарушение. Оно проявляет себя, в частности, тем, что мозг савантов функционирует иначе, чем у здоровых людей. Например, они имеют доступ Explaining and inducing savant skills: privileged access to lower level, less‑processed information к так называемой низкоуровневой, малообработанной информации, которую хранят нейроны, и недоступной большинству в сознательном состоянии.
Также у многих савантов повреждено или вовсе отсутствует мозолистое тело, которое соединяет два полушария мозга. Пытаясь скомпенсировать недостаток, мозг устанавливает новые нейронные связи, и в некоторых случаях это оборачивается появлением феноменальной памяти.
Иногда синдром саванта возникает после тяжёлой травмы The savant syndrome: an extraordinary condition. A synopsis: past, present, future головы с повреждением левой передней височной доли мозга. Существуют исследования, в которых учёные временно отключали эту область, и у добровольцев проявлялись некоторые признаки савантизма. В частности, увеличивался объём запоминаемых ими цифр или улучшались навыки рисования по памяти.
Есть вероятность The savant syndrome: an extraordinary condition. A synopsis: past, present, future , что способности саванта можно пробудить в каждом здоровом человеке — временно заглушая те или иные зоны мозга, стимулируя создание новых нейронных цепей с помощью таблеток, других фармацевтических препаратов, медитаций и прочих способов. Но пока существуют лишь единичные исследования на этот счёт. Нет уверенности, что, экспериментируя с мозгом, можно улучшить его состояние, а не навредить.
Можно ли вылечить синдром саванта
Сам по себе синдром саванта не является заболеванием и не входит Mental Health and Mental Disorders: An Encyclopedia of Conditions в настольную книгу психиатров — «Диагностическое и статистическое руководство по психическим расстройствам» (DSM‑5).
Поэтому лечат Savant Syndrome FAQs не собственно савантизм, а лишь основное заболевание — например, аутизм, синдром Аспергера, болезни ЦНС, нарушения, связанные с травмами мозга. Если его удаётся скорректировать, признаки синдрома саванта могут исчезнуть сами собой.
Нередко после реабилитации таланты сохраняются и даже помогают людям социализироваться, найти применение своей гениальности в обществе.
Синдром Прадера-Вилли ( Синдром гипотонии-ожирения )
Синдром Прадера-Вилли - это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
МКБ-10

Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 - 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.
Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% - наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом - полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек - недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Диагностика
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения - ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза - показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации - основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития - синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция - увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии - бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия - прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина - карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике - предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
1. Синдром Прадера-Вилли у детей: новое в этиологии, патогенезе и лечении/ Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В.// Российский вестник перинатологии и педиатрии - 1999 - №4.
2. Синдром Прадера-Вилли в Беларуси: генетическая структура и фенотипическая характеристика/ Хурс О.И., Политыко А.Д., Румянцева Н.В. и др.//Известия Национальной Академии наук Беларуси - 2010 - № 1.
3. Clinical report-health supervision for Children with Prader-Willi Syndrome, the Committee on Genetics/ McCandless S.E.// Pediatrics - 2011 - №1.
4. Prader-Willi syndrome: clinical and genetic fi ndings/ Butler M.G., Thompson T.// Endocrinologist - 2000 - №10.
Синдром саванта: нужно ли лечить?

Саванты, савантизм. Что это такое, кто эти люди? Чем они отличаются от всех остальных? Читайте в статье MedAboutMe.
Савант-синдром: начало
Термин «синдром саванта» появился в литературе благодаря доктору Дж. Лэнгдону Дауну, автору еще одного известного термина — «синдром Дауна». Вернее, доктор Даун использовал понятие idiot savants, «саванты-идиоты». Это было в конце 19 века, в 1887 году. Слово «идиот» было не ругательным, а вполне официальным обозначением людей с низким (ниже 25) IQ. А в французском языке savant означает человека ученого, эрудированного. Парадоксальное сочетание отлично обрисовывало особенности некоторых пациентов доктора Дауна, весьма ограниченных во многих отношениях, и необыкновенно одаренных в чем-то другом.
Один из савантов, наблюдаемых доктором, мог с точностью определять время без часов. Другие были одаренными музыкантами или превосходно рисовали. Был пациент, запомнивший слово в слово шеститомный исторический труд, он мог безошибочно пересказывать его что с начала до конца, что от конца к началу, что с любого места в любом направлении. Но при этих несомненных талантах в других областях эти же пациенты были совершенно неразвиты и беспомощны.
В формулировке Дауна савант-синдром обозначался как наличие особых способностей при выраженной задержке умственного развития.
Дальнейшие исследования этого явления показали, что саванты все-таки не идиоты: уровень IQ у них хоть и низкий, но все-таки выше 40.
Потом заметили, что савантизм часто связан с расстройствами аутистического спектра (РАС), и вместо idiot savants стали употреблять термин autistic savant. Но и это оказалось некорректным: как минимум половина савантов не являются аутистами, хотя у них часто имеются другие формы расстройств психики. А аутизм вовсе не обязательно сочетается с савантизмом: среди людей с РАС далеко не каждый является савантом. В результате савантизм стал самостоятельным определением.
Сегодня синдром саванта не входит в перечень психических расстройств, то есть заболеванием не считается, и этот термин используется только в качестве неофициального определения.
В 1988 году вышел фильм «Человек дождя», получивший сразу 4 «Оскара». Один из главных героев в нем — савант Раймонд, прекрасно и тонко сыгранный Дастином Хоффманом. Для понимания сути савантизма этот фильм может быть хорошей иллюстрацией, хотя посмотреть его стоит в любом случае.
Причины возникновения савантизма

До сих пор нет единого мнения ученых по этому поводу. Есть некий набор фактов, которые пока не укладываются ни в одну достаточно стройную и убедительную теорию.
Исследование мозга савантов с помощью различных новых технологий показывают наличие различных аномалий в его строении, причем они могут быть как приобретенными, так и врожденными. Отдельные части мозга могут быть как больше обычного размера, так и уменьшены или вовсе отсутствовать. Вероятно, наличие подобных аномалий побуждает остальные части мозга брать на себя «повышенные обязательства» и компенсировать по мере возможности нарушение функций.
Многие из людей с синдромом саванта обладают знаниями и способностями, которым они никогда не обучались. При этом они могут быть сильно ограниченными в других областях. Объяснить с точки зрения науки наличие подобных врожденных талантов, порой граничащих с гениальностью, не представляется возможным, если только не принять идею о генетической передаче знаний.
Это первое, что приходит в голову, когда речь идет о случаях врожденного савантизма. У человека с глубокими когнитивными нарушениями остаются «островки гениальности», наличие которых трудно объяснимо.
После черепно-мозговой травмы или перенесенных заболеваний головного мозга возможно развитие приобретенного савантизма. В крайне редких случаях проявляется внезапный савантизм — спонтанное и необъяснимое появление уникальных способностей, которым не предшествовали ни травмы, ни болезни, и не наблюдалось никаких особенных способностей. Мне известно всего 11 случаев спонтанного савантизма.
Как проявляется синдром саванта?
Вариантов достаточно много, но чаще всего встречаются отдельные необычно развитые навыки или склонности. Их называют «осколочными». Например, савант может обладать способностью запоминать что-то с удивительной точностью и в огромном объёме. Это могут быть телефонные или автомобильные номера, тексты любого типа и объёма, цифровые, звуковые последовательности. При феноменальной памяти человек может быть совершенно беспомощным и умственно ограниченным.
Реже встречается талант в определенной узкой области, который проявляется не только в запоминании: савант может проявлять яркие творческие способности — в изобразительном искусстве, музыке, математике, игре в шахматы или в решении логических задач.
Наиболее редко появляются по-настоящему гениальные саванты, способности которых абсолютно уникальны. На всем земном шаре их не более 50 человек.
Известные люди с синдромом саванта

Главный герой «Человека дождя» был «списан» с Лоуренса Кима Пика, самого, наверное, известного гениального саванта современности. Этот уникальный человек не только обладал редкостной способностью к мгновенному запоминанию колоссальных объёмов информации: он мог читать одновременно два разных текста — один правым глазом, другой — левым. Он мог безошибочно назвать любую календарную дату за последние 2000 лет, назвать расстояние в километрах между любыми населенными пунктами (достаточно крупными, чтобы быть нанесенными на карту), ответить на вопрос из области географии, истории, искусства или спорта. Но при этом Ким Пик не мог самостоятельно даже умыться, одеваться или обслуживать себя в быту. Хотя трудностей в общении у него не было, и поставленный первоначально диагноз «аутизм» был снят, Ким Пик проявлял признаки умственной отсталости. Например, он был неспособен к абстрагированию, не понимал переносного смысла выражений. Ученым даже не удалось определить уровень его IQ, так как савант все время показывал разные результаты — от глубокого слабоумия до нормальных показателей. В среднем IQ этого уникального человека был около 87.
Дэниел Тэммет проявил необычные математические способности после дебюта эпилепсии в 4 года. Затем у него «прорезались» способности к изучению языков: исландский язык, например, он выучил за неделю до уровня свободного общения. А еще он помнил 22514 цифр числа π после запятой. В 25 лет у Тэммета был диагностирован синдром Аспергера, но особенных трудностей с социализацией Дэниел не испытывал.
Исследователь синдрома саванта, невролог Джон Р. Хьюз, еще в 2012 году писал в своей статье, опубликованной в Advances in Experimental Medicine and Biology о возможных механизмах связи между эпилепсией и развитием савантизма. Хьюз предпологал, что в результате травмы или заболевания головного мозга с поражением левой лобно-височной доли происходит подавление деятельности левого полушария, что позволяет правому полушарию развивать необычные навыки.
Джейсон Паджетт внезапно приобрел математические способности после черепно-мозговой травмы (ЧМТ), полученной, когда на мужчину напали грабители. До травмы Паджетт не только не проявлял к точным наукам никакого интереса — он даже колледж окончить не смог. Но удар по голове кардинально изменил мировосприятие Джейсона: окружающая действительность предстала перед ним в виде формул. Паджетт не только получил впоследствии математическое образование, но и сам стал преподавать математику. Кроме того, Паджетт ведет научную работу в области теории чисел.
После получения ЧМТ в 2017 году проснулся талант к рисованию у Дайаны де Авила, а ранее она к изобразительному искусство, да и к творчеству вообще, была совершенно равнодушна. Сейчас Дайана — известный художник, участник многих выставок.
Кстати, среди савантов мужчин в 6 раз больше, чем женщин.
Лесли Лемке не повезло с самого рождения: он появился на свет раньше срока, с целым «букетом» врожденных аномалий. У малыша был поврежден головной мозг, из-за глаукомы и ретинопатии ему удалили глаза. Мать от него отказалась, и заботу о ребёнке взяла на себя медсестра из роддома, в котором Лесли появился на свет. Самостоятельно глотать мальчик начал в годовалом возрасте, стоять научился к 12 годам, после этого стал с трудом передвигаться. Тогда же у него проявились его необычные способности к музыке. Один раз услышав концерт Чайковского, он сумел сыграть его так, словно с отличием окончил консерваторию по классу фортепиано. После этого Лемке много лет давал концерты по всему миру, пока состояние его здоровья не ухудшилось.
История Дерека Паравиччини похожа: он также родился недоношенным и вскоре потерял зрение. Интеллект мужчины и в зрелом возрасте не отличается от уровня развития 4-летнего ребёнка, он совершенно беспомощен в бытовом плане. Но со своих 4 лет Дерек начал давать концерты: он обладает уникальной музыкальной памятью и не менее уникальной способностью к импровизации.Музыкальные критики называют его одним из наиболее ярких музыкантов современности.
Нужно ли лечить савантизм?

Вероятнее всего, ответ на этот вопрос будет отрицательным. Зачем лечить то, что можно называть подарком судьбы? Даже если дар саванта заключается всего лишь в уникальной способности к запоминанию цифр или звуков. А если открывается настоящий талант — то тем более.
Случаи исчезновения савантизма зафиксированы.
Например, были два брата-близнеца с серьезными психическими нарушениями и способностью феноменально оперировать с числами. Мальчики общались преимущественно друг с другом, были буквально одним целым. Они не только одинаково выглядели, но и одинаково двигались и реагировали на происходящее синхронно и аналогично. И играли они тоже странно, по им одним понятным правилам: называли друг другу простые числа в только им одним ведомой последовательности. Детей возили с выступлениями по стране, и они срывали овации, демонстрируя удивительные способности к счету. Но впоследствии было решено разлучить мальчиков, чтобы побудить их к развитию и социализации. И они действительно смогли овладеть новыми навыками и стали общаться с окружающими. Но их математические способности исчезли, когда связь между ними была разрушена.
О других необычных детях читайте в статье «24 удивительных ребёнка: быть «не таким».
Читайте далее

Мозг ребенка и жиры пищи: зачем нужны омега-3 кислоты?
Питание многих детей к школьному возрасту существенно отличается от здорового, сбалансированного рациона.
Синдром Прадера-Вилли
Синдром Прадера-Вилли - это заболевание генетического плана, встречающееся крайне редко. Его развитие обусловлено тем, что семь генов, либо их частей, располагающиеся на 15 отцовской хромосоме отсутствуют, либо не могут нормально функционировать. Синдром был описан в 1956 году учёными А. Прадером, Г. Вилли, А. Лабхартом, Э. Зиглером и Г. Фанкони.
Согласно статистике, синдром выявляют у 1 новорождённого из 10-25 тысяч. На манифестацию патологии оказывает влияние отцовский генетический материал, так как нарушенная часть 15 хромосомы подвержена явлению импринтинга. То есть, копия лишь одного гена среди генов данного региона будет работать в полном объёме.
Симптомы синдрома Прадера-Вилли
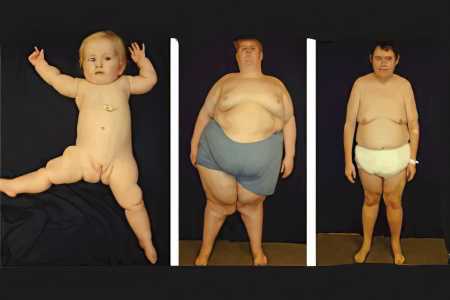
Выставить больному верный диагноз позволяют следующие симптомы синдрома Прадера-Вилли, даже если они проявляются не в полном объёме:
Во время внутриутробного развития снижена двигательная активность плода, женщины часто страдают от многоводия, обнаруживается предлежание плода.
Во время рождения ребёнок находится в ягодичном предлежании, наблюдается летаргия и гипотония. Слабый мышечный тонус оказывает влияние на сосательный рефлекс, провоцируя трудности во время грудного вскармливания. Ребенок может испытывать проблемы с дыханием. Синдром гипогонадизма характерен для данного генетического заболевания.
В раннем детстве на первый план выходит отставание в физическом развитии, наблюдаются проблемы в интеллектуальном плане. Ребенок быстро утомляется, склонен к сонливости. Нередко развивается косоглазие. Именно для раннего детства характерен сколиоз, который в младенчестве не диагностируется.
Для более старших детей характерны нарушения в речевом плане, наблюдается избыточная масса тела. В возрасте от 2 до 8 лет ребёнок начинает проявлять склонности к перееданию, нарушается физическая координация, страдает сон. Сколиоз продолжает прогрессировать.
В пубертатном периоде у подростков часто наблюдается задержка полового созревания, рост таких детей ниже их сверстников. Гибкость аномально высокая, масса тела превышает норму.
К 18 годам люди с синдромом Прадера-Вилли страдают от невозможности зачать ребёнка, так как являются бесплодными. Волосы на интимной зоне жидкие, продолжает прогрессировать гипогонадизм. Ожирение, низкое давление, проблемы в интеллектуальной сфере, трудности в обучении - всё это симптомы, вызванные имеющимися генетическими нарушениями. Кроме того, в период совершеннолетия человеку уже может быть выставлен диагноз «сахарный диабет», спровоцированный имеющейся генной патологией.
Взрослые люди с симптомом Прадера-Вилли имеют следующие внешние характеристики: нос у них широкий и большой, лоб высокий, веки опущены, глаза миндалевидные, верхние и нижние конечности маленькие, пальцы на них узкие, масса тела избыточная. По сравнению с остальными близкими родственниками, волосы и кожа больного члена семьи несколько светлее. Половое и моторное развитие человека нарушено, на коже образуются стрии, имеется склонность к дерматилломания (выщипыванию участков кожи).
Нейро-когнитивные нарушения
В 1992 году Курф и Фрим исследовали уровень интеллекта у людей с синдром Прадера-Вилли. Было установлено, что большая часть больных (39%) имеют незначительную умственную отсталость, по 27% пациентов имели умеренную умственную отсталость, или находились на границе интеллектуальной деятельности, выдавая уровень интеллекта в диапазоне от 70 до 85. С низким средним уровнем интеллекта было выявлено 5% людей, а с тяжёлой умственной отсталостью 1%. При этом глубокая умственная отсталость наблюдалась менее, чем в 1% случаев.
Установлено, что в детском возрасте у больных наблюдаются способности к чтению, они имеют обширный словарный запас. Тем не менее, речевые нарушения снижают их понимание. С трудом таким людям даётся математика и письмо, страдает концентрация внимания, зрительная и краткосрочная память. Поэтому, даже несмотря на уровень развития интеллекта, с возрастом проблемы в указных сферах не пропадают.
Поведенческие нарушения
Повышенный аппетит приводит к развитию ожирения.
Компульсивное поведение выражается в повышенном уровне тревожности и в дерматилломания.
Нарушения в психическом развитии проявляются в депрессиях, паранойе, галлюцинациях.
Часто, именно поведенческие расстройства приводят к тому, что больных госпитализируют.
Эндокринные нарушения
У людей с синдромом Прадера-Вилли наблюдается нехватка в организме гормона роста, что приводит к развитию ожирения, повышению плотности костной ткани.
Гипогонадизм является причиной того, что яички недоопускаются у мужчин в мошонку, а у женщин наблюдается раннее половое оволосение (адренархе). Однако, оба этих состояния можно откорректировать хирургическим и консервативным путём.
Причины синдрома Прадера-Вилли
Причины синдрома Прадера-Вилли кроются в генетических нарушениях. В результате определённых мутаций, происходит потеря участка 15 хромосомы отцовской копии генов.
Ещё причиной хромосомных перестроек, кроме мутации генов являются:
Унаследование двух пар хромосом только от матери (материнская униотцовская дисомия);
Нарушения, возникшие в результате разрыва хромосомы или по причине неравного кроссинговера (делеция);
Нарушения в результате хромосомных транслокаций с переносом участка хромосомы в негомологичную хромосому.
При этом риск того, что в семье второй ребёнок родится с синдромом Прадера-Вилли, зависит от того, что стало причиной перестройки. Так, униотцовкая диссомния и делеция сводят риск лишь к 1%, хромосомные транслокации повышают его до 25%, а мутации на фоне импринтинга до 50%. Паренатальное тестирование все эти варианты позволяет просчитать.
Диагностика синдрома Прадера-Вилли
Своевременная диагностика синдрома Прадера-Вилли позволяет не только выявить имеющиеся нарушения, но и начать незамедлительное лечение, которое намного улучшает прогноз развития патологии.
Если ранее диагноз выставлялся лишь на основе клинических признаков, то современная медицина использует метод генетического тестирования. Его обязательно необходимо провести всем новорождённым с гипотонией.
Что касается дифференциальной диагностики, то данную генетическую патологию необходимо отличать от синдрома Дауна.
Лекарств, позволяющих полностью избавиться от генетических нарушений, не существует. Однако, препараты для лечения синдрома Прадера-Вилли, а вернее для купирования его симптомов, находятся на стадии разработки.
Поэтому врачи рекомендуют в обязательном порядке всем детям с обнаруженным синдромом Прадера-Вилли проходить физиотерапию, которая направлена на повышение мышечного тонуса. Процесс обучения также должен быть адаптирован под интеллектуальные возможности ребёнка.
Большой проблемой остаётся вопрос ожирения, поэтому таким детям рекомендовано каждый день делать инъекции рекомбинантного гормона роста. Это позволяет контролировать аппетит, а также даёт возможность оказать поддержку в увеличении мышечной массы, а не жировой ткани.
Чтобы исключить обструктивное апноэ сна, больным на постоянной основе рекомендуют использовать аппарат для вспомогательной вентиляции лёгких.
Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность - "Лечебное дело" в 1991 году, в 1993 году "Профессиональные болезни", в 1996 году "Терапия".
Наши авторы
Читайте также: