Синдром Видеманна (Wiedemann) - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 22.01.2026
Видемана - Ленца синдром, по именам немецких педиатра H. R. Wiedemann и медицинского генетика W. D. Lenz) - талидомидная эмбриопатия, связанная с приемом на 4-10-й неделе беременности препарата талидомида: легкого седативного и снотворного средства, синтезированного в 1957 в ФРГ и изъятого из продажи в 1962 вследствие его тератогенного действия. Характеризуется множественными аномалиями развития конечностей различного характера и степени выраженности: от небольшого дефекта до отсутствия плечевых и бедренных костей, костей предплечья и голени (кисти и стопы непосредственно связаны с плечевым и тазовым поясом соответственно) или полным отсутствием конечностей; характерны также аномалии наружного и внутреннего уха, глухота или тугоухость; микрофтальмия; атрезия пищевода и двенадцатиперстной кишки, гипоплазия слепой кишки, желчного пузыря; поражения черепных нервов, умственная отсталость, эпилепсия, дислексия, дисфагия, аутизм, иногда гиперкинезы; аномалии сердца и крупных сосудов, дистрофия почек, врожденный гидронефроз; двурогая матка, атрезия влагалища, атрезия заднепроходного отверстия и др.
H. R. Wiedemann. Hinweis auf eine derzeitige Haufung hypo- und aplastischer Fehlbildungen der Gliedmassen. Die Medizinische Welt, Stuttgart, 1961; 2: 1863-1866.
W. Lenz, K. Knapp. Die Thalidomid-Embryopathie. Deutsche medizinische Wochenschrift, Stuttgart, 1962; 87: 1232-1242.
Синдром Видемана - Штайнера - Wiedemann-Steiner syndrome
Содержание
Признаки и симптомы
Признаки, описанные при синдроме Видеманна - Штайнера, включают: [5]
- Невысокий рост
- Отставание в развитии
- Низкий мышечный тонус (гипотония ) особенно в младенчестве
- Характерные черты лица
- Волосатые локти (гипертрихоз кубити )
Синдром Видеманна-Штайнера может быть связан с глобальными задержками в развитии, проблемами со сном, проблемами с кормлением и пищеварением, необычными чертами лица, низким / миниатюрным ростом, гипотонией, проблемами с зубами, волосатыми локтями, длинными ресницами и т. Д. [6]
Причина
Синдром Видеманна-Штайнера возникает в результате мутаций в MLL (также известный как KMT2A ) ген на длинной руке хромосома 11. [4] Ген кодирует фермент модификации гистонов, то есть помогает изменять экспрессию других генов. [7] Состояние является аутосомно-доминантным, что означает, что для того, чтобы у человека был синдром, необходима только одна аномальная копия гена. На сегодняшний день в большинстве случаев мутация произошла de novo, то есть ни один из родителей не пострадал, и мутация носит спорадический характер. Потомки тех, у кого есть WSS, имеют 50% шанс получить WSS. [6]
Механизм, с помощью которого мутации в гене MLL вызывают фенотип синдрома Видемана-Штайнера, пока не известен. [ нужна цитата ]
Диагностика
При подозрении на синдром Видемана - Штайнера можно провести анализ гена MLL. В противном случае это может быть диагностировано целиком.секвенирование экзома или же секвенирование всего генома.
В этой области проводится ограниченное диагностическое тестирование. Стандартные скрининговые тесты, которые проводятся во время беременности и могут диагностировать такие синдромы, как синдром Дауна, не диагностируют WSS. Кроме того, базовые генетические диагностические тесты, проводимые после рождения, не включают тестирование на WSS. Полное секвенирование экзома было использовано для идентификации большинства людей с WSS. Часто медицинские работники не предлагают вариант тестирования всего экзома, или связанные с этим расходы не покрываются страховкой, или требуют большой доплаты, ограничивающей людей от проведения тестирования. Часто пациентам дают другие неверные медицинские объяснения или менее конкретный и более широкий диагноз, например: аутизм и Синдром Рубинштейна - Тайби. Кроме того, как только человек достигает определенного возраста или этапа в своей жизни, когда ему поставили неправильный диагноз или ему не поставили диагноз, он / она может перестать искать ответы на свои медицинские испытания и невзгоды, что означает, что он никогда не столкнется с формальным диагнозом WSS. [6] Также были пациенты с синдромом Видемана-Штайнера, которым изначально неправильно поставили диагноз: Синдром Кабуки. [8]
Специфического лечения или лечения синдрома Видеманна - Штайнера не существует. Детям с этим заболеванием может быть полезен ряд поддерживающих методов лечения, таких как физиотерапия, Логопедия, дополнительное питание при плохом питании и специальная образовательная поддержка. [ нужна цитата ]
Люди, страдающие синдромом Видеманна-Штайнера, часто получают физическую, профессиональную, речевую, кормящую и / или поведенческую терапию. Иппотерапия и музыкальная терапия также были полезны для людей, страдающих от WSS. Дети школьного возраста, затронутые WSS, могут получить пользу от индивидуальных помощников, модифицированного обучения и / или специальных дневных классов. [6]
Эпидемиология
Синдром Видемана-Раутенштрауха ( Неонатальный прогероидный синдром )
Синдром Видемана-Раутенштрауха - это врожденное заболевание, которое характеризуется преждевременным старением и проявляется с неонатального периода. Развитие болезни связывают с генетическими мутациями POLR3A, LMNA, ERCC8. Болезнь манифестирует в периоде новорожденности, проявляется старческой внешностью, множественными врожденными пороками, отставанием в росте и психомоторном развитии. Для диагностики патологии назначают кариотипирование, нейровизуализацию, ЭКГ и УЗИ сердца. Специфическое лечение синдрома находится на этапе разработки, поэтому пациентам назначают симптоматические препараты, проводят реконструктивные операции.
МКБ-10



Общие сведения
Болезнь Видемана-Раутенштрауха названа в честь двух немецких педиатров, которые в 1977-1979 гг. независимо друг от друга описали типичную клиническую картину синдрома. В медицинской литературе патологию также называют неонатальным прогероидным синдромом. Заболевание встречается крайне редко: на сегодня известно только 15 случаев с подтвержденным диагнозом и еще 36 пациентов с подозрением на синдром Видемана-Раутенштрауха (СВР). Статистика распространенности патологии на 100 тыс. населения неизвестна.

Причины
Этиология и патогенез заболевания пока точно не установлены. При расширенном генетическом исследовании у больных детей выявляются мутации генов POLR3A, LMNA и ERCC8. Синдром не является строго наследственным заболеванием. Однако в современной генетике считается, что риск повторения болезней в семье около 25% - это указывает на возможный аутосомно-рецессивный тип передачи мутантных генов.
Патогенез
Ученым не удалось выявить механизмы развития заболевания. Некоторые специалисты связывают его прогрессирование с нарушениями гормонального и липидного метаболизма, в частности с высоким уровнем пролактина и триглицеридов при концентрации холестерина на верхней границе нормы. Нарушения физического развития обусловлено низким содержанием ростового гормона и инсулиноподобного фактора роста. Поражение ЦНС связывают с процессами демиелинизации.
Симптомы
Внешние признаки синдрома Видемана-Раутенштрауха заметны с момента рождения. У младенца наблюдается низкая масса тела, задержка антенатального развития, псевдогидроцефалия. Характерным признаком заболевания является сухая морщинистая кожа и выраженные вены на черепе, что в сочетании с треугольным лицом создает старческий вид. Подбородок выступает, глаза глубоко посажены, брови и ресницы скудные. Волосы на голове редкие и тусклые. С возрастом внешность ребенка практически не меняется.
Для синдрома Видемана-Раутенштрауха типичен широко открытый большой родничок, гипотония скелетных мышц, недостаток жировой клетчатки на лице и на конечностях. Липоатрофия не затрагивает боковые поверхности туловища и ягодицы, которые имеют нормальный объем и детскую пухлость. У 70% новорожденных присутствуют молочные зубы. Зачастую встречаются недоразвитие и укорочение дистальных фаланг пальцев рук и ног.
У части детей с синдромом Видемана-Раутенштрауха наблюдаются пупочные грыжи, вентрикуломегалия, крипторхизм. Патологии опорно-двигательного аппарата проявляются остеопенией, суставными контрактурами, дисплазией тазобедренного сустава. Поражение дыхательной системы представлено слабым криком, размягчением хрящей трахеи, приступами удушья. Вовлечение в процесс пищеварительной системы проявляется гастроэзофагеальным рефлюксом и хроническими запорами.
Осложнения
Самым опасным при синдроме Видемана-Раутенштрауха является поражение сердца. Инфаркты в раннем возрасте и декомпенсация врожденных пороков — основные причины летального исхода. У пациентов наблюдаются проблемы со зрением, вызванные помутнением роговицы, врожденной глаукомой, поражением сетчатки глаза. Большую опасность представляет патология базальных ганглиев, которая проявляется дисрегуляцией вегетативных функций, мышечного тонуса, высшей нервной деятельности.
При синдроме Видемана-Раутенштрауха встречается задержка психомоторного развития, ее интенсивность варьирует от соответствия нижней границе возрастной нормы до умственной отсталости. По мере роста у ребенка наблюдается сильный дефицит длины и массы тела, из-за чего старческие черты лица еще больше выделяются. У некоторых больных отмечается неврологический дефицит в виде атаксии, тремора, поражения черепных нервов.
Обследование ребенка с типичным фенотипом и пороками развития проводится педиатром совместно с врачом-генетиком. Диагностика начинается с детального осмотра пациента, выявления внешних признаков недоразвитости и стигм эмбриогенеза, после чего переходят к анализу беременности и родов, изучению семейного анамнеза. В процессе диагностики используются следующие методы исследования:
- МРТ головного мозга. С ее помощью удается определить поражение базальных ядер, признаки гидроцефалии, последствия перенесенного внутримозгового кровоизлияния Исследование также назначается при подозрении на инсульт. У новорожденных детей активно используется нейросонография как более быстрый и доступный вариант нейровизуализации.
- Рентгенография костей скелета. Диагностика проводится при видимых костно-суставных деформациях, чтобы определить вид и степень нарушения. Для прицельного исследования пораженных суставов применяется эхосонография, КТ и МРТ.
- УЗИ сердца. С помощью эхокардиографии определяют врожденные пороки сердца и магистральных сосудов, оценивают скорость и характер кровотока, измеряют фракцию выброса. Диагностика дополняется электрокардиографией, КТ сердца, ангиографией.
- Анализы крови. Для синдрома Видемана-Раутенштрауха характерно повышение уровня глюкозы натощак, дислипидемия с увеличением количества атерогенных фракций липопротеидов. При гормональной диагностике определяется снижение инсулиноподобного ростового фактора и соматотропного гормона.
- Кариотипирование. Генетическая диагностика необходима для исключения других наследственных болезней, вызванных аномалиями числа или формы хромосом. При синдроме Видемана-Раутенштрауха определяется нормальный мужской или женский кариотип без выраженных хромосомных нарушений.
Дифференциальная диагностика
Диагностика синдрома Видемана-Раутенштрауха затруднена вследствие его редкой встречаемости и неясной этиологической картины. При постановке диагноза проводится дифференциация с другими синдромами, сопровождающимися прогерией. Необходимо исключить болезнь Хатчинсона-Гилфорда, признаки которой возникают только на втором году жизни, и синдром Вернера, манифестирующий в подростковом возрасте.
Лечение синдрома Видемана-Раутенштрауха
Консервативная терапия
На сегодня в медицине нет препаратов, которые могли бы излечить заболевание. Пациентам назначаются симптоматические лекарства. Для предупреждения кардиоваскулярных кризов используют статины, антикоагулянты и гипотензивные препараты. Большой проблемой остается малый вес тела и плохое физическое развитие детей, поэтому назначаются препараты рекомбинантного гормона роста. При тугоподвижности суставов проводится физиотерапия.
Хирургическое лечение
Помощь детских хирургов требуется при тяжелых врожденных пороках, которые нарушают жизнедеятельность ребенка. Чаще всего операция осуществляется для коррекции сердечно-сосудистых аномалий и проводится на первом году жизни пациента. Своевременное выполнение коронарного шунтирования или ангиопластики может замедлить развитие кардиоваскулярной патологии и продлить жизнь пациенту.
Экспериментальное лечение
С 2006 года в США разрабатываются препараты на основе ингибиторов фарнезилтрансферазы. Изначально они были предназначены для онкологических больных, чтобы замедлить процесс старения клеток и вернуть им нормальную форму. Позже была выявлена способность лекарств действовать на механизм прогерии и замедлять ее развитие. Пока разработка находится на стадии лабораторных исследований, препараты не применяются на людях в рамках клинических исследований.
Прогноз и профилактика
Средняя продолжительность жизни ребенка с СВР не превышает 6 лет. В литературе описаны в случае гибели еще в младенческом возрасте, также есть информация о 3-х пациентах, доживших до подросткового возраста. Причиной смерти в основном выступают инфаркты и инсульты. Этиопатогенетическое лечение болезни пока не разработано, поэтому прогноз для жизни неблагоприятный. Профилактические мероприятия отсутствуют ввиду крайней редкости болезни и сложностей с определением ее этиологии.
1. POLR3A-ассоциированная гипомиелинизированная лейкодистрофия: описание клинического случая и обзор литературы/ А.Ф. Мутарзина// Нервно-мышечные болезни. - 2021. - №4.
2. Синдром Видемана-Раутенштрауха (неонатальный прогероидный синдром): описание клинического случая и краткий обзор литературы/ Е.В. Большова, О.А. Вишневская// Международный эндокринологический журнал. - 2017. - №8.
3. Wiedemann-Rautenstrauch syndrome with bilateral tarsal kink: three sutures for correction/ M. Batur, E. Seven, A. Zinal, T. Yaеar// J. Craniofac. Surg. — 2017. — Vol. 28, № 3.
Синдром Видемана-Раутенштрауха
Синдром Видемана-Раутенштрауха (СВР), также известный как неонатальный прогероидный синдром, представляет собой очень редкое генетическое заболевание, характеризующееся старением при рождении, задержкой роста до и после рождения (пренатальная и послеродовая задержка роста), а также недостаточностью или отсутствие жирового слоя под кожей (подкожная липоатрофия). Предполагается, что у большинства больных с СВР сократилась продолжительность жизни. Лишь немногие из них дожили до подросткового возраста, а еще меньше — до 20 лет.
СВР характеризуется старением при рождении и недостаточностью или отсутствием слоя жира под кожей (подкожная липоатрофия). В результате кожа может выглядеть необычно тонкой, хрупкой, сухой, блестящей, морщинистой и состарившейся. Некоторые вены и мышцы могут чрезмерно выступать, особенно на лбу. По неизвестным причинам с возрастом пострадавших младенцев аномальные отложения жира могут накапливаться под кожей в нижних (каудальных) областях тела, особенно вокруг ягодиц, областей вокруг гениталий и ануса (аногенитальная область), и области между ребрами и бедрами (боками). Кроме того, у младенцев и детей с этим заболеванием живот может казаться необычно большим и выпуклым.
У больных с синдромом Видемана-Раутенштрауха задержка роста может произойти до рождения (задержка внутриутробного развития), особенно в течение последних трех месяцев (третий триместр) развития плода. Задержка роста будет продолжаться и после рождения (послеродовой период). Пациенты с СВР также плохо набирают вес и не могут развиваться в течение всей жизни. Кроме того, в некоторых случаях пораженные младенцы могут испытывать трудности с глотанием (дисфагия) и кормлением, что может способствовать задержке роста и развития.
При СВР может наблюдаться прогрессирующее неврологическое ухудшение. Конкретные симптомы могут варьироваться от человека к человеку, поскольку у больных могут быть не все симптомы, перечисленные ниже.
Младенцы и дети с СВР также имеют характерные черепно-лицевые аномалии. У многих больных мягкое пятно в передней части черепа может быть аномально большим и широким, и его закрытие может быть отсроченным. Фиброзные промежутки между другими костями черепа (черепные швы) также могут быть аномально широкими. Кроме того, у младенцев с СВР лобные кости и боковые части черепа (теменные кости) чрезмерно выступают, а лицевые кости необычно малы и недоразвиты (гипоплазия).
Такие отклонения могут привести к тому, что голова будет выглядеть необычно большой (псевдогидроцефалия). У пораженных младенцев и детей характерные лицевые аномалии могут включать:
- необычно маленький рот (микростомия);
- выступающий подбородок;
- низко посаженные уши, которые сильно наклонены к затылку (назад).
Черты лица обычно кажутся необычно маленькими по сравнению с большим лбом и боковыми сторонами черепа. Кроме того, у пораженных младенцев может быть необычно маленький, отчетливый «клювовидный» нос, который становится более выраженным с возрастом.
У большинства младенцев и детей с синдромом Видемана-Раутенштрауха также присутствуют дополнительные черепно-лицевые аномалии. У пораженных младенцев может быть от 2 до 4 передних зубов (неонатальных резцов), которые выпадают в раннем младенчестве. Последующее развитие зубов (прикуса) задерживается и нарушается. Кроме того, у младенцев и детей с заболеванием нижние веки могут опускаться или поворачиваться наружу (эктропион), обнажая тонкие и нежные слизистые оболочки, выстилающие веки, а также часть глазных яблок (конъюнктиву). У одного пациента также был описан спастический заворот, состояние, при котором веко поворачивается внутрь, так что ресницы и кожа трутся о поверхность глаза. Интересной особенностью в некоторых случаях является то, что нижние веки могут закрывать больше, чем нижнюю половину глазного яблока, как если бы веки располагались выше, чем ожидалось. У больных младенцев и детей также необычно редкие волосы на коже головы, брови и ресницы (гипотрихоз). В семье с тремя затронутыми братьями и сестрами также были отмечены различные глазные аномалии, включая:
- ;
- помутнение роговицы;
- перфорацию роговицы;
- микрофтальм (необычно маленький размер глаза).
Младенцы и дети с СВР могут также иметь отличительные аномалии, поражающие кисти, ступни, руку и ноги (конечности). Руки и ноги ненормально тонкие, кисти и стопы непропорционально большие; пальцы рук и ног длинные, с необычно маленькими, не полностью развитыми (атрофическими) или утолщенными (дистрофическими) ногтями. Суставы толстые и жесткие, особенно в плечах, локтях и коленях. Недавние исследования МРТ (магнитно-резонансной томографии) подтвердили наличие нормального количества подкожного жира в туловище и заметную потерю жира на лице и дистальных отделах конечностей. Истончение костей (остеопения) может предрасполагать к переломам костей. Нарушается также трансформация костных клеток-предшественников в кости (остеобласты) и хрящевые клетки (хондроциты). Отсутствие способности к клеточной дифференцировке у пациентов СВР может быть причиной клинического проявления и симптомов этого редкого заболевания.
Большинство младенцев и детей с синдромом Видемана-Раутенштрауха также имеют разную степень умственной отсталости, которая может варьироваться от легкой до тяжелой. Однако некоторые дети продемонстрировали почти нормальное умственное развитие. В младенчестве у больных могут начаться прогрессирующие неврологические и нервно-мышечные нарушения. У большинства пациентов наблюдается серьезная задержка в приобретении навыков, требующих координации физической и умственной деятельности (психомоторная отсталость). Кроме того, во многих случаях младенцы и дети с расстройством не контролируют голову, демонстрируют пониженный мышечный тонус (гипотония) и имеют нарушенную способность координировать произвольные движения в области грудной клетки и брюшной полости (атаксия туловища). Например, они могут испытывать трудности с контролем диапазона движений во время определенных мышечных действий и могут испытывать ритмичный непроизвольный тремор при выполнении определенных движений (намеренный тремор). Младенцы и дети с этим заболеванием также могут испытывать быстрые непроизвольные горизонтальные движения глаз (горизонтальный нистагм) и ограниченную остроту зрения. У младенцев может быть дисфония, а у детей постарше — необычно высокий голос.
Кроме того, исследователи сообщили, что неврологическое ухудшение, наблюдаемое у некоторых больных с СВР, может быть связано с потерей миелиновой оболочки нервных волокон (демиелинизация) в белом веществе мозга (например, чистая суданофильная лейкодистрофия). Миелин — это беловатое жирное вещество, которое образует защитную оболочку или «чехол» вокруг определенных нервных волокон (аксонов) и служит электрическим изолятором, обеспечивая эффективную передачу нервных импульсов. «Белое вещество» головного и спинного мозга (центральной нервной системы) в основном состоит из пучков миелинизированных нервных волокон. У большинства пациентов с СВР в указанном возрасте не было лейкодистрофии. Сообщалось о мальформации Денди-Уокера и вентрикуломегалии, кальцификации базальных ганглиев и агенезии мозолистого тела.
Отсутствие подкожной жировой ткани побудило исследователей сравнить СВР с синдромом генерализованной липодистрофии (синдром Берардинелли-Сейпа). Однако лабораторные исследования в этих изученных случаях не показали повышения уровня глюкозы, липидов или инсулина натощак, как можно было бы ожидать при синдроме Берардинелли-Сейпа. Однако у некоторых пациентов был повышенный уровень триглицеридов. Жировые подушечки локализуются на боку, а не на ягодицах, что характерно для этого синдрома, но также может наблюдаться при врожденном нарушение гликозилирования. У больных с СВР также может развиться аномальное искривление позвоночника из стороны в сторону (сколиоз). Кроме того, младенцы и дети с СВР часто предрасположены к рецидивирующим респираторным инфекциям, которые могут привести к опасным для жизни осложнениям.
В одном из немногих случаев, когда проводилась вскрытие, было обнаружено почти полное отсутствие брыжейки, ткани, которая фиксирует тонкий кишечник на задней стороне брюшной стенки, и отсутствие мезоколона, ткани, которая фиксирует поперечную часть толстого кишечника.
Причины и факторы риска
Синдром Видемана-Раутенштрауха, скорее всего, передается по наследству как аутосомно-рецессивное генетическое заболевание.
Рецессивные генетические нарушения возникают, когда человек наследует две копии аномального гена одного и того же признака, по одной от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он является носителем болезни, но обычно бессимптомным. Риск заражения ребенка от двух родителей-носителей составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей, составляет 25%. Риск одинаков для мужчин и женщин.
У некоторых больных с СВР были родители, которые были кровными родственниками.
Все люди несут несколько аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют более высокий шанс, чем родители, не являющиеся кровными родственниками, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с редким рецессивным генетическим заболеванием.
Конкретный основной дефект, ответственный за заболевание, остается неизвестным. Однако некоторые исследователи предполагают, что определенную роль могут играть нарушения созревания костей, гормонального и жирового (липидного) обмена.
Затронутые группы населения
В некоторых случаях нарушение роста, макроцефалия и/или другие характерные признаки, указывающие на синдром Видемана-Раутенштрауха, могут быть обнаружены до рождения (пренатально) с помощью ультразвука.
У большинства пациентов синдром Видемана-Раутенштрауха диагностируется вскоре после рождения на основании тщательной клинической оценки и выявления характерных физических признаков (например, низкого роста, характерных черепно-лицевых и скелетных пороков развития, отсутствия или дефицита подкожного жира и т.д.). В некоторых случаях могут также проводиться специализированные тесты для выявления определенных отклонений, потенциально связанных с заболеванием. Например, рентгеновские исследования могут выявить и/или подтвердить широкие черепные швы и/или другие аномалии костей черепа. Кроме того, возможно, что компьютерная томография (КТ), магнитно-резонансная томография (МРТ) и/или другие специальные исследования могут выявить широкую потерю жировых покрытий (миелиновой оболочки) на нервных волокнах (демиелинизация) в белом веществе головного мозга (чистая суданофильная лейкодистрофия) или другие аномалии, как описано выше.
Стандартные методы лечения
Лечение синдрома Видемана-Раутенштрауха направлено на устранение конкретных симптомов, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий команды специалистов. Педиатрам, специалистам, которые оценивают и лечат расстройства нервной системы (неврологи), физиотерапевтам и/или другим медицинским работникам, может потребоваться систематическое и всестороннее планирование лечения пострадавшего ребенка.
Специфические методы лечения синдрома Видемана-Раутенштрауха являются симптоматическими и поддерживающими. В некоторых случаях, если пораженные младенцы и дети испытывают затруднения при глотании и кормлении и не могут правильно питаться через рот, в желудок или часть тонкой кишки может быть хирургическим путем введена трубка для обеспечения надлежащего питания (кормление через зонд). Кроме того, следует тщательно наблюдать за пораженными младенцами и детьми, чтобы не допустить респираторных инфекций. Генетическое консультирование будет полезно для пострадавших и их семей.

Высшее образование (Кардиология). Врач-кардиолог, терапевт, врач функциональной диагностики. Хорошо разбираюсь в диагностике и терапии заболеваний дыхательной системы, желудочно-кишечного тракта и сердечно-сосудистой системы. Закончила академию (очно), за плечами большой опыт работ.
Синдром Беквита - Видемана

Синдром Беквита - Видемана — нарушение физического развития человека. Заболевание может проявляться яркими симптомами или протекать незаметно. Чаще обращают внимание на внешние признаки, которые хорошо видны визуально: когда ребенок при рождении имеет массу тела свыше четырех килограммов, рост — от пятидесяти шести сантиметров. При синдроме различные части тела непропорциональны и даже асимметричны: часто увеличен язык, большие уши, слишком пышные щеки.
Есть видимые дефекты брюшины, например грыжа. Детки склонны к новообразованиям, врожденным порокам сердца. Диагностируются увеличенные внутренние органы и низкий уровень сахара в крови (гиперинсулинизм), что может вызывать судороги. Если патология слишком тяжелая, возможен летальный исход.
Часто люди даже не знают о такой патологии, как синдром Беквита-Видемана, но болезнь диагностируют нередко: согласно статистическим данным, на каждые четырнадцать тысяч новорожденных рождается один ребенок с патологией.
Заболевание наследственного характера, развивается из-за изменений в одиннадцатой хромосоме. Выявить патологию в пубертатном периоде нельзя. В девяноста процентах случаев в семейном анамнезе родителей генетических отклонений не было. Остальные двадцать процентов передают болезнь по наследству.
Этиология
Появиться синдром Беквита-Видемана может из-за таких причин:
- генетический фактор;
- спонтанная мутация.
В случае наследования передача происходит по аутосомно-доминантному типу: патология передается как мальчикам, так и девочкам. Симптомы заболевания не всегда будут проявляться у одного из родителей. Специалисты объясняют это как мозаичную форму, когда половина клеток с нормальным генотипом, а половина — изменены.
Бывает и спонтанное появление синдрома по невыясненным пока причинам, которые оказывают влияние на клетки зародыша еще в самом начале беременности.
Симптоматика
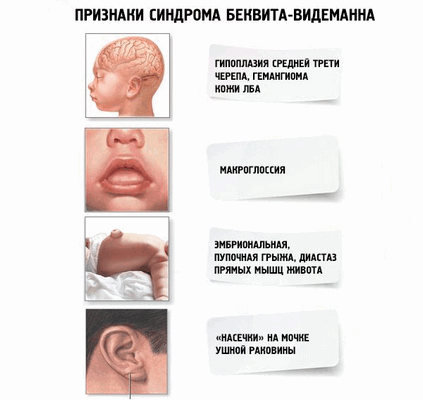
Патология имеет как внешние, так и внутренние признаки, указывающие на ее развитие:
- ребенок начинает быстро расти с раннего детства — темп роста начинает замедляться после восьми лет;
- в некоторых случаях аномальное развитие частей тела происходит только с одной стороны, поэтому возникает асимметричность внешнего вида;
- у некоторых детей слишком увеличивается язык, что вызывает трудности при глотании и дыхании;
- сильно увеличиваются органы брюшной полости;
- наблюдаются увеличенные ушные раковины;
- аномальное развитие почек;
- низкий уровень сахара в крови.
У детей повышен риск развития онкологии. Новообразованиям подвержены почки, могут возникнуть гепатобластомы и опухоль Вильмса.
При обращении будущих родителей к доктору будет проводиться генетическое тестирование, которое покажет возникшую мутацию. Это сложная процедура, помогающая диагностировать синдром в 80 % случаев.
Консультацию должен провести врач-генетик, который подскажет, какое обследование необходимо пройти, ведь процедура назначается индивидуально.
Выявить синдром Беквита-Видемана можно и у беременной женщины. Признаки патологии проявляются как осложнения при беременности:
- большое количество околоплодных вод;
- слишком высокий риск преждевременной родовой деятельности.
Выявить патологии можно во время проведения УЗИ.
Лечение
Здоровье ребенка необходимо постоянно контролировать — если понадобится, оказать ему врачебную помощь.
- в индивидуальном порядке необходимо следить за уровнем сахара в крови;
- в течение жизни больные люди должны внимательно следить за здоровьем, потому что высок риск возникновения новообразований.
Необходимо отметить, что взрослые люди, которым в раннем детстве ставили такой диагноз, вполне нормально живут, не испытывая дискомфорта.
Если у новорожденного слишком увеличен язык, назначают оперативное вмешательство. В ходе проведения операции выполняют коррекцию формы языка. Избежать хирургического вмешательства не удастся, потому что дефект будет создавать проблемы во время кормления ребенка и при дыхании. В будущем речь будет искажена.
Проблему с увеличенными внутренними органами и пупочной грыжей решают с помощью хирургического вмешательства.
Консультацию проводят иммунолог и ортопед.
Таким детям очень нужна забота родителей. Ребенок не должен находиться на сквозняке, переохлаждаться. К тому же его необходимо оберегать от инфекционных заболеваний, ведь у больных детей часто низкий иммунитет, когда даже обычное респираторное заболевание может оказаться большой проблемой. Родители даже при небольших проблемах со здоровьем должны обращаться за помощью к специалисту.
Профилактика
В качестве профилактики еще до планирования беременности можно пройти специальные тесты, которые помогут обнаружить изменения хромосом у будущих родителей. Об этом задумываются редко, узнавая об отклонении уже после рождения ребенка с таким диагнозом.
Заболевание чаще передается по наследству, поэтому если в семье кто-то болен, вероятность рождения ребенка с патологией составляет 55 %.
На сегодняшний день, к сожалению, нет медикаментозного лечения синдрома. Если пара, где один из родителей или оба больны, хочет завести детей, единственным методом рождения здорового ребенка будет искусственное оплодотворение.
Прогноз для жизни вполне благоприятен, если вовремя выявить болезнь и оказать ребенку медицинскую помощь. За новорожденным должны хорошо ухаживать родители. Спустя некоторое время темп роста приходит в норму, а пропорции тела восстанавливаются.
Читайте также: