Синдром Шильдера (Schilder) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Шильдера болезнь I Ши́льдера боле́знь (Р.F. Schilder, амер. невропатолог и психиатр, 1886—1940; синонимы: периаксиальный диффузный лейкоэнцефалит, симметричный интерглобулярный склероз, диффузный склероз нервной системы, диффузный периаксиальный энцефалит Шильдера)
Этиология и патогенез неизвестны. Предполагается инфекционно-аллергическая природа лейкоэнцефалита. Дискутируется вопрос о связи рассеянного склероза (Рассеянный склероз) с Ш. б., которая рассматривается как вариант рассеянного склероза в детском возрасте.
Заболевание развивается в любом возрасте, но чаще у детей и лиц молодого возраста. Ему обычно предшествуют черепно-мозговые травмы, профилактические прививки, инфекции. У взрослых и детей клинические проявления Ш. б. значительно отличаются. У взрослых заболевание манифестирует психическими нарушениями по шизофреноподобному типу с изменениями личности (см. Шизофрения), поведения, расстройствами высших корковых функций, галлюцинаторным и психотическим синдромами. Затем развиваются нарушения зрения (гомонимная Гемианопсия), центральное снижение слуха, судорожные припадки (часто джексоновского типа), центральный тетрапарез, Псевдобульбарный паралич. В терминальной стадии наступает обездвиженность, слепота, глухота, Кахексия.
У детей начало заболевания нередко бывает острым с подъемом температуры тела, головной болью, рвотой. Затем ребенок становится вялым, обедняется речь, возникают нарушения поведения, приступы возбуждения сменяются периодами заторможенности. Снижается слух, нарушается зрение, развиваются мозжечковые симптомы, спастический гемипарез, локальные судороги, а затем миоклонические судороги всего тела.
Течение неуклонно прогрессирующее, у детей чаще подострое с длительностью до одного года, у взрослых — хроническое (до 10—15 лет).
Диагноз при жизни поставить трудно. Следует обращать внимание на прогрессирующий характер заболевания, многообразие неврологических симптомов, выявление участков пониженной плотности в веществе головного мозга (чаще в лобных и затылочных долях) при компьютерной томографии. Дифференциальный диагноз проводят с опухолями мозга, вирусным энцефалитом, подострым склерозирующим панэнцефалитом.
Специфического лечения нет. Наряду с симптоматическим лечением применяют стероидные гормоны, иммунокорректоры (тималин, тактивин).
Библиогр.: Болезни нервной системы, под ред. П.В. Мельничука, т. 1, с. 277, М., 1982; Маркова Д.А. и Леонович А.Л. Рассеянный склероз, с. 91, М., 1978.
II Ши́льдера боле́знь (P.F. Schilder, 1886—1940, американский невропатолог и психиатр; син.: аплазия аксонов диффузная экстракортикальная, лейкоэнцефалит периаксиальный диффузный, склероз интерглобулярный симметричный, склероз нервной системы диффузный, Шильдера диффузный периаксиальный энцефалит, энцефалит периаксиальный)
прогрессирующее демиелинизирующее заболевание головного мозга, характеризующееся ранней дистрофией аксонов, проявляющееся центральными параличами и парезами, гиперкинезами, эпилептиформными припадками, расстройствами зрения, психическими нарушениями с развитием слабоумия.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
ШИЛЬДЕРА БОЛЕЗНЬ
ШИЛЬДЕРА БОЛЕЗНЬ (P. F.Schilder, американский невропатолог и психиатр, 1886—1940; синоним: периаксиальный диффузный лейкоэнцефалит, внутримозговой центролобарный симметричный склероз, аксиальная экстракортикальная диффузная аплазия, прогрессирующая лейкопатия мозга, детский периваскулярный некроз и склероз мозга, склерозирующий периаксиальный диффузный прогрессирующий энцефалит, склерозирующая прогрессирующая энцефалолейкопатия) — воспалительное заболевание центральной нервной системы, относящееся к демиелинизирующим болезням, к группе прогрессирующих лейко энцефалитов.
Впервые описана и выделена как самостоятельная болезнь нервной системы воспалительной природы Шильдером в 1912 году, который наблюдал в течение 19 недель девочку 14 лет с прогрессирующей деменцией и симптомами нарастания внутричерепного давления. При патологоанатомическом исследовании в этом случае были обнаружены обширные очаги де-миелинизации в белом веществе обоих полушарий головного мозга, массивные периваскулярные инфильтраты, состоящие из лимфоцитов и плазмоцитов, большое число фагоцитирующих клеток нейроглии (см.), пролиферация и гипертрофия глиоцитов. При этом осевые (аксиальные) цилиндры нервных волокон были относительно сохранны, в связи с чем Шильдер назвал заболевание периаксиальным диффузным энцефалитом. В последующие годы данные гистохимического и биохимического исследования позволили отнести ряд случаев подобного заболевания к различным типам наследственных лейкодистрофий (см.). Углубленное изучение патоморфологической картины болезни дало основание некоторым исследователям рассматривать Шильдера болезнь как атипичный вариант рассеянного склероза (см.).
Этиология и патогенез не установлены. Предполагается, что Шильдера болезнь, как и другие лейкоэнцефалиты (см.), является заболеванием инфекционно-аллергической природы. Обсуждается роль вирусов как пусковых факторов гиперергического аутоиммунного процесса.
Патологическая анатомия. При макроскопическом исследовании паутинная оболочка головного мозга обычно отечна, иногда утолщена, сосуды поверхности мозга полнокровны, отдельные извилины или доли мозга уплощены, желудочки расширены. На срезах полушарий головного мозга в белом веществе уже при макроскопическом исследовании видны множественные очаги (в редких случаях — единичные) округлой или неправильной формы, четко отграниченные, местами сливающиеся между собой, имеющие розовато-серый цвет, мягкие на ощупь, а также очаги желтовато-серого цвета, плотноватые, пористые (иногда с кистами) диаметром (на плоскости среза мозга) от нескольких миллиметров до 1 см и более. Эти очаги демиелинизации в разных стадиях процесса организации локализуются в белом веществе долей головного мозга, часто в симметричных участках обоих полушарий, иногда вблизи коры головного мозга; при этом дугообразные волокна головного мозга остаются сохранными. Имеются описания случаев локализации очагов также в базальных ядрах, зрительном перекресте (хиазме), мозжечке, стволе головного мозга и в спинном мозге.
При гистологическом исследовании очагов демиелинизации обнаруживают изменения, характерные для разных стадий распада миелина и процесса организации этих очагов. В области свежих очагов (макроскопически они розовато-серые, мягкие) видны зернистые шары, а также астроциты (см. Нейроглия) с гипертрофированной цитоплазмой (тучные клетки Ниссля), гигантские («монстрозные») глиоциты (см. Ганглии) с одним или двумя крупными гиперхромными ядрами неправильной формы; встречаются и гигантские глиоциты с несколькими бледно окрашивающимися, пузырьковидными ядрами, расположенными эксцентрично. Осевые цилиндры нервных волокон в очагах демиелинизации относительно сохранны, многие из них набухшие, извиты, местами утолщены и гиперхромны, фрагментированы. По мере организации очага демиелинизации количество зернистых шаров и гигантских глиоцитов уменьшается, появляются волокнообразующие аетроциты, количество которых постепенно увеличивается. Выявляются также интенсивно импрегнирующиеся при специальных окрасках микроглиоциты с узловатыми отростками и крупными ядрами — «клетки-корнеплоды». Постепенно на месте очага демиелинизации формируется глиальный рубец, нередко с полостями (макроскопически он желтовато-серого цвета, пористый, плотноватый). Нейроны, аксоны которых оказались в пределах очагов демиелинизации, подвергаются ретроградной дегенерации. В паутинной оболочке и вокруг сосудов мозга видны инфильтраты, состоящие из лимфоцитов, немногочисленных плазмоцитов, зернистые шары, наблюдается также пролиферация клеток стенок мелких сосудов, гиалииоз и склеротические изменения стенок артерий и вен. Во всех отделах мозга и особенно вблизи очагов демиелинизации часто обнаруживают «дренажные» (отечные) олигодендроглиоциты.
Клиническая картина. Заболевание может развиться в любом возрасте, но чаще заболевают дети и лица молодого возраста. Частота заболевания у мужчин и женщин одинакова. Начало Шильдера болезни постепенное, реже острое и даже инсультоподобное. Течение неуклонно прогрессирующее, иногда относительно стационарное с ремиссиями и обострениями.
Клинические проявления в начале болезни разнообразны. У взрослых первыми симптомами обычно являются изменения личности, нарушения поведения, снижение работоспособности, расстройства высших корковых функций, приводящие к социальной дезадаптации больных. На этом фоне возникают приступы возбуждения с галлюцинаторным синдромом и психотические состояния, которые нередко дают основание думать об остром начале шизофрении (см.). Частым ранним симптомом являются расстройства зрения в виде гомонимной или квадрантной гемианопсии (см.), а также центральное снижение слуха. По мере развития болезни снижается интеллект, нарастает апатия, которая может сменяться приступами возбуждения. В последующем развивается амавроз (см. Слепота) вследствие ретробульбарного неврита или диффузной демиелинизации затылочных долей головного мозга. Наблюдаются полиморфные судороги, чаще джексоновского типа с потерей сознания (см. Джексоновская эпилепсия). При исследовании черепномозговых (черепных, Т.) нервов выявляется псевдобульбарный синдром (см. Псевдобульбарный паралич), центральный паралич мышц лица (см. Лицевой нерв, Тройничный нерв). Брюшные рефлексы, как правило, исчезают. Двигательные нарушения, обычно начинающиеся с ног, проявляются повышением тонуса мышц ног по пирамидному типу, в связи с чем походка становится спастико-атакти-ческой, а затем распространяются на руки. Характерны центральные (пирамидные) тетрапарезы и тетраплегии (см. Параличи, парезы) с тенденцией к нарастанию мышечного тонуса. Наряду с тяжелыми двигательными нарушениями отмечается тремор рук, хореоатетоз (см. Гиперкинезы), гиперкинезы в мышцах языка, асинергия (см. Мозжечок).
ШШльдера болезнь может протекать с преобладанием психических расстройств, дизартрией (см.), атаксией (см.), с изменением тонуса мышц (см. Тонус) по пластическому (экстрапирамидному) типу. В терминальной стадии больные Шильдера болезнью полностью обездвижены, резко выражена кахексия (см.).
У детей Шильдера болезнь нередко начинается остро, инсультоподобно. Отмечаются подъем температуры, головная боль, тошнота, иногда боли в области шеи. Ребенок становится вялым, речь бедная с элементами псевдо-бульбарной дизартрии (см.). Появляются нарушения поведения, приступы неадекватного возбуждения, которые сменяются заторможенностью. Характерно снижение слуха и нарушение зрения. В отличие от взрослых, у детей двигательные нарушения чаще проявляются статической и динамической атаксией (см.), а центральные параличи чаще носят характер моноплегий и гемиплегий (см. Гемиплегия). По мере развития болезни возникают парциальные судороги с потерей сознания, преобладанием тонико-клонического компонента. Прогредиентное течение болезни приводит к психическим и тяжелым двигательным нарушениям, в терминальной стадии появляются частые миоклонические судороги во всем теле. В отдельных случаях у детей Шильдера болезнь может протекать с симптомами повышения внутричерепного давления и по клинической картине напоминать опухоль головного мозга.
При исследовании глазного дна выявляется негрубый симметричный отек дисков зрительных нервов. Характерным для Шильдера болезни является центральный амавроз — слепота при сохранении реакции зрачков на свет. Часто определяется гомонимная или квадрантная гемианопсия. На ЭЭГ (см. Электроэнцефалография) регистрируется диффузная симметричная дезорганизация ритма биоэлектрической активности головного мозга. При эхоэнцефалографии (см.) смещения срединных структур мозга не отмечается. Давление цереброспинальной жидкости нормальное, наблюдается незначительное повышение содержания белка, изменение реакции Ланге (см. Цереброспинальная жидкость). При компьютерной томографии (см. Томография компьютерная) головного мозга иногда определяются участки понижения плотности в области лобных и затылочных долей, соответствующие локализации крупных очагов демиелинизации.
Многообразие клинических проявлений обусловлено диффузным характером демиелинизирующего процесса в головном мозге, различными размерами очагов демиелинизации и их локализацией, а также неодинаковой степенью выраженности воспалительных явлений.
Диагноз установить трудно в связи с многообразием клинических проявлений Шильдера болезни, исключительной редкостью заболевания. Клиническая картина Шильдера болезни может имитировать многие другие заболевания центральной нервной системы, в том числе опухоль головного мозга (см.), туберкулезный менингит (см.), болезнь Коновалова — Вильсона (см. Гепато-церебралъная дистрофия), лейко-дистрофии (см.), поствакцинальные энцефалиты (см.), сифилис нервной системы (см. Сифилис) и др.
Лечение. Специфического лечения нет. В отдельных случаях процесс стабилизируется при лечении стероидными гормонами в сочетании с гипосенсибилизирующей и рассасывающей терапией.
Библиогр.: Гусев Е. И. Лейкоэнце-фалит Шильдера, в кн.: Болезни нервной системы, под ред. П. В. Мельничука, т. 1„ с. 277, М., 1982; Марков Д. А. и Леонович А. Л. Рассеянный склероз, с. 91, М., 1978; С а в е н к о С. Н. Рассеянный склероз и диффузный периак-сиальный энцефалит, Киев, 1966; Ф и н-к е л ь И. И. Диффузный периаксиаль-ный энцефалит Шильдера, Журн. невропат. и психиат., т. 60, № 9, с. 1089, 1960; Ч а л и с о в И. A. Leucoencephalitis. diffusa scleroticans atypica (типа Schil-der’a), Сов. психоневрол., № 6, с. 33,. 1932; Adams R. D. a. K u b i k C. S. Symposium on multiple sclerosis and demye-linating diseases, morbid anatomy of demye-linative diseases, Amer. J. Med., v. 12, p. 510, 1952; Adams R. D. a. Victor M. Principles of neurology, N. Y. a. o.« 1977; Brain W. R. Brain’s clinical neurology, p. 465, Oxford a. o., 1978; Ferrer I. a. o. Schilder’s disease, Child’s Brain, v. 8, p. 294, 1981; Gr i-sold W., Jellinger K. u. V о 1 1-m e r R. Morbus Schilder bei 54 jahringer Frau mit klinischer Remission, Nervenarzt* Bd 53, S. 164, 1982; Handbook of clinical neurology, ed. by P. J. Vinken a. Bruyn,. v. 9, p. 469, Amsterdam a. o., 1975; Lhermitte F. e. a. Les formes cavi-taires de la sclerose en plaques et de la maladie de Schilder, Rev. neurol., t. 137, p. 589, 1981; Schilder P. Zur Kenn-tnis der sogenannten diffusen Sklerose, Z„ ges. Neurol. Psychiat., Bd 10, S. 1, 1912. Л. О. Бадалян; В. А. Моргунов (пат. ан.).
ЛЕЙКОЭНЦЕФАЛИТ
Лейкоэнцефалит (leukoencephalitis; греч. leukos белый + enkephalos головной мозг + -itis) — воспалительно-дистрофическое поражение белого вещества головного мозга. Лейкоэнцефалит относятся к демиелинизирующим заболеваниям (см.).
Впервые заболевание из группы Лейкоэнцефалитов описал Дансон (J. Danson) в 1933 г. под названием «подостро» форма летаргического энцефалита». В 4939 г. Петте и Деринг (H. Pette, G. Doring) сообщили об энцефалите с хроническим прогрессирующим течением, несколько отличающимся по клиническим и патоморфологическим проявлениям, назвав его узелковым панэнцефалитом. В 1945 г. это же заболевание описано Ван-Богартом (L. Van Bogaert) как «подострый склерозирующий лейкоэнцефалит». В дальнейшем Ван-Богарт тщательно изучил клинику и морфологию этой хронической прогрессирующей формы энцефалита. С группой Лейкоэнцефалита также сходны описанный в 1912 г. Шильдером (Р. F. Schilder) диффузный периаксиальный энцефалит и геморрагический Лейкоэнцефалит, о к-ром сообщил Херст (E. W. Hurst) в 1941 г.
Содержание
Этиология и патогенез
Предполагается, что Лейкоэнцефалит являются заболеваниями инфекционно-аллергической природы. Дискутируется роль миксовирусов, вирусов кори, бешенства и Herpes zoster как пусковых факторов гиперергического аутоиммунного процесса.
Классификация
Выделяют следующие клинико-морфол, формы Л.: подострый склерозирующий лейкоэнцефалит Ван-Богарта, периаксиальный лейкоэнцефалит Шильдера, острый геморрагический Л. При Л. демиелинизирующий процесс обычно сочетается с поражением нейронов в той или иной степени, поэтому для некоторых его форм употребляется также термин «панэнцефалит».
Патологическая анатомия
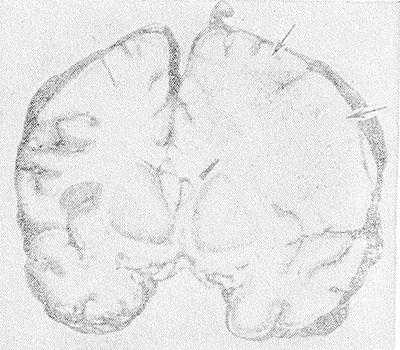
Рис. 1. Фронтальный срез головного мозга больного, умершего от геморрагического лейкоэнцефалита: белое вещество справа более отечное, усеяно мелкими петехиальными кровоизлияниями (указаны стрелками), прилежащие участки коры нечетко очерчены.
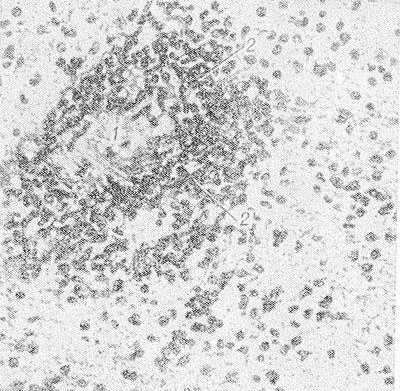
Рис. 2. Микропрепарат ткани белого вещества мозга больного, умершего от геморрагического лейкоэнцефалита: стенка кровеносного сосуда (1) инфильтрирована и окружающая нервная ткань пропитана полиморфно-ядерными лейкоцитами (2).
Макроскопическое исследование мозга при Л. выявляет расширение борозд и атрофию извилин. На срезе полушарий определяются различного размера участки деструкции и демиелинизации во всех отделах мозга, преимущественно в белом веществе, но захватывающие различные участки серого вещества коры (рис. 1). В наиболее пораженных отделах мозг имеет губчатую консистенцию, желудочки мозга умеренно расширены.
Гистол, картина характеризуется диффузной подострой воспалительной реакцией с периваскулярной инфильтрацией лимфоцитами и плазмоцитами и очаговой демиелинизацией (рис. 2). Воспалительные изменения преимущественно локализованы в белом веществе мозга, иногда в коре, подкорковых ганглиях, мозговых оболочках. Разрушается нормально сформированный миелин (миелинокластический тип поражения). Степень демиелинизации и деструкции нервной ткани варьирует в различных очагах. Отдельные мелкие очаги могут сливаться. У краев очага демиелинизации олигодендроциты увеличены, содержат амфофильные включения, в более пораженных участках они полностью исчезают. Кроме того, встречается много больших причудливой формы астроцитов с гиперхроматическими многодольчатыми или несколькими ядрами. Аксоны остаются относительно сохранными на ранних стадиях процесса, позднее в них могут быть дистрофические изменения. Нейроны коры полушарий большого мозга могут содержать включения двух типов: сферические частицы диам. 30—40 мкм и продолговатые, или тубулярные, структуры несколько меньшего диаметра. Включения чаще встречаются при небольшой длительности заболевания. Гистохимические исследования обнаруживают во включениях большое количество белка. В большинстве случаев находят пролиферативную реакцию глии. Глиоз может быть мелкоузелковый или в виде крупных очагов (псевдоопухоль). Диффузное разрастание волокнистой глии приводит иногда к уплотнению мозгового вещества, так что мозг на разрезе имеет хрящевидную консистенцию. Стенки артерий и вен утолщены, с избытком ретикулярных волокон в адвентиции.
Клиническая картина
Нервно-психические нарушения являются наиболее ранним проявлением заболевания. Вначале отмечаются жалобы на повышенную утомляемость, вялость, раздражительность, неустойчивость настроения. Постепенно круг нервно-психических расстройств расширяется. Появляется злобность, эффективность, жадность, эгоистичность, жестокость, недисциплинированность, инертность мышления. Больные часто совершают немотивированные поступки, теряют навыки опрятности.
На фоне психических нарушений постепенно в течение нескольких недель или месяцев прогрессирует очаговая неврологическая симптоматика: апрактические расстройства, приводящие к потере навыков самообслуживания (см. Апраксия); гностические нарушения (см. Агнозия); возникают расстройства чтения, письма, счета. У некоторых больных наблюдаются зрительные, слуховые галлюцинации. Двигательные нарушения вначале представлены преимущественно экстрапирамидными расстройствами: выявляется ригидность, феномен «зубчатого колеса» (см. Дрожательный паралич). Наблюдаются полиморфные гиперкинезы мышц лица, конечностей, туловища — тремор (см. Дрожание), торсионный спазм (см. Торсионная дистония), гемибаллизм (см. Гиперкинезы), миоклонии (см.). Пирамидные нарушения в типичных случаях развиваются на более поздних стадиях в виде моно-, геми- или тетрапарезов и параличей (см. Параличи, парезы). К часто встречающимся симптомам очагового поражения относятся статическая и локомоторная атаксия (см.) мозжечкового или лобного типа. Двустороннее поражение корково-ядерных путей приводит к нарушениям фонации, глотания. Бульбарный паралич развивается довольно редко.
Постоянным признаком заболевания являются судороги (см.). Они могут появляться на разных стадиях болезни. Наиболее характерны малые и абортивные судорожные припадки, реже генерализованные большие припадки. В поздней стадии заболевания развиваются трофические и вегетативные расстройства: кахексия, пролежни, нарушения терморегуляции, профузный пот и т. д. В терминальной стадии больные обездвижены, иногда наблюдается децеребрационная ригидность (см.).
Течение Лейкоэнцефалита может быть неуклонно прогрессирующим или ремиттирующим. В последнем случае клин, картина может напоминать рассеянный склероз (см.).
При электроэнцефалографии регистрируют периодическую пароксизмальную активность с интервалом 5 — 15 сек. одновременно в большинстве отведений в виде медленных (1—2 в 1 сек.) высоковольтажных волн.
В крови определяется лейкоцитоз, повышение фракции гамма-глобулина, обычно повышен титр антител к коревому вирусу или к миксовирусам (вирус jc, sv-40).
В цереброспинальной жидкости в большинстве случаев не наблюдается цитоза и увеличения содержания белка. Однако при электрофоретическом исследовании белков обнаруживают, что гамма-глобулин составляет до 40 и более процентов от общего количества белка, а фракция альбумина снижена. Коллоидные реакции дают максимальную флоккуляцию в первых пробирках (паралитический тип реакции Ланге).
Лечение
Лечение должно быть комплексным. Показана гормональная и симптоматическая терапия. Положительный эффект получают при назначении кортикостероидов. Лечение глюкокортикоидами (преднизолоном) следует начинать в ранней стадии патологического процесса с учетом ритма гормональной деятельности надпочечников. Гормональная терапия дополняется противоаллергическими (димедрол, пипольфен, супрастин, диазолин) и противосудорожными препаратами. Показаны препараты, снижающие мышечный тонус (мидокалм, амедин, мидантан, циклодол и др.), витамины группы В и другие симптоматические средства. Применение активной терапии может задерживать течение болезни и способствовать ремиссиям на несколько лет.
Прогноз
При неуклонно прогрессирующем течении больные погибают через 2—12 мес. после появления первых симптомов. При ремиттирующем течении заболевание длится до 3 лет и более, а ремиссии могут продолжаться от нескольких месяцев до нескольких лет, в течение которых симптомы заболевания почти или полностью отсутствуют.
Особенности отдельных форм лейкоэнцефалита
Подострый склерозирующий лейкоэнцефалит Ван-Богарта. При патоморфол, исследовании мозга больных с этой формой Л., как правило, обнаруживаются внутриклеточные включения. Степень поражения уменьшается в направлении от коры к филогенетически более древним образованиям, но чаще, чем при других формах, поражается ствол и спинной мозг.
Клинической особенностью этой формы Лейкоэнцефалита является раннее проявление и преобладание экстрапирамидных нарушений (Гиперкинетическая форма), к к-рым лишь на поздних этапах присоединяются пирамидные симптомы. Эпилептические припадки не характерны.
Периаксиальный диффузный лейкоэнцефалит Шильдера. Патоморфол, особенностью по сравнению с другими Л. и рассеянным склерозом является относительно ранняя дистрофия аксонов. Эта форма отличается от предыдущей преобладанием пирамидных симптомов и частыми эпилептическими припадками. Обычно наблюдаются большие припадки. Характерно развитие ретробульбарного неврита зрительных нервов или центральной формы слепоты, связанной с демиелинизацией затылочных долей (см. Шильдера болезнь).
Острый геморрагический лейкоэнцефалит. По клин, и патоморфол, признакам эта форма Л. сходна с вирусными и поствакцинальными энцефалитами. При патологоанатомическом исследовании выявляют отек мозга, на срезах в веществе мозга — большие очаги мягкой розовато-серой или желтоватой окраски с множественными точечными кровоизлияниями. Гистол. картина характеризуется фибринозным некрозом стенок мелких сосудов, в основном венул, окруженных экссудатом фибрина, воспалительными клетками и кольцевидными геморрагическими зонами. В этих же периваскулярных зонах— демиелинизация с умеренной или выраженной деструкцией аксонов. На самых ранних стадиях периваскулярные инфильтраты представлены гл. обр. нейтрофилами, однако в более старых очагах находят много лимфоцитов и плазмоцитов.
Клиника острого геморрагического Лейкоэнцефалита характеризуется чрезвычайно острым началом, молниеносным нарастанием тяжести симптомов поражения мозга. Заболевают лица обоего пола в возрасте от 20 до 40 лет. Длительность течения от 2 дней до 2 нед. Развернутой клин, картине предшествуют катаральные явления в зеве, лихорадка с лейкоцитозом в периферической крови. Через 2—4 дня появляется головная боль, ригидность мышц шеи, нарушается сознание, иногда развивается кома. Характерны фокальные или генерализованные судороги, двигательные нарушения в виде геми- или тетраплегии, псевдобульбарный паралич. На глазном дне — отек диска (соска) зрительного нерва. Редко наблюдаются подострые и хронические формы. С помощью ЭЭГ и артериографии могут быть обнаружены фокальные изменения. В цереброспинальной жидкости — выраженный плеоцитоз за счет полиморфно-ядерных лейкоцитов, встречаются также лимфоциты; содержание белка повышено до 1 г/л и более; часто выявляется ксантохромия цереброспинальной жидкости, микроскопически можно обнаружить единичные эритроциты.
Исход обычно летальный.
Библиография: Маркова Е. Д. и др. Клинико-анатомические и вирусологические данные в случае подострого склерозирующего панэнцефалита, Журн, невропат. и психиат., т. 77, № 7, с. 100 7, 1977, библиогр.; Цукер М. Б. Клиническая невропатология детского возраста, М., 1978, библиогр.; Чумаков М. Вирусологические аспекты изучения этиологии некоторых хронических заболеваний нервной системы (подострый склерозирующий панэнцефалит, вилюйский энцефаломиелит, множественный склероз), в кн.: Демиелинизирующие заболевания нервн. системы в Эксперим, и клин., под ред. А. И. Булыгина, с. 79, Минск, 1975; Balakоva H., Kvicalа V. Radiologicke a scintigraficke nalezy u akutnich zanetlivech procesii CNS, Cs. Neurol. Neurochir., sv. 40, s. 350, 1977; Gilroy J. a. Meyer J. S. Medical neurology, N. Y., 1975; Lhermitte F. Les leucoencephalites, P., 1950, bibliogr.; Pette H. u. Doring G. Uber einheimische Panencephalomyelitis vom charakter der Encephalitis japonica, Dtsch. Z. Nervenheilk., Bd 149, S. 7, 1939.
Синдром Геллера
Синдром Геллера - первазивное расстройство психического развития, характеризующееся внезапной утратой сформированных функций и навыков. Дебютирует в период с 2 до 10 лет. Ребенок утрачивает речь, способность решать интеллектуальные задачи, выполнять бытовые ритуалы. Не пытается использовать невербальные средства коммуникации, не интересуется играми. Становится раздражительным, тревожным, непослушным, гиперактивным. Диагностика проводится методом беседы, наблюдения, психологического исследования когнитивной сферы. Специфическое лечение не разработано, назначаются коррекционные занятия, симптоматическая медикаментозная терапия.
Общие сведения
Синдром впервые был описан австрийским педагогом Т. Геллером, в честь которого получил название. Синонимы - детское дезинтегративное расстройство, детская деменция, дезинтегративный психоз, симбиотический психоз, синдром Крамера-Польнова. Заболевание встречается редко, по различным данным, частота составляет 0,001-0,0017%. До недавнего времени считалось, что распространенность не зависит от пола, но развитие методов диагностики позволило различать случаи болезни Геллера с синдромом Ретта у девочек, в результате было установлено, что эпидемиологические показатели среди мальчиков выше в 4 раза. В МКБ-10 заболевание отнесено к рубрике «другие дезинтегративные расстройства детского возраста».
Причины синдрома Геллера
Этиологические факторы остаются неизвестными. Данные последних исследований указывают на связь патологического процесса с нейробиологическими механизмами центральной нервной системы. По результатам электроэнцефалографического обследования почти у 50% больных детей обнаруживается изменение электрической активности в головном мозге. Продолжается изучаться связь синдрома с судорогами, лейкодистрофией, болезнью Шильдера. Выдвигается предположение об инфекционном происхождении болезни, существовании фильтрующегося вируса - возбудителя малых размеров, недоступного исследованию под микроскопом.
Патогенез
Патогенетическая основа синдрома Геллера неизвестна, однако выделены закономерности развития патологических процессов. Заболеванию предшествует не менее двух и не более десяти лет нормального развития: ребенок частично или полностью овладевает речью, понимает обращения взрослых, использует социальные навыки. Внезапно возникают первые симптомы - гиперактивность, эмоциональные нарушения. В течение 6-12 месяцев распадается большинство приобретенных навыков, интеллектуальное развитие снижается до степени глубокой умственной отсталости (идиотии), теряется контроль опорожнения мочевого пузыря, кишечника. Затем регресс останавливается, состояние стабилизируется. Дальнейшее развитие, восстановление утраченных навыков происходит медленно при массивной психолого-педагогической помощи.
Симптомы синдрома Геллера
В продромальном периоде наблюдаются эмоциональные отклонения: ребенок проявляет своенравность, раздражительность, гневливость, тревожность. К аффективной вспыльчивости добавляется гиперактивность. Становятся недоступными сложные виды деятельности, требующие концентрации и распределения внимания, усидчивости, выполнения действий по образцу. Практически утрачиваются ранее существовавшие способности: сбор сложных моделей конструктора, рисование, разукрашивание, участие в сюжетно-ролевых играх («больница», «магазин»). Ребенок неусидчив, злится, отказывается от занятия при ошибках, затруднениях. На данном этапе признаки интеллектуального снижения отсутствуют, определить начало синдрома Геллера невозможно.
После нескольких месяцев эмоциональной неустойчивости, гиперактивности развиваются более специфические симптомы. Речь прогрессивно обедняется: сокращается словарный запас, развернутые фразы заменяются простыми, структурированные предложения - командами и односложными ответами («дай», «иди», «да», «нет»). В конечном итоге речь распадается, утрачивается разговорный язык, понимание обращений. Одновременно снижается интерес к сотрудничеству и общению. Ребенок замкнут, аутичен, не участвует в играх с другими людьми, не проявляет желания выполнять совместную деятельность. Исчезает стремление познавать окружающий мир, обедняются эмоции, упрощаются игровые навыки, развивается своеобразное «равнодушие».
Сложные двигательные навыки распадаются, заменяются стереотипными действиями. Больной неспособен выполнять ежедневные ритуалы - умываться, чистить зубы, надевать одежду, убирать игрушки, принимать пищу, ходить в туалет. Усиливаются проявления гиперактивности. Нарушается контроль мочеиспускания, опорожнения кишечника. Зачастую к признакам синдрома добавляются симптомы неврологической патологии. Спустя год после начала заболевания ребенок полностью утрачивает речевые, социальные, бытовые навыки.
Осложнения
После интенсивного прогрессирования болезни наступает стабильный период. Осложнения соматического и психического характера отсутствуют, но становится фактически невозможной социальная адаптация. При синдроме Геллера дети нуждаются в специальном обучении. Они не могут получать образование в средних и профессиональных учебных заведениях, не овладевают профессией, не создают семьи. Развитие медленное, поэтому им необходим постоянный посторонний уход, который в благоприятных случаях заменяется контролем. Заболевание ребенка изменяет социальное функционирование родителей, большинству приходиться отказываться от профессиональной деятельности, увлечений.
Диагностика
Диагностика синдрома Геллера зачастую начинается с консультации педиатра и невролога. Родители обращаются к специалистам на этапе постепенной утраты сформированных ранее навыков. Из-за редкости заболевания в первую очередь проводится осмотр ребенка и инструментальные обследования, позволяющие выявить более вероятные неврологические патологии - эпилепсию, опухоль и травму головного мозга. После исключения этих диагнозов ребенок направляется к врачу-психиатру. Специфическая диагностика включает:
- Беседу. Опрашивая родителей, врач выясняет характерные особенности течения болезни: период правильного развития, прогрессивный распад существующих функций, регресс более чем двух сфер. Отмечается нарушение языковых, социальных, бытовых, игровых, двигательных навыков.
- Наблюдение. В ходе консультации специалист фиксирует особенности эмоциональных реакций и поведения ребенка. Для синдрома Геллера характерна гиперактивность в сочетании с аутистическими проявлениями - стереотипиями, отсутствием интереса к общению, нежеланием использовать мимику, пантомимику, жесты как инструменты коммуникации.
- Психологическое тестирование. Психолог проводит исследование интеллектуальных способностей ребенка. Набор методик определяется глубиной дефекта, возрастом пациента, его способностью устанавливать и удерживать продуктивный контакт. Используется тест прогрессивных матриц Равена, невербальная часть теста Векслера, «коробка форм», пирамидка.
Синдром Геллера дифференцируют с ранним детским аутизмом, болезнью Ретта, детской шизофренией. Основные диагностические критерии: период обычного развития, предшествующий болезни, не меньше 2 лет; появление симптомов до десятилетнего возраста; быстрый прогрессирующий распад навыков (от 6 до 12 мес.). В клинической картине преобладает дефицитарность, обеднение психических функций.
Лечение синдрома Геллера
Терапия детского дезинтегративного расстройства имеет общее направление с лечением раннего детского аутизма - основное внимание уделяется ранним и интенсивным мероприятиям, методы основаны на бихевиоральном походе, имеют высокую степень структурированности. Эффективность медикаментозного лечения не доказана, лекарства используются на первоначальном этапе для купирования выраженных поведенческих нарушений. Программа развития составляется индивидуально, к процессу реабилитации подключаются врачи, психологи, специальные педагоги, родители. В комплексный подход включены:
- Развивающие и коррекционные мероприятия. Занятия по восстановлению речи, интеллектуальных функций проводятся индивидуально специалистами различных профилей - психологами, олигофренопедагогами, дефектологами, логопедами. Мероприятия, направленные на формирование социальных навыков, коррекцию эмоциональных нарушений осуществляются в небольших группах. Дети учатся сотрудничать, помогать, принимать помощь.
- Семейное консультирование, психотерапия. Работа с родителями ориентирована на обучение уходу за детьми, информирование об особенностях заболевания, прогнозе. В рамках групповых занятий реализуется психотерапевтическая помощь. Встречи родителей, имеющих детей с болезнью Геллера и РДА, позволяют уменьшить изоляцию семей, получить эмоциональную поддержку, практический опыт организации быта, досуга, развивающих домашних занятий.
- Социальная реабилитация. Усилия педагогов нацелены на формирование практических полезных навыков. Дети обучаются одеваться, умываться, использовать столовые приборы, держать карандаш и рисовать, лепить из глины, пластилина. Параллельно происходит коррекция поведенческих и эмоциональных отклонений - развивается усидчивость, концентрация внимания.
Прогноз и профилактика
Прогноз для детей, имеющих синдром Геллера, неблагоприятный, потерянные навыки остаются утраченными либо восстанавливаются крайне медленно, не полностью. При ранней интенсивной терапии около 20% пациентов приобретают способность изъясняться простыми фразами, осваивают самообслуживание, базовые бытовые и трудовые навыки, становятся социально активными в семье, реабилитационных и лечебных заведениях, которые посещают. Меры профилактики данного заболевания не разработаны, так как не установлены его причины и патогенетическая база. Исследования продолжаются.
Лейкоэнцефалит Шильдера
Лейкоэнцефалит Шильдера — дегенеративно-демиелинизирующее поражение головного мозга, сопровождающееся образованием крупных или сливных зон демиелинизации. Имеет неуклонно прогрессирующее течение с неспецифичной и полиморфной клинической картиной, которая может включать психические нарушения, пирамидный и экстрапирамидный синдромы, когнитивный дефицит, поражение черепно-мозговых нервов, эписиндром. Диагностируется лейкоэнцефалит Шильдера по клиническим критериям и результатам МРТ после исключения другой патологии с подобными проявлениями. Терапия осуществляется глюкокортикостероидами, антиконвульсантами, миорелаксантами и психотропными средствами. Однако лечение малоэффективно.
МКБ-10
Лейкоэнцефалит Шильдера впервые был рассмотрен в качестве самостоятельной нозологии в 1912 г. психоневрологом, имя которого прочно закрепилось в названии заболевания, хотя сам автор обозначил описанную им патологию термином «периаксиальный диффузный лейкоэнцефалит». Позже различными исследователями были представлены описания других клинических форм лейкоэнцефалита: в 1941 г. - геморрагического лейкоэнцефалита, в 1945 г. - подострого склерозирующего лейкоэнцефалита. Поскольку основной патоморфологический субстрат болезни составляют диффузные зоны демиелинизации белого вещества, лейкоэнцефалит Шильдера входит в группу демиелинизирующих заболеваний.
Преимущественный возраст манифестации болезни Шильдера до сих пор остается спорным вопросом. Зарубежные специалисты в области неврологии считают характерным дебют в возрастном периоде от 7 до 12 лет, а отдельные авторы предлагают относить заболевание к детской форме рассеянного склероза. Наблюдения отечественных неврологов, напротив, свидетельствуют о равной степени поражения лиц различной возрастной категории.
Причины лейкоэнцефалита Шильдера
Этиопатогенез болезни Шильдера находится в стадии изучения. Из названия заболевания видно, что первоначально подразумевалась воспалительная этиология церебрального поражения, т. е. энцефалит. Предполагается вирусная теория заболевания по типу медленных инфекций. Среди возможных инфекционных агентов дискутируется роль кори, герпетической инфекции, миксовирусов, которые, возможно, запускают процесс аутоиммунного церебрального воспаления. Однако безуспешные попытки выделения возбудителя привели к возникновению иной этиопатогенетической теории. Последняя предполагает связь лейкоэнцефалита Шильдера с дисфункцией регуляторных механизмов липидного обмена, что сближает заболевание с наследственными лейкодистрофиями.
Морфологические изменения заключаются в образовании в белом церебральном веществе полушарий значительных зон демиелинизации, имеющих четкие заостренные очертания и зачастую асимметрично расположенных. В ряде случаев подобные очаги формируются в мозжечке и мозговом стволе. У пациентов, заболевших в пубертатном периоде и во взрослом возрасте, описаны случаи, когда наряду с зонами обширной демиелинизации наблюдаются округлые бляшковидные очаги, напоминающие бляшки рассеянного склероза.
Симптомы лейкоэнцефалита Шильдера
Заболевание отличается наличием неспецифичной и полиморфной симптоматики. Может манифестировать исподволь развивающимися психическими расстройствами: лабильностью настроения, апатией, нарушением поведения, эпизодами возбуждения с галлюцинаторным синдромом. Интеллектуальное снижение прогрессирует вплоть до деменции. Наблюдаются аграфия, акалькулия, алексия, агнозия, апраксия. Вследствие демиелинизации черепных нервов возникает неврит зрительного нерва, офтальмоплегия, тугоухость, снижение зрения, бульбарные расстройства. При поражении мозжечка появляется мозжечковая атаксия, скандированная речь, интенционный тремор. Поражение зрительной зоны коры проводит к гемианопсии, корковому амаврозу. Возможны экстрапирамидные нарушения в виде гиперкинезов, торсионной дистонии и т. п. Пирамидные расстройства обычно наблюдаются на поздних этапах лейкоэнцефалита в виде моно-, геми- и тетрапарезов. Зачастую присутствует судорожный синдром (по типу джексоновкой эпилепсии или с генерализованными эпиприступами), характеризующийся отсутствием специфической ЭЭГ-картины.
Вариативность сочетаний различных симптомокомплексов настолько выражена, что не позволяет выделить типичный вариант течения болезни Шильдера. В ряде случаев клиника сходна с прогредиентным вариантом рассеянного склероза, в других - имеет псевдотуморозный характер, в третьих — напоминает психиатрическую патологию. В последнем случае пациенты могут проходить лечение у психиатра вплоть до развития явной неврологической симптоматики.
Диагностика лейкоэнцефалита Шильдера
Прижизненно диагностировать лейкоэнцефалит Шильдера весьма затруднительно. Эта задача требует от невролога тщательного сопоставления анамнестических, клинических и томографических данных, внимательного проведения дифдиагностики со схожими заболеваниями. С целью обследования зрительного и слухового анализаторов к консультациям могут привлекаться офтальмолог и отоларинголог.
Электроэнцефалография выявляет признаки диффузного церебрального поражения: снижение альфа-активности и дезорганизацию ритма; зачастую определяется эпилептиформная активность. При исследовании цереброспинальной жидкости обнаруживается повышение уровня гамма-глобулина на фоне снижения удельного веса альбуминовой фракции. Наиболее информативным способом инструментальной диагностики выступает МРТ головного мозга. Болезнь Шильдера подтверждает наличие как минимум одного большого или пары сливных очагов демиелинизации в белом церебральном веществе.
Для установления окончательного диагноза многие неврологи руководствуются критериями C.M. Poser 1985 г.: наличие по данным МРТ 1-2 округлых зон демиелинизации величиной не менее 2х3 см; отсутствие патологии надпочечников; исключение любой иной церебральной патологии (внутримозговой опухоли, рассеянного энцефаломиелита, инсульта и пр.); соответствие норме уровня жирных кислот в сыворотке крови; выявление на аутопсии зон диффузного хронического склероза. В некоторых случаях отличить лейкоэнцефалит Шильдера от лейкодистрофии позволяют лишь гистологические исследования церебральных тканей пораженной зоны.
Лечение и прогноз лейкоэнцефалита Шильдера
Отсутствие ясных представлений об этиопатогенезе болезни Шильдера пока не позволило разработать более или менее эффективные методы ее лечения. Отмечен некоторый эффект глюкокортикостероидной терапии, в связи с чем многим пациентам назначают метилпреднизолон, вначале парентерально в ударной дозе, а затем внутрь с постепенным снижением дозы. Параллельно проводится курс нейропротекторной, антиоксидантной и сосудистой терапии, при необходимости назначаются антиконвульсантное лечение (карбамазепин, диазепам), миорелаксанты (амантадин, толперизон, амидин), противоотечные мероприятия (фуросемид, ацетазоламид, магния сульфат), психотропные фармпрепараты.
Своевременно начатое лечение способно лишь несколько задержать прогрессирование патологии. Однако, не смотря на его проведение, все пациенты погибают. Время наступления летального исхода варьирует от нескольких месяцев до 3 лет с момента дебюта лейкоэнцефалита.
Читайте также:
- Как подобрать солнцезащитные очки? Критерии выбора
- Эфирные масла. Польза для души и тела
- Циклические нуклеотиды при эпилепсии. Роль кальция в развитии эпилепсии
- Барьерные методы контрацепции: презервативы, шеечные колпачки, спермициды, диафрагма
- Диагностика локализованной фиброзной опухоли плевры (ЛФОП) на рентгене, КТ, МРТ