Синдром Вилларе (Villaret) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 27.01.2026
Синдром Прадера-Вилли- врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 25000-10000 новорожденных. Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание. Заболевание характеризуется выраженной мышечной гипотонией при рождении, сохраняющейся в течение первого года жизни ребенка. Сосательный и глотательный рефлексы снижены, что затрудняет кормление ребенка. Из-за гипотонии у таких детей задерживается развитие двигательных функций: они с трудом учатся держать голову, сидеть и т. д. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает.
Позднее, к второму-четвертому году жизни появляются постоянное чувство голода и отсутствие насыщения, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей. Из-за тяжелого ожирения грозным осложнением является обструктивное апноэ (остановка дыхания) во сне.
Рост больных нередко снижен. Часто отмечается долихоцефалия (удлиненная форма головы), миндалевидный разрез глаз, низко расположенные ушные раковины, широкая переносица, маленький рот с тонкой верхней губой. Стопы и кисти больных диспропорционально маленькие (акромикрия). У 75% детей наблюдается слабая пигментация кожи, волос и радужки.
У мальчиков при рождении отмечается недоразвитие полового члена, мошонки,крипторхизм, а у девочек недоразвитие половых губ, иногда и матки. В дальнейшем заболевание проявляется задержкой или отсутствием полового созревания, бесплодием.
Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед.). Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарем, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, косоглазие.
Продолжительность жизни больных может достигать 60 лет и более. Нередко у таких детей развивается сахарный диабет.
Лечение
Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным.
Младенцы со сниженным мышечным тонусом должны получать массаж и другие виды специальной терапии. Комплекс лечебных мероприятий включает также диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). Рекомендуется терапия гормоном роста.
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Медико-генетическое консультирование
Родителям ребенка с синдромом Прадера-Вилли рекомендуется пройти генетическое обследование, прежде чем планировать дальнейшую беременность, поскольку существует риск того, что следующий ребенок у тех же родителей родится также с синдромом Прадера-Вилли, что зависит от механизма, вызвавшего генетический сбой.
Синдром Виларе
Данный лор синдром характеризуется рядом симптомов характерных поражению задней группы черепно-мозговых нервов, что возможно в результате получения травм головы и шеи, сдавления тканей новообразованием, развития выраженных воспалительных явлений провоцирующих отёк тканей.
Клиническая картина
Самым основным симптомом данного синдрома является паралич глотки с нарушением акта глотания. Второй по выраженности симптом - односторонний паралич мягкого нёба, с нарушением его подвижности, односторонний парез или паралич грудино-ключично-сосцевидной и трапециевидной мышц, парестезии с нарушением чувствительности в описанных выше анатомических зонах.
Диагностика
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
ВИЛЛАРЕ СИНДРОМ
сочетание синдрома Горнера и одностороннего паралича мягкого нёба, глотки, гортани, голосовой мышцы с парезом или параличом грудино-ключично-сосцевидной и трапециевидной мышц, обусловленное односторонним поражением языкоглоточного, блуждающего, добавочного, подъязычного нервов и шейного симпатического нерва при воспалительных процессах, травмах или новообразованиях в околоушной области.
Вилларе синдром (М. Villaret, 1877-1946, франц. невропатолог) — сочетание синдрома Горнера и одностороннего паралича мягкого неба, глотки, гортани, голосовой мышцы с парезом или параличом грудино-ключично-сосцевидной и трапециевидной мышц, обусловленное односторонним поражением языкоглоточного, блуждающего, добавочного, подъязычного нервов и шейного симпатического нерва при воспалительных процессах, травмах или новообразованиях в околоушной области.
(М. Villaret, 1877-1946, франц. невропатолог) сочетание синдрома Горнера и одностороннего паралича мягкого неба, глотки, гортани, голосовой мышцы с парезом или параличом грудино-ключично-сосцевидной и трапециевидной мышц, обусловленное односторонним поражением языкоглоточного, блуждающего, добавочного, подъязычного нервов и шейного симпатического нерва при воспалительных процессах, травмах или новообразованиях в околоушной области.
Синдром Прадера-Вилли: как распознать патологию развития?

Синдром Прадера-Вилли — генетическая патология, диагностируемая с частотой от 1:10 000 до 1:25 000 случаев. Причем количество людей с данным нарушением различается не только в зависимости от распространенности заболевания в различных регионах, но и напрямую коррелирует с развитостью медицинского обслуживания. При отсутствии генетика в клинике, стертых симптомах родители и окружающие нередко считают, что ребёнок просто «хорошо кушает», не любит активные игры и немного запаздывает в развитии, возможно, ему нужно больше внимания и дополнительных занятий. А к моменту поступления в школу выясняется, что к лишнему весу и отставанию в умственном развитии привели не социальные факторы, а генетическая патология, коррекцией которой надо было начинать заниматься с самого рождения.
Синдром Прадера-Вилли

Заболевание названо по именам исследователей, впервые описавших симптомокомплекс. Швейцарские педиатры Андреа Прадер и Генрих Вилли выделили из массы маленьких пациентов группу детей со схожими нарушениями в развитии и описали ее в середине прошлого века как отдельное заболевание.
Позднее было выявлено, что к возникновению данного синдрома приводит отсутствие или нарушение работы 7 генов (сегмент 11.2-q13) на хромосоме №15, причем из-за различия в симптомах при отсутствии или частичном нарушении выраженность заболевания может значительно варьироваться — от тяжелых случаев, диагностируемых при рождении, до стертых форм.
Изначальная причина нарушений в геноме не ясна, а вот наследуется эта патология всегда по мужской линии при наличии соответствующей генетической идеосомии у материи, и передается от отца к детям, хотя здоровые копии генов могут приводить к скрытому наследованию, и малыши с таким заболеванием рождаются и у людей без наличия синдрома.
Генетическая предрасположенность — дополнительный фактор, повышающий возраст постановки диагноза у малышей до предшкольного (и даже более позднего) обследования: внешние проявления синдрома Прадера-Вилли, достаточно нехарактерные для любой другой семьи, при явном генетическом наследовании выглядят очень логично: ребёнок просто похож на папу, и внешностью, и аппетитом, и характером.

Синдром Прадера-Вилли — достаточно редкая патология развития, однако опасна она не только своими проявлениями, но и неизвестностью. Характерная внешность детей, поведенческие нарушения, отставания в развитии могут сопутствовать не только множеству иных заболеваний, но и наследоваться от родителей. При такой картине понятно, что обращение к специалистам может затянуться на весь предшкольный период, а порой дети остаются без правильного диагноза и необходимого лечения, просто потому, что очень похожи на папу, а это считается нормой.
В моей практике был мальчик Володя, которого привели в развивающий центр в 8 лет с установкой «подготовить к школе». Предшкольное тестирование весной ребёнок не прошел из-за несоответствия уровня развития (память, внимание, мыслительные процессы) требованиям общеобразовательного учреждения, но родители считали, что всему виной то, что Вова не посещал детский садик, а проводил время в основном со старенькой бабушкой в деревне. Она же его и «раскормила» на козьем молоке, лишнего веса у мальчика было достаточно.
Володя действительно «проваливал» тесты на развитие по возрасту, отличался хорошим аппетитом, отличной зрительной памятью и склонностью к играм в лего-конструкторы, иногда упрямился и «взрывался». В целом он соответствовал уровню пятилетнего ребёнка. Довольно долго, месяца три после начала занятий, наши специалисты также считали причиной проблем мальчика социально-педагогическую запущенность. Но время шло, буквы не запоминались, а потом забирать Володю пришел папа с еще более выраженной внешней симптоматикой: невысокий рост, полнота, выраженная переносица, миндалевидные глаза, акромикрия и т. п. Вот с этого момента картина начала проясняться.
После длительных переговоров с мамой и уговоров папы Вова получил наконец консультацию у генетика и направление в школу специального вида. Стало ясно, что общеобразовательная школа пока не готова к обучению ребёнка, отсутствует класс коррекции, и выбрасывать деньги на изучение букваря в частном центре в данный момент смысла не имеет, нужны логопеды-дефектологи, эндокринолог, специальная диета и много работы, чтобы наверстать упущенное время и предупредить сопутствующие заболевания.
Интеллект, страдающий у больных синдромом Прадера-Вилли, в случае семейного наследования заболевания осложняет коррекцию. Нередко отец не хочет принять, что и сам он болен, и ребёнок нуждается в лечении. Частая смена настроения, повышенная ригидность, агрессивность, которые также являются симптомами болезни, приводят к необходимости длительной предварительной работы с членами семьи перед началом непосредственных занятий с детьми. Парам также необходима консультация генетика перед зачатием следующих детей.
Проявления генетической патологии у детей

Вероятность развития патологии можно заподозрить еще в процессе вынашивания. На необходимость консультации и обследования у генетика могут указывать пониженная активность плода, его неправильное положение в полости матки, многоводие, а также несоответствующий срокам уровень хорионического гонадотропина человека у беременной женщины.
Дети появляются на свет в срок, с симптомами незначительной внутриутробной асфиксии, в среднем каждый третий ребёнок рождается с ягодичным предлежанием. У мальчиков часто отмечают недоразвитие полового члена, мошонки, явления крипторхизма. У девочек — недоразвитие внешних половых органов или всей репродуктивной системы.
Первые годы жизни характеризуются отставанием в психомоторном развитии. Мышечная дистония различной степени тяжести приводит как к запаздыванию появления навыков держать головку, сидеть, ползать, так и может быть выражена нарушениями сосательного, глотательного рефлексов, в тяжелых случаях детей приходится кормить через зонд и подключать к аппарату искусственной вентиляции легких.
Явления дистонии проходят в среднем к 7-8 годам. Но высокой двигательной активностью дети не отличаются, чему способствует еще один фактор: ожирение.
В период 2-4 лет у малышей развивается постоянное чувство голода и отсутствует сигнал о насыщении, что приводит к быстрому образованию лишнего веса, откладывающемуся в основном на туловище и проксимальных областях конечностей. Ожирение может доходить до тяжелых стадий и грозить остановкой дыхания во сне (ночным апноэ).
К частым внешним проявлениям относят также низкий рост, долихоцефалию (удлиненную форму черепа), миндалевидную форму глаз, близорукость, миопию, низкое расположение ушей по сравнению с нормой, выраженную переносицу, тонкую верхнюю губу, акромикрию (непропорциональное развитие ступней и кистей рук, выглядящих слишком маленькими по сравнению с туловищем).
Ранее считалось, что синдром Прадера-Вилли обязательно вызывает значительную умственную отсталость и коэффициент интеллекта у взрослых составляет от 20 до 85, сопровождаясь идиотией.
В соответствии с исследованиями специалистов, установлено, что, в зависимости от выраженности симптоматики и коррекционной работы, только 40% больных отличаются интеллектом ниже среднего.
Ученые, работавшие с подростками, выявили еще более утешительные цифры: IQ может быть выше 85 баллов (нормой считается 90-115) у 5% больных, у 27% контрольной группы IQ составляет от 70 до 85 баллов, что соответствует невыраженной умственной отсталости, 39% детей отличались коэффициентом в 50-70 баллов (незначительная умственная отсталость), 27% — выраженная форма, и только 1% соответствовал критериям глубокой умственной отсталости.
Дети с данным синдромом характеризуются нестандартным развитием когнитивных способностей: имея развитые навыки визуального восприятия, чтения, они редко могут полностью выразить мысли или пересказать смысл прочитанного из-за неразвитости речи, то есть понимание превышает экспрессию.
Страдает восприятие информации на слух, логическое мышление, мелкая и крупная моторика, зрительная память. Однако специалисты утверждают, что, используя сильные стороны и корректируя слабые, можно добиться значительного улучшения всех мыслительных процессов.
Лечение, коррекция и осложнения заболевания

Специфической терапии, позволяющей изменить геном или устранить последствия хромосомных нарушений, не существует. Поэтому при работе с маленькими пациентами специалисты выбирают симптоматическое, коррекционное направление и профилактику осложнений.
Для снижения выраженности эндокринных нарушений необходима строгая диета (для детей-дошкольников менее ограниченная, но хорошо сбалансированная, для детей старше с калорийностью не более 1000 ккал в день), режим питания, контроль вплоть до запирания продуктов в отдельных шкафах. По назначению проводят гормонотерапию, инъекции рекомбинантного гормона роста и стимуляцию полового развития в случае нарушений полового развития.
Эндокринолог необходим детям с симптомом Прадера-Вилли и по причине вероятности развития сахарного диабета из-за снижения толерантности к глюкозе.
К подростковому периоду может возникнуть необходимость в наблюдении у психиатра: дети с синдромом Прадера-Вилли нередко страдают обсессивно-компульсивным расстройством, могут проявлять самоагрессию, отличаться высокой тревожностью.
Узкие специалисты, о которых нельзя забывать:
- стоматолог, из-за вязкости слюны наблюдается раннее возникновение кариеса;
- окулист, часто синдром сопровождается миопией и косоглазием;
- ортопед: гипотония мышц вызывает ортопедические нарушения, сколиоз;
- андролог и гинеколог: синдром Прадера-Вилли может сопровождаться нарушениями развития органов половой сферы.
Необходимо помнить, что из-за своих особенностей и дети, и взрослые с синдромом Прадера-Вилли могут испытывать затруднения в определении своего самочувствия, не уметь выразить вербально свои мысли, чувства, не обращаться вовремя к родителям или за медицинской помощью, из-за чего процент хронических соматический заболеваний в данной группе катастрофически высок.
Следить за здоровьем ребёнка с данным диагнозом надо с раннего младенчества, при этом обучая обращать внимание на признаки заболеваний, сообщать о них, понимать, когда и какая помощь требуется. При неполном нарушении работы генов и курсе коррекции дети с синдромом Прадера-Вилли вырастают во взрослых, способных к самостоятельной жизни, работе, образованию семьи и воспитанию собственных здоровых детей при условии своевременной консультации у генетика.
Синдром Прадера-Вилли - это заболевание генетического плана, встречающееся крайне редко. Его развитие обусловлено тем, что семь генов, либо их частей, располагающиеся на 15 отцовской хромосоме отсутствуют, либо не могут нормально функционировать. Синдром был описан в 1956 году учёными А. Прадером, Г. Вилли, А. Лабхартом, Э. Зиглером и Г. Фанкони.
Согласно статистике, синдром выявляют у 1 новорождённого из 10-25 тысяч. На манифестацию патологии оказывает влияние отцовский генетический материал, так как нарушенная часть 15 хромосомы подвержена явлению импринтинга. То есть, копия лишь одного гена среди генов данного региона будет работать в полном объёме.
Симптомы синдрома Прадера-Вилли
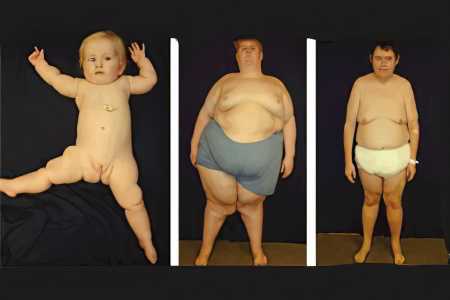
Выставить больному верный диагноз позволяют следующие симптомы синдрома Прадера-Вилли, даже если они проявляются не в полном объёме:
Во время внутриутробного развития снижена двигательная активность плода, женщины часто страдают от многоводия, обнаруживается предлежание плода.
Во время рождения ребёнок находится в ягодичном предлежании, наблюдается летаргия и гипотония. Слабый мышечный тонус оказывает влияние на сосательный рефлекс, провоцируя трудности во время грудного вскармливания. Ребенок может испытывать проблемы с дыханием. Синдром гипогонадизма характерен для данного генетического заболевания.
В раннем детстве на первый план выходит отставание в физическом развитии, наблюдаются проблемы в интеллектуальном плане. Ребенок быстро утомляется, склонен к сонливости. Нередко развивается косоглазие. Именно для раннего детства характерен сколиоз, который в младенчестве не диагностируется.
Для более старших детей характерны нарушения в речевом плане, наблюдается избыточная масса тела. В возрасте от 2 до 8 лет ребёнок начинает проявлять склонности к перееданию, нарушается физическая координация, страдает сон. Сколиоз продолжает прогрессировать.
В пубертатном периоде у подростков часто наблюдается задержка полового созревания, рост таких детей ниже их сверстников. Гибкость аномально высокая, масса тела превышает норму.
К 18 годам люди с синдромом Прадера-Вилли страдают от невозможности зачать ребёнка, так как являются бесплодными. Волосы на интимной зоне жидкие, продолжает прогрессировать гипогонадизм. Ожирение, низкое давление, проблемы в интеллектуальной сфере, трудности в обучении - всё это симптомы, вызванные имеющимися генетическими нарушениями. Кроме того, в период совершеннолетия человеку уже может быть выставлен диагноз «сахарный диабет», спровоцированный имеющейся генной патологией.
Взрослые люди с симптомом Прадера-Вилли имеют следующие внешние характеристики: нос у них широкий и большой, лоб высокий, веки опущены, глаза миндалевидные, верхние и нижние конечности маленькие, пальцы на них узкие, масса тела избыточная. По сравнению с остальными близкими родственниками, волосы и кожа больного члена семьи несколько светлее. Половое и моторное развитие человека нарушено, на коже образуются стрии, имеется склонность к дерматилломания (выщипыванию участков кожи).
Нейро-когнитивные нарушения
В 1992 году Курф и Фрим исследовали уровень интеллекта у людей с синдром Прадера-Вилли. Было установлено, что большая часть больных (39%) имеют незначительную умственную отсталость, по 27% пациентов имели умеренную умственную отсталость, или находились на границе интеллектуальной деятельности, выдавая уровень интеллекта в диапазоне от 70 до 85. С низким средним уровнем интеллекта было выявлено 5% людей, а с тяжёлой умственной отсталостью 1%. При этом глубокая умственная отсталость наблюдалась менее, чем в 1% случаев.
Установлено, что в детском возрасте у больных наблюдаются способности к чтению, они имеют обширный словарный запас. Тем не менее, речевые нарушения снижают их понимание. С трудом таким людям даётся математика и письмо, страдает концентрация внимания, зрительная и краткосрочная память. Поэтому, даже несмотря на уровень развития интеллекта, с возрастом проблемы в указных сферах не пропадают.
Поведенческие нарушения
Повышенный аппетит приводит к развитию ожирения.
Компульсивное поведение выражается в повышенном уровне тревожности и в дерматилломания.
Нарушения в психическом развитии проявляются в депрессиях, паранойе, галлюцинациях.
Часто, именно поведенческие расстройства приводят к тому, что больных госпитализируют.
Эндокринные нарушения
У людей с синдромом Прадера-Вилли наблюдается нехватка в организме гормона роста, что приводит к развитию ожирения, повышению плотности костной ткани.
Гипогонадизм является причиной того, что яички недоопускаются у мужчин в мошонку, а у женщин наблюдается раннее половое оволосение (адренархе). Однако, оба этих состояния можно откорректировать хирургическим и консервативным путём.
Причины синдрома Прадера-Вилли
Причины синдрома Прадера-Вилли кроются в генетических нарушениях. В результате определённых мутаций, происходит потеря участка 15 хромосомы отцовской копии генов.
Ещё причиной хромосомных перестроек, кроме мутации генов являются:
Унаследование двух пар хромосом только от матери (материнская униотцовская дисомия);
Нарушения, возникшие в результате разрыва хромосомы или по причине неравного кроссинговера (делеция);
Нарушения в результате хромосомных транслокаций с переносом участка хромосомы в негомологичную хромосому.
При этом риск того, что в семье второй ребёнок родится с синдромом Прадера-Вилли, зависит от того, что стало причиной перестройки. Так, униотцовкая диссомния и делеция сводят риск лишь к 1%, хромосомные транслокации повышают его до 25%, а мутации на фоне импринтинга до 50%. Паренатальное тестирование все эти варианты позволяет просчитать.
Диагностика синдрома Прадера-Вилли
Своевременная диагностика синдрома Прадера-Вилли позволяет не только выявить имеющиеся нарушения, но и начать незамедлительное лечение, которое намного улучшает прогноз развития патологии.
Если ранее диагноз выставлялся лишь на основе клинических признаков, то современная медицина использует метод генетического тестирования. Его обязательно необходимо провести всем новорождённым с гипотонией.
Что касается дифференциальной диагностики, то данную генетическую патологию необходимо отличать от синдрома Дауна.
Лечение синдрома Прадера-Вилли
Лекарств, позволяющих полностью избавиться от генетических нарушений, не существует. Однако, препараты для лечения синдрома Прадера-Вилли, а вернее для купирования его симптомов, находятся на стадии разработки.
Поэтому врачи рекомендуют в обязательном порядке всем детям с обнаруженным синдромом Прадера-Вилли проходить физиотерапию, которая направлена на повышение мышечного тонуса. Процесс обучения также должен быть адаптирован под интеллектуальные возможности ребёнка.
Большой проблемой остаётся вопрос ожирения, поэтому таким детям рекомендовано каждый день делать инъекции рекомбинантного гормона роста. Это позволяет контролировать аппетит, а также даёт возможность оказать поддержку в увеличении мышечной массы, а не жировой ткани.
Чтобы исключить обструктивное апноэ сна, больным на постоянной основе рекомендуют использовать аппарат для вспомогательной вентиляции лёгких.
Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность - "Лечебное дело" в 1991 году, в 1993 году "Профессиональные болезни", в 1996 году "Терапия".
Наши авторы
Читайте также: