Талассемии у детей: клиника, диагностика, лечение
Добавил пользователь Morpheus Обновлено: 22.01.2026
Талассемии представляют собой гетерогенную группу гемоглобинопатии, в основе которых лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина А. Талассемия - это мишеневидноклеточная анемия с нарушенным соотношением НЬА и HbF по биохимическим показателям; при этом возможна частичная недостаточность определенной цепи или ее полное отсутствие при преобладании другой цепи. Так, при нарушении синтеза ß-цепи будут преобладать а-цепи и наоборот. Бета-талассемия обусловлена снижением продукции ß-цепей гемоглобина. Неповрежденные а-цепи избыточно накапливаются в клетках эритропоэза, что ведет к повреждению мембраны и разрушению как клеток эритроидного ряда в костном мозге, так и эритроцитов в периферической крови; развиваются неэффективный эритропоэз и гемолиз с гипохромией эритроцитов, ибо содержание гемоглобина в эритроцитах недостаточно. Первыми описали ß-талассемию американские педиатры Кули и Ли в 1925 г. Тяжелая гомозиготная форма ß-талассемии получила название болезни Кули, или большой талассемии. Кроме того, по выраженности анемии и других клинических симптомов выделяют промежуточную, малую и минимальную талассемию. Помимо стран Средиземноморья талассемия встречается во Франции, Югославии, Швейцарии, Англии, Польше, а также у жителей Закавказья и Средней Азии, где в некоторых регионах частота носительства достигает 10-27 %.
Что провоцирует / Причины Талассемии:
При талассемии нарушается синтез одной из четырёх цепей глобина. Наследование патологии от одного (гетерозиготность) или обоих родителей (гомозиготность), тип нарушенной цепи определяют выраженность клинических проявлений. Причины повышенной гибели эритроцитов связаны с нарушенной структурой клетки из-за неправильного соотношения цепей глобина в гемоглобине. Кроме укорочения жизни эритроцитов при данном заболевании происходит гибель клеток предшественников эритроцитов в костном мозге.
Патогенез (что происходит?) во время Талассемии:
Патогенез ß-талассемии связан с мутацией в локусе ß-глобина на 11-й паре хромосом, нарушающей синтез ß-глобиновой цепи. Вследствие неадекватного синтеза гемоглобина развивается гипохромная анемия. Преципитаты избыточного количества а-цепей удаляются из эритроцитов и эритрокариоцитов клетками ретикулогистиоцитарной системы; при этом клетки повреждаются и быстрее разрушаются. Таков механизм неэффективного эритропоэза и гемолиза эритроцитов и ретикулоцитов; гибель последних происходит в селезенке. При ß-талассемии накапливается также HbF, обладающий большим сродством к кислороду; однако отдача его тканям затруднена, что приводит к их гипоксии. Неэффективный эритропоэз способствует расширению плацдарма кроветворения, что отражается на структуре скелета; вместе с тем деструкция эритрокариоцитов в костном мозге ведет к повышенному всасыванию железа и патологической перегрузке организма железом. Гематологические признаки ß-талассемии иногда выявляются у больных анемией среди русских.
Симптомы Талассемии:
Клиника большой талассемии проявляется уже в детстве. У больных детей своеобразный башенный череп, монголоидное лицо с увеличенной верхней челюстью. Ранний признак болезни Кули - сплено- и гепатомегалия, развивающиеся за счет экстрамедуллярного кроветворения и гемосидероза. Со временем у них формируются цирроз печени, сахарный диабет в результате фиброза поджелудочной железы, а гемосидероз миокарда приводит к застойной сердечной недостаточности. Гомозиготная бета-талассемия (большая талассемия, анемия Кули) характеризуется резким снижением образования HbA1, значительным увеличением содержания HbF, низким, нормальным или повышенным содержанием НbA2. Содержание НbF может колебаться от 30 до 90 %, иногда бывает ниже 10%. Течение заболевания характеризуется тяжелой гемолитической анемией, проявляющейся к концу первого года жизни ребенка, гепато- и спленомегалией, монголоидиостью лица и башенным черепом, отставанием ребенка в физическом развитии, нередко желтушностью и бледностью кожных покровов. У части больных развиваются язвы в области голеней. Рентгенологически обнаруживают симптом «ежика» или «щетки», который положителен при увеличении содержания HbF, отрицателен при увеличении процента НbA2. У детей в возрасте от 6 мес. до 1 года в мелких костях стоп и кистей выявляется истончение коркового слоя со вздутием кости и образованием грубосетчатой структуры костного мозга. Начиная с 1-го года жизни ребенка отмечается нарушение развития костей, быстро прогрессирующее до периода полового созревания. Длительно продолжающийся гемолиз (ретикулоцитоз, увеличение свободной фракции билирубина сыворотки крови, уробилинурия, гиперсидеремия), частые переливания эритроцитной массы приводят к развитию гемосидероза печени и селезенки. Нередко происходит образование билирубиновых камней в желчных путях. Уровень гемоглобина достигает 30-50 г/л, цветовой показатель 0>5 и ниже. В мазках крови обнаруживают мишеневидные эритроциты, отличающиеся малым содержанием гемоглобина и укорочением продолжительности жизни, анизопойкилоцитоз, эритро- и нормобласты. Отмечается повышение осмотической стойкости эритроцитов, лейкопения (в период гемолитического криза). В костном мозге - раздражение эритро-нормобластического ростка. Иногда возникают апластический криз или явления гиперспленизма. При тяжелой гомозиготной талассемии больные умирают на первом году жизни, при сравнительно более спокойной форме заболевания они могут дожить до взрослого возраста. Гетерозиготная бета-талассемия протекает в виде как бессимптомной, так и манифестной форм с незначительно увеличенной селезенкой, специфическими костными изменениями, нередко выраженной гипохромной анемией, часто анизоцитозом, пойкилоцитозом и мишеневидностью эритроцитов, повышенной их осмотической резистентностью увеличением количества НbA2 (примерно до 8% от общего гемоглобина), у части больных - HbF (до 5%). При гетерозиготной дельтабета-талассемии (F) отмечается высокое содержание HbF при нормальном уровне НbA2. Клинические признаки и гематологические сдвиги аналогичны встречающимся при гетерозиготной бета-талассемии. Гомозиготные формы дельтабета-талассемии (F) проявляются почти теми же клинико-гематологическими нарушениями, что и гомозиготная бета-талассемия. У больных с этой формой заболевания обнаруживается только HbF. Среди больных талассемией удается выделить лиц с гетеро-и гомозиготными формами А2F-талассемии, которые по признакам, характеризующим их течение, по существу мало отличаются от бета-талассемии. В группе больных бета-талассемией случаи большой талассемии с выраженными клиническими проявлениями встречаются реже, чем промежуточные и малые формы. При обследовании родственников больных чаще обнаруживается минимальная форма бета-талассемии. Выделяют следующие формы а-талассемии: водянка плода с гемоглобином Bart's (у4). гемоглобинопатия Н (бета4), а-талассемия-1 и а-талассемия-2. Водянка плода представляет собой гомозиготное состояние (по генам а-th-l), несовместимое с жизнью. Беременность в подобных случаях непроизвольно прерывается и у плода выявляют водянку мозга, гепатомегалию. Электрофоретическим исследованием гемоглобина обнаруживается Hb Bart's (80-90%, сочетающийся со следами НbН. Гемоглобинопатия Н - один из вариантов а-талассемии - проявляется гемолитической анемией, увеличением селезенки, тяжелым течением костных изменений. Картина периферической крови характеризуется понижением содержания гемоглобина, анизо- и пойкилоцитозом, гипохромией и множественными включениями в эритроцитах (выпавший в осадок гемоглобин Н). Гетерозиготные формы а-талассемии выявляются у родственников больных гемоглобинопатией Н. а-Талассемия-1 (малая форма заболевания) возникает при сочетании гена а-th-l с нормальным геном a-цепочкового синтеза. Она характеризуется небольшой анемией, умеренным анизо- и пойкилоцитозом, внутриэритроцитарными включениями, повышенной осмотической резистентностью эритроцитов. У взрослых больных а-талассемией-1 гемоглобиновые фракции бывают в пределах нормы, у новорожденных выявляется Hb Bart's (5-10%). а-Талассемия-2 (минимальная форма заболевания) развивается при сочетании гена a-th-2 с нормальным геном а-цепочкового синтеза. Клинические проявления отсутствуют.
Диагностика Талассемии:
При анализе крови определяется гипохромная гиперрегенераторная анемия разной степени тяжести. В мазке крови обнаруживают гипохромные эритроциты малых размеров, мишеневидные, различной формы; много нормоцитов. В биохимическом анализе крови выявляются гипербилирубинемия за счет свободной фракции, гиперсидеремия, снижение ОЖСС, повышение активности ЛДГ. В эритроцитах повышен уровень фетального гемоглобина. Альфа-талассемия распространена преимущественно в Юго-Восточной Азии, Китае, Африке и в Средиземноморье. Синтез а-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. В то же время агрегаты из ß-цепей, количество которых при а-талассемии обнаруживают в избытке, более растворимы, чем агрегаты из а-цепей, поэтому гемолиз при а-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен. Следовательно, клинические и лабораторные данные при а-талассемии выражены менее отчетливо, чем при ß-талассемии; их основное отличие в биохимическом составе гемоглобина эритроцитов: при а-талассемии уменьшено содержание а-цепей гемоглобина. Пренатальная (дородовая) диагностика Если оба родителя страдают талассемией, целесообразно исследование плода в период беременности на предмет заболевания талассемией с целью возможного своевременного прерывания беременности. Обнаружение т.н. «гомозиготных» (более тяжелых) форм талассемии у плода- показание для прерывания беременности. Применяется 2 основных метода- фетоскопия и амниоцентез. Оба они связаны с получением клеток плода с помощью пункции через переднюю брюшную стенку (первая из них делается под контролем УЗИ) с последующим медико-генетическим исследованием полученных клеток. Врач-генетик определит предпочтительный метод исследования в зависимости от срока беременности, данных УЗИ и индивидуальных особенностей беременной. Оба метода имеют свой риск, в первую очередь - преждевременные роды. Имеется также, хотя и очень небольшой, риск присоединения инфекции и даже гибели плода (по данным литературы- около 3%). Для решения вопроса о планировании семьи людям, имеющим родственников, больных талассемией, обязательно следует обращаться к генетику и он назначит Вам, если нужно, необходимое дородовое обследование.
Лечение Талассемии:
Профилактика Талассемии:
Профилактика талассемии основывается на выявлении подверженных риску лиц посредством программ скрининга носителей или изучения историй семьи и предоставления адекватной информации о риске и о возможностях сокращения такого риска. Бета-талассемия обладает уникальным свойством: здоровых носителей можно определять простым, недорогим и точным анализом крови. Таким образом, можно выявлять пары носителей и информировать о генетическом риске до того, как они создадут семью. Скрининг - это недорогой и доступный способ выявления носителей, который можно предлагать в широкой гамме ситуаций в различных условиях: в средней школе, перед вступлением в брак или в женских консультациях. Пары выявленных таким образом носителей информируются о генетическом риске и о возможных вариантах его снижения, которые обычно включают дородовую диагностику. Большинство пар, подверженных риску талассемии, обращаются за дородовой диагностикой гемоглобинопатии. Стандартный метод диагностики - это взятие пробы ворсинок хорионов и анализ ДНК при сроке беременности 10-12 недель. Программы скрининга и консультирование могут привести к серьезному сокращению подверженных болезни новорожденных. Эти программы можно анализировать благодаря беседам с родителями, имеющими больных детей, хотя у них был доступ к скринингу и консультированию. В большинстве случаев рождение затронутых болезнью детей является результатом неспособности систем здравоохранения адекватно информировать родителей о возможном риске и мерах профилактики, а не тем, что они отвергают тестирование плода. Выбор соответствующей стратегии для введения профилактики талассемии зависит от конкретных условий. В некоторых обществах начинать можно с обеспечения дородовой диагностики для тех пар, которые уже знают о существовании риска либо благодаря скринингу, либо потому, что у них больной ребенок. Такой подход значительно сокращает число новорожденных с заболеванием. С другой стороны в обществах, где дородовая диагностика еще не обеспечена, можно предложить скрининг для лиц репродуктивного возраста. Эта стратегия ведет к меньшему сокращению числа детей с заболеванием, однако обычно стимулирует спрос на службы дородовой диагностики. В настоящее время во многих странах имеются примеры эффективного применения методов профилактики талассемии на основе различных программ скрининга носителей. Например, в Греции, на Кипре, в Исламской Республике Иран и Италии скрининг на талассемию до вступления в брак является стандартной; большинство пар, подверженных риску, определяется достаточно своевременно, чтобы предложить раннюю диагностику на первой беременности. Большинство таких пар пользуются этой услугой и имеют здоровых детей. В Соединенном Королевстве Великобритании и Северной Ирландии и других странах северо-западной Европы, где дородовая диагностика широко распространена, скрининг предлагается во время беременности. Программы скрининга нуждаются в поддержке в виде просвещения общественности и регламентарных структур, с тем чтобы отдельные лица могли принимать обоснованные решения и чтобы люди были защищены от дискриминации на основании результатов их тестов. Некоторые национальные программы, цель которых - содействие скринингу носителей, в свою очередь стимулировали социальные изменения, включая принятие во многих странах прекращения беременности в тех случаях, когда доказано, что плод страдает серьезным генетическим расстройством. Это привело к разработке соответствующих технологий и служб в Бахрейне, Исламской Республике Иран и Саудовской Аравии. Масштабы принятия и внедрения программ профилактики расширяются во многих частях Азии, например в Индии, Индонезии, Китае, Малайзии, на Мальдивских Островах, в Сингапуре и Таиланде. Генетическое консультирование играет важную роль в защите автономии индивидуума или пары и в осуществлении ими своего права на максимальную информацию о болезни и имеющихся возможностях. Эффективность служб по борьбе против талассемии зависит от учета их сотрудниками культурной практики и принятия действий, соответствующих данному социальному контексту. При консультировании также необходимо учитывать культурные, религиозные и этические взгляды личности или пары. Успех генетического консультирования в значительной степени определяется его просветительным, добровольным и непредписывающим характером.
К каким докторам следует обращаться если у Вас Талассемия:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Талассемии, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Талассемия
Талассемия - наследственные гемоглобинопатии, характеризующиеся угнетением синтеза цепочечных белковых молекул, образующих структуру гемоглобина. Это приводит к повреждению мембраны эритроцитов и разрушению красных клеток крови с развитием гемолитических кризов. Признаками талассемии служат характерные костные изменения, гепатоспленомегалия, анемический синдром. Диагноз талассемии подтверждается клиническими и лабораторными данными (исследованием гемограммы, гемоглобина, миелограммы, электрофоретическим методом). Возможна пренатальная диагностика талассемии. В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
МКБ-10



Общие сведения
Талассемия - группа генетически детерминированных болезней крови, развивающихся при нарушении синтеза a- или β-цепей гемоглобина, сопровождающихся гемолизом, гипохромной анемией, микроцитозом. В гематологии талассемия относится к наследственным гемолитическим анемиям - количественным гемоглобинопатиям.
Талассемия широко распространена среди населения Средиземноморского и Черноморского региона; название заболевания буквально переводится как «анемия морского побережья». Также случаи талассемии нередки в странах Африки, Ближнего Востока, Индии и Индонезии, Средней Азии и Закавказья. С синдромом талассемии каждый год в мире рождается 300 тыс. детей. В зависимости от формы патологии течение талассемии может быть тяжелым, фатальным или легким, бессимптомным. Так же, как серповидно-клеточная анемия, талассемия играет роль защитного фактора против малярии.

Причины талассемии
Талассемия является генетическим заболеванием с аутосомно-рецессивным наследованием. Непосредственной причиной патологии выступают различные мутационные нарушения в гене, кодирующем синтез той или иной цепи гемоглобина. Молекулярную основу дефекта могут составлять синтез аномальной матричной РНК, делеции структурных генов, мутации регуляторных генов либо их неэффективная транскрипция. Следствием подобных нарушений служит снижение или отсутствие синтеза одной из полипептидных гемоглобиновых цепей.
Так, при b-талассемии бета-цепи синтезируются в недостаточном количестве, что приводит к избытку альфа-цепей, и наоборот. Избыточно продуцируемые полипептидные цепи откладываются в клетках эритроидного ряда, вызывая их повреждение. Это сопровождается деструкцией эритрокариоцитов в костном мозге, гемолизом эритроцитов в периферической крови, гибелью ретикулоцитов в селезенке. Кроме этого, при b-талассемии в эритроцитах накапливается фетальный гемоглобин (НbF), не способный транспортировать кислород в ткани, что вызывает развитие тканевой гипоксии. Вследствие костномозговой гиперплазии развивается деформация костей скелета. Анемия, тканевая гипоксия и неэффективный эритропоэз в той или иной степени нарушают развитие и рост ребенка.
Для гомозиготной формы талассемии характерно наличие двух дефектных генов, унаследованных от обоих родителей. При гетерозиготном варианте талассемии пациент является носителем мутантного гена, унаследованного от одного из родителей.
Классификация
С учетом поражения той или иной полипептидной цепи гемоглобина различают:
- a-талассемию (с подавлением синтеза альфа-цепей HbA). Данная форма может быть представлена гетерозиготным носительством манифестного (α-th1) или немого (α-th2) гена; гомозиготной a-талассемией (водянкой плода с гемоглобином Бартса); гемоглобинопатией Н
- b-талассемию (с подавлением синтеза бета-цепей HbA). Включает в себя гетерозиготную и гомозиготную β-талассемию (анемию Кули), гетерозиготную и гомозиготную δβ-талассемию (F-талассемию)
- γ-талассемию (с подавлением синтеза гамма-цепей гемоглобина)
- δ-талассемию (с подавлением синтеза дельта-цепей гемоглобина)
- талассемию, обусловленную нарушением структуры гемоглобина.
Симптомы талассемии
Признаки большой (гомозиготной) b-талассемии проявляются уже в течение 1-2-го года жизни ребенка. Больные дети имеют характерное монголоидное лицо, седловидную переносицу, башенный (четырехугольный) череп, гипертрофию верхней челюсти, нарушение прикуса, гепато- и спленомегалию. Проявлениями анемизации служат бледный или землисто-желтушный цвет кожных покровов.
Поражение трубчатых костей сопровождается отставанием в росте и патологическими переломами. Возможно развитие синовита крупных суставов, калькулезного холецистита, язв нижних конечностей. Фактором, осложняющим течение b-талассемии, выступает гемосидероз внутренних органов, приводящий к развитию цирроза печени, фиброза поджелудочной железы и, как следствие, - сахарного диабета; кардиосклероза и сердечной недостаточности. Больные восприимчивы к инфекционным заболеваниям (кишечным инфекциям, ОРВИ и др.), возможно развитие тяжелых форм пневмонии и сепсиса.
Малая (гетерозиготная) b-талассемия может протекать бессимптомно или с минимальными клиническими проявлениями (умеренным увеличением селезенки, незначительно выраженной гипохромной анемией, жалобами на повышенную утомляемость). Аналогичная симптоматика сопровождает течение гетерозиготной формы a-талассемии.
При гомозиготной форме a-талассемии альфа-цепи полностью отсутствуют; фетальный гемоглобин у плода не синтезируется. Данная форма талассемии несовместима с жизнью, что приводит к внутриутробной гибели плода вследствие развивающегося синдрома водянки или самопроизвольному прерыванию беременности. Течение гемоглобинопатии Н характеризуется развитием гемолитической анемии, спленомегалии, тяжелых костных изменений.
Диагностика
Талассемию следует заподозрить у лиц с семейным анамнезом, характерными клиническими признаками и лабораторными показателями. Больные талассемией нуждаются в консультации гематолога и медицинского генетика.
Типичными гематологическими изменениями служат снижение уровня гемоглобина и цветового показателя, гипохромия, наличие мишеневидных эритроцитов, повышение уровня железа сыворотки крови и непрямого билирубина. Электрофорез Hb на ацетат-целлюлозной пленке используется для определения различных гемоглобиновых фракций. При изучении пунктата костного мозга обращает внимание гиперплазия красного кроветворного ростка с высоким числом эритробластов и нормобластов. Молекулярно-генетические исследования позволяют выявить мутацию в локусе a- или β-глобина, нарушающую синтез полипептидной цепи.
На краниограммах при большой b-талассемии выявляется игольчатый периостоз (феномен «волосатого черепа»). Характерна поперечная исчерченность трубчатых и плоских костей, наличие мелких очагов остеопороза. С помощью УЗИ брюшной полости обнаруживается гепатоспленомегалия, камни желчного пузыря.
При подозрении на талассемию требуется исключить железодефицитную анемию, наследственный микросфероцитоз, серповидно-клеточную анемию, аутоиммунную гемолитическую анемию. В семьях, имеющих больных талассемией, рекомендуется проведение генетического консультирования супругов и инвазивной дородовой диагностики (биопсии хориона, кордоцентеза, амниоцентеза) для выявления гемоглобинопатии на ранних сроках беременности. Подтверждение гомозиготных форм талассемии у плода служит показанием для искусственного прерывания беременности.
Лечение талассемии
Лечебная тактика при различных формах талассемии неодинакова. Так, пациенты с малой b-талассемией в лечении не нуждаются. С другой стороны, больным с гомозиготной b-талассемией с первых месяцев жизни требуется проведение гемотрансфузионной терапии (переливание размороженных или отмытых эритроцитов), введение хелатирующих препаратов, связывающих железо (дефероксамина), глюкокортикоидов при возникновении гемолитических кризов. При всех формах талассемии показан прием препаратов фолиевой кислоты и витаминов группы В.
При гиперспленизме (особенно на фоне гемоглобиноза Н) требуется удаление селезенки (спленэктомия). Из-за склонности к присоединению инфекционных осложнений больным рекомендуется обязательная вакцинация против пневмококковой инфекции. Многообещающим методом лечения талассемии служит трансплантация костного мозга от гистосовместимого донора.
Прогноз
Прогноз больших форм талассемии неблагоприятный; больные погибают в младенческом или молодом возрасте. При гетерозиготной бессимптомной форме талассемии продолжительность и качество жизни в большинстве случаев не страдают. Первичная профилактика талассемии включает предупреждение браков между гетерозиготными носителями генов заболевания, а при высоком генетическом риске рождения больного потомства - отказ от деторождения.
Гемолитическая анемия
Гемолитическая анемия - патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для данной группы заболеваний типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.



Гемолитическая анемия (ГА) - малокровие, обусловленное нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Данная группа анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных когортах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. Патология характеризуется укорочением жизненного цикла эритроцитов и их распадом (гемолизом) раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).

Причины
Этиопатогенетическую основу наследственных гемолитических синдромов составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфофункциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды, среди которых:
- Аутоиммунные процессы. Образование антител, агглютинирующих эритроциты, возможно при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии). Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода.
- Токсическое действие на эритроциты. В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Вызывать разрушение клеток крови может прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков).
- Механическое повреждение эритроцитов. Гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Патогенез
Центральным звеном патогенеза ГА является повышенное разрушение эритроцитов в органах ретикулоэндотелиальной системы (селезенке, печени, костном мозге, лимфатических узлах) или непосредственно в сосудистом русле. При аутоиммунном механизме анемии происходит образование антиэритроцитарных АТ (тепловых, холодовых), которые вызывают ферментативный лизис мембраны эритроцитов. Токсические вещества, являясь сильнейшими окислителями, разрушают эритроцит за счет развития метаболических, функциональных и морфологических изменений оболочки и стромы красных кровяных телец. Механические факторы оказывают прямое воздействие на клеточную мембрану. Под влиянием этих механизмов из эритроцитов выходят ионы калия и фосфора, а внутрь поступают ионы натрия. Клетка разбухает, при критическом увеличении ее объема наступает гемолиз. Распад эритроцитов сопровождаются развитием анемического и желтушного синдромов (так называемой «бледной желтухой»). Возможно интенсивное окрашивание кала и мочи, увеличение селезенки и печени.
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные. Наследственные ГА включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз - болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) - анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) - анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии- анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные ГА подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия - б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные) - обусловлены воздействием антител
- токсические - анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- механические - анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Симптомы
Наследственные мембранопатии, ферментопении и гемоглобинопатии
Наиболее распространенной формой данной группы анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке. Манифестация микросфероцитарной ГА возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом - обтурационная желтуха. При микросфероцитозе во всех случаях увеличена селезенка, а у половины пациентов - еще и печень. Кроме наследственной микросфероцитарной анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Энзимопенические анемии связаны с недостатком определенных ферментов эритроцитов (чаще - Г-6-ФД, глутатион-зависимых ферментов, пируваткиназы и др). Гемолитическая анемия может впервые заявлять о себе после перенесенного интеркуррентного заболевания или приема медикаментов (салицилатов, сульфаниламидов, нитрофуранов). Обычно заболевание имеет ровное течение; типична «бледная желтуха», умеренная гепатоспленомегалия, сердечные шумы. В тяжелых случаях развивается ярко выраженная картина гемолитического криза (слабость, рвота, одышка, сердцебиение, коллаптоидное состояние). В связи с внутрисосудистым гемолизом эритроцитов и выделением гемосидерина с мочой последняя приобретает темный (иногда черный) цвет. Особенностям клинического течения гемоглобинопатий - талассемии и серповидно-клеточной анемии посвящены самостоятельные обзоры.
Приобретенные гемолитические анемии
Среди различных приобретенных вариантов чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер. Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка. При некоторых формах аутоиммунных анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Токсические анемии протекают с прогрессирующей слабостью, болями в правом подреберье и поясничной области, рвотой, гемоглобинурией, высокой температурой тела. Со 2-3 суток присоединяется желтуха и билирубинемия; на 3-5 сутки возникает печеночная и почечная недостаточность, признаками которых служат гепатомегалия, ферментемия, азотемия, анурия. Отдельные виды приобретенных гемолитических анемий рассмотрены в соответствующих статьях: «Гемоглобинурия» и «Тромбоцитопеническая пурпура», «Гемолитическая болезнь плода».
Осложнения
Каждый вид ГА имеет свои специфические осложнения: например, ЖКБ - при микросфероцитозе, печеночная недостаточность - при токсических формах и т.д. К числу общих осложнений относятся гемолитические кризы, которые могут провоцироваться инфекциями, стрессами, родами у женщин. При остром массивном гемолизе возможно развитие гемолитической комы, характеризующейся коллапсом, спутанным сознанием, олигурией, усилением желтухи. Угрозу жизни больного несут ДВС-синдром, инфаркт селезенки или спонтанный разрыв органа. Неотложной медицинской помощи требуют острая сердечно-сосудистая и почечная недостаточность.
Определение формы ГА на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки. Лабораторный диагностический комплекс включает:
- Исследование крови. Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. При аутоиммунных анемиях большое диагностическое значение имеет положительная проба Кумбса.
- Анализы мочи и кала. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина.
- Миелограмму. Для цитологического подтверждения выполняется стернальная пункция. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, порфирии, гемобластозы. Пациента консультируют гастроэнтеролог, клинический фармаколог, инфекционист и другие специалисты.
Лечение
Различные формы ГА имеют свои особенности и подходы к лечению. При всех вариантах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости - гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной анемии выполняется спленэктомия. Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям - введение антидотов.
Прогноз и профилактика
Течение и исход зависят от вида анемии, тяжести протекания кризов, полноты патогенетической терапии. При многих приобретенных вариантах устранение причин и полноценное лечение приводит к полному выздоровлению. Излечения врожденных анемий добиться нельзя, однако возможно достижение длительной ремиссии. При развитии почечной недостаточности и других фатальных осложнений прогноз неблагоприятен. Предупредить развитие ГА позволяет профилактика острых инфекционных заболеваний, интоксикаций, отравлений. Запрещается бесконтрольное самостоятельное использование лекарственных препаратов. Необходимо тщательная подготовка пациентов к гемотрансфузиям, вакцинации с проведением всего комплекса необходимых обследований.
4. Клинические рекомендации по диагностике и лечению аутоиммунных гемолитический анемий/ Цветаева Н.В., Никулина О.Ф. - 2014.
Наследственный микросфероцитоз ( Болезнь Минковского-Шоффара )
Наследственный микросфероцитоз - это гемолитическая анемия, обусловленная генетическим дефектом мембран эритроцитов и характеризующаяся постоянным гемолизом. Клинические признаки включают бледность, желтушность кожи, слизистых, боли в левой части живота за счет увеличения селезенки, а также деформацию скелета. В раннем возрасте развивается желчнокаменная болезнь. Диагностика осуществляется с помощью общего анализа крови, определения осмотической резистентности эритроцитов. Иногда требуется проведение электрофореза мембранных белков. Основным лечением является удаление селезенки (спленэктомия).



Наследственный микросфероцитоз (НМС, болезнь Минковского-Шоффара) - врожденное гематологическое заболевание из группы мембранопатий. Впервые болезнь подробно была описана немецким терапевтом Оскаром Минковским в 1900 г., спустя 7 лет французский терапевт Анатоль Шоффар установил снижение осмотической резистентности красных кровяных телец при НМС. Распространенность данной патологии в среднем составляет 1: 2500 человек, несколько чаще она встречается в Японии, странах Африки, Северной Европы. Клиническая манифестация может произойти в любом возрасте, но чаще наступает в юношеском или зрелом возрасте. Больше страдают лица мужского пола.

В основе наследственного микросфероцитоза лежит мутация гена, кодирующего синтез одного из белков цитоскелета мембраны эритроцитов. В разных семьях обнаруживаются мутации различных генов. Ген альфа-цепи спектрина расположен на 1 хромосоме (локус Iq21), ген бета-цепи - на 14 хромосоме (локус q22-q23), а ген анкирина - на 8 хромосоме (локус 8p 11.2). Болезнь характеризуется аутосомно-доминантным типом наследования.
К предрасполагающим факторам можно отнести наличие среди близких родственников больного наследственным микросфероцитозом или бессимптомного носителя мутантных генов. Спровоцировать резкое обострение (гемолитический криз) или первое проявление НМС у лиц с легкой формой могут инфекционные патологии, вакцинация, сильный стресс. У женщин обострения нередко возникают при наступлении беременности.
Вследствие постоянной деструкции красных клеток компенсаторно усиливаются процессы костномозгового кроветворения. Из-за хронического гемолиза высвобождается большое количество неконъюгированного билирубина, который направляется в печень для секреции в желчь. Поэтому желчный пузырь начинает заполняться пигментными камнями. При патологоанатомическом исследовании обнаруживают гиперплазию эритроидного ростка костного мозга трубчатых, плоских костей. Кровенаполнение пульпы резко выражено. Также нередко отмечается гемосидероз внутренних органов.
Яркость клинической картины зависит от того, дефицит какого белка наблюдается у пациента, и является он гетерозиготным или гомозиготным носителем мутантных генов. По этим критериям различают следующие степени тяжести наследственного микросфероцитоза:
- Легкая. Небольшой гемолиз, развивающийся у взрослых людей под влиянием провоцирующих факторов. Селезенка увеличена незначительно. Уровень гемоглобина 100-120 г/л.
- Средняя. Умеренный гемолиз и спленомегалия. Кожа желтушной окраски. Уровень гемоглобина 80-100 г/л.
- Тяжелая. Редкая форма. Выраженный гемолиз, большие размеры селезенки, скелет деформирован. Характерно кризовое течение с большим количеством осложнений и вероятностью летального исхода. Уровень гемоглобина 60-80 г/л. Имеется потребность в постоянных гемотрансфузиях.
- Бессимптомная (латентная). При этой разновидности человек даже не подозревает, что болен. Данная форма характерна для гетерозиготных лиц. Единственным признаком может быть наличие небольшого количества микросфероцитов, высокий ретикулоцитоз. Истинная частота распространенности неизвестна.
Симптомы наследственного микросфероцитоза
Начало заболевания обычно постепенное. При латентной и легкой форме усиленное костномозговое кроветворение компенсирует постоянное разрушение эритроцитов, что позволяет поддерживать уровень гемоглобина на должном уровне. Тяжесть клинической картины определяется степенью гемолиза. На первый план обычно выступает желтушность кожных покровов, склер, слизистой оболочки рта с лимонно-шафрановым оттенком. Долгое время желтуха может быть единственным признаком наследственного микросфероцитоза.
Анемичный синдром проявляется бледностью кожи, слизистых, симптомами пониженного артериального давления (слабостью, головокружением, тахикардией). Практически всегда увеличена селезенка, из-за чего больной испытывает тянущую или ноющую боль в левом подреберье. Желтуха, спленомегалия, анемия составляют гемолитическую триаду. Нередко увеличена печень, но не так сильно, как селезенка, поэтому тяжесть и боль в правом подреберье незначительны.
Если заболевание манифестирует с раннего детского возраста, то развивается деформация костного скелета (стигмы дизэмбриогенеза) - башенный череп, укорочение мизинцев, широкая переносица и т. д. У взрослых больных с тяжелой формой НМС, которым не была проведена спленэктомия, иногда наблюдаются трофические язвы нижних конечностей (область голени, лодыжек), что обусловлено ухудшением микроциркуляции.
Особо яркую клинику имеет гемолитический криз, возникающий под влиянием различных провоцирующих факторов. Вследствие массивного гемолиза у больного повышается температура тела, нарастает интенсивность желтухи. Присоединяются симптомы билирубиновой интоксикации (потеря аппетита, рвота, боли в мышцах, суставах). Из-за резкого увеличения селезенки боли усиливаются, приобретают распирающий характер. Уровень гемоглобина падает до критических значений, пациент может потерять сознание.
Наиболее частыми осложнениями (50%) считаются желчнокаменная болезнь и калькулезный холецистит, возникающие по причине высвобождения из разрушенных эритроцитов большого количества билирубина, секретирующегося в желчь. Длительные переливания крови могут привести к перегрузке железом, вторичному гемохроматозу (цирроз печени, кардиомиопатия, сахарный диабет 2 типа). Трофические язвы ног в редких случаях способствуют развитию бактериальных воспалительных процессов в подкожной, межмышечной клетчатке (флегмона, некротизирующий фасциит).
Самое опасное состояние, которое возникает при наследственном микросфероцитозе, - апластический криз, вызванный инфицированием парвовирусом В19. В костном мозге прекращаются процессы кроветворения, резко снижается содержание в крови всех форменных элементов (эритроцитов, лейкоцитов, тромбоцитов). Появляются кровотечения, глубокая анемия, высокая чувствительность к инфекционным агентам.
Пациенты с болезнью Минковского-Шоффара подлежат обследованию у врача-гематолога. При осмотре пациента учитывается степень выраженности гемолитической триады, наличие деформации лицевого скелета. Уточняется, есть ли данное заболевание у кого-либо из близких родственников. С целью подтверждения диагноза назначается дополнительное обследование, которое включает:
- Анализы крови. В общем анализе крови обнаруживаются снижение уровня эритроцитов, гемоглобина, увеличение ретикулоцитов (до 20%). Размер эритроцитов уменьшен (меньше 7 мкм). При морфологической оценке мазка крови выявляются микросфероциты. Биохимический анализ крови показывает признаки гемолиза - высокую концентрацию непрямого билирубина, лактатдегидрогеназы.
- Верифицирующие тесты. Выявляется уменьшение осмотической устойчивости эритроцитов в виде повышенной чувствительности к лизису в гипотонических растворах NaCl (0,4-0,6%). Также характерна слабая способность красных клеток крови к фиксации флуоресцентного красителя эозин-5-малеимида при ЭМА-тесте. Электрофорез мембранных белков эритроцитов позволяет точно определить дефицит конкретного протеина (спектрин, анкирин).
- Инструментальные исследования. При УЗИ органов брюшной полости у пациента с любой формой наследственного микросфероцитоза обнаруживается увеличение селезенки, а иногда и печени. Часто находят камни в желчном пузыре. На рентгенографии костей черепа, трубчатых костей видны признаки разрастания костного мозга - расширение костномозгового канала, участки остеопороза, истончение кортикального слоя.
Спектр исключаемых патологий при болезни Минковского-Шоффара довольно широк. Наиболее часто приходится дифференцировать НМС от аутоиммунных гемолитических анемий. С этой целью для исключения иммунной природы гемолиза проводят антиглобулиновый тест (реакцию Кумбса). При НМС результат отрицательный. Легкие формы, сопровождающиеся лишь желтухой и небольшой спленомегалией, нужно отличать от доброкачественных гипербилирубинемий (синдрома Жильбера). Перегрузку железом дифференцируют с первичным (наследственным) гемохроматозом.
Лечение наследственного микросфероцитоза
Пациенты с легкой и бессимптомной формой не нуждаются в лечении. Им необходимо лишь регулярно посещать врача, сдавать клинический анализ крови. Больные средне-тяжелым и тяжелым НМС должны проходить лечение в отделении гематологии. Развитие гемолитического, апластического кризов из-за большого риска летального исхода являются показанием для перевода в отделение реанимации и интенсивной терапии.
Консервативная терапия
При уровне гемоглобина ниже 70 г/л производится переливание эритроцитарной массы, взвеси или отмытых эритроцитов. При гемоглобине меньше 50 г/л прибегают к переливанию цельной крови. Гемотрансфузию стоит выполнять медленно во избежание гемолитических реакций. При длительных гемотрансфузиях с целью выведения избытка железа обязательно используется хелатирующая терапия - дефероксамин, аскорбиновая кислота.
Для поддержания ремиссии (предотвращения кризов) больным тяжелым и средне-тяжелым НМС назначается постоянный прием профилактической дозы фолиевой кислоты. При апластическом кризе требуется дополнительное введение тромботического концентрата, стимуляторов лейкопоэза (филграстим), антибиотиков широкого спектра действия (цефтриаксон). Трофические язвы обрабатываются антисептическими растворами (фурацилин), мазями, содержащими антибиотики.
Хирургическое лечение
Основной радикальный вид лечения, обеспечивающий выздоровление больного, - тотальная спленэктомия (полное удаление селезенки). Она показана пациентам с частыми гемолитическими кризами, глубокой анемией, выраженной гипербилирубинемией и спленомегалией. Оптимальный возраст для операции - 6 лет. Предпочтение отдается лапароскопическому вмешательству как менее травматичному. Субтотальная резекция и эмболизация селезеночной артерии не рекомендуются, так как ассоциированы с высокой частотой рецидивов. При желчнокаменной болезни показана холецистэктомия.
Профилактика и прогноз
В целом наследственный микросфероцитоз является доброкачественным заболеванием. Подавляющее число пациентов имеет легкую или бессимптомную форму с незначительной спленомегалией и компенсированным гемолизом. Летальные исходы крайне редки (1-2%) и связаны с кризами (гемолитическими и апластическими). После спленэктомии продолжительность жизни не отличается от таковой у общей популяции. Первичная профилактика не разработана.
Отсутствие селезенки увеличивает риск инфицирования инкапсулированными микроорганизмами. Поэтому перед операцией (за 2-3 недели) обязательно проведение вакцинации против пневмококка, менингококка и гемофильной палочки. Дети до 6 лет должны получать профилактические дозы пенициллиновых антибиотиков (амоксициллин). Также с целью предотвращения постспленэктомического тромбоза назначаются антикоагулянты (низкомолекулярные гепарины).
Талассемия — болезнь, вызванная гемоглобином

Талассемия — это не одна, а группа похожих болезней, передающихся по наследству и вызванных нарушенным синтезом белковой части гемоглобина. Заболевание достается ребенку с вероятностью в 100%, если один из родителей содержит измененный ген. Поэтому оно считается самой распространенной наследственной патологией.
Немного истории
Болезнь была впервые описана в 1925 году американскими врачами-педиатрами, которые при лечении эмигрантов из Италии выявили одинаковую клиническую картину у детей с тяжелым малокровием, увеличением печени и селезенки, изменениями костей.
Затем появились работы, описывающие более легкое течение болезни у взрослых пациентов. Термин «талассемия» предложили в 1936 году. Дословно он означает «болезнь морского побережья». Высказана мысль о связи патогенеза заболевания с нарушением синтеза глобиновых цепочек.
Почему разрушаются эритроциты?
Патогенез талассемии достаточно изучен. Первичные изменения начинаются с нарушенного синтеза гемоглобина, а именно белковых цепочек вещества. Известно, что гемоглобин, входящий в состав 90% массы эритроцитов, — единственное вещество, способное связывать молекулы кислорода и разносить их из легочной ткани по всему организму.
Его структура состоит из пигмента (гема), включающего железо, и набора из двух пар белковых цепочек. Их называют по типичному расположению аминокислот альфа- и бета-цепями. При нарушенном синтезе одного из типов полипептидов накапливается другой вид.
В результате разрушаются все клетки эритроцитарного ряда (сами эритроциты и их предшественники), из них выходит гемоглобин. Малокровие (анемия) развивается по гипохромному типу. Это подтверждается низким цветовым показателем.
«Виновниками» разрушений являются гены, ответственные за построение белковой части гемоглобина. Их мутация нарушает способность составлять необходимый набор аминокислот в цепи. Причину изменений связывают с возбудителем малярии — плазмодием. Доказаны его мутирующие способности. Талассемия по территории распространенности совпадает с эпидемическими зонами малярии.

Основные четыре цепочки белков, связывающие гем и влияющие на эритроциты
Дети могут получить болезнь путем наследования от родителей в двух видах:
- гомозиготном — ген-мутант передается от обоих родителей;
- гетерозиготном — ген болезни передается только от матери или отца, носителем может быть один из родителей.
Соответственно, называются формы талассемии.
При гетерозиготном носительстве выделяют:
- «немой» ген (α-th2);
- «манифестный» ген (α-th1).
Разновидности заболевания
Все виды заболевания связаны с кислородной недостаточностью, наступившей вследствие разрушения эритроцитов — единственных клеток, обеспечивающих газообмен в органах и тканях.
В зависимости от сбоя синтеза полипептидной цепочки различают:
Наиболее распространена бета-талассемия. Она характеризуется избыточным накоплением α-цепочек глобина.
Выявлены также редкие формы болезни, которые названы гамма- и дельта-талассемией. В группу включены:
- некоторые гемоглобинопатии;
- гомозиготная альфа-талассемия с водянкой плода;
- смешанный вид β- и δ-талассемии, когда поражаются одновременно дельта- и бета-цепи.
Каждая форма имеет свои типичные клинические проявления и отличия. Но в зависимости от тяжести течения принято выделять:
- хроническую легкую — люди доживают до старости;
- хроническую среднетяжелую — пациенты не переживают детский возраст;
- тяжелую — ребенок погибает в период новорожденности.
Клинические проявления большой талассемии
Симптомы талассемии зависят по клинике и времени проявления от вида генной мутации.
Большая талассемия (болезнь Кули) развивается при гомозиготной передаче от родителей. Ее проявления заметны у детей сразу после рождения.
У новорожденного замечают:
- удлиненный вверх череп («башенный»);
- более развитую верхнюю челюсть;
- монголоидный тип лицевого скелета.

Типичная внешность и изменения структуры костей черепа используются в диагностике
К 12-месячному возрасту проявляется:
- расширенная носовая перегородка, «приплюснутый» нос;
- костные наросты на стопах;
- нарушенный прикус;
- желтушность кожи (в связи с поражением селезенки).
Кислородное голодание тканей приводит к органным нарушениям. При осмотре отмечаются:
- увеличение печени, край плотный на ощупь (развивается ранний цирроз);
- возникновение сердечной недостаточности (лишнее железо откладывается в миокарде и поражает сократимость сердечной мышцы);
- отставание в физическом и умственном развитии от сверстников.
У малыша развивается сахарный диабет из-за фиброзирования поджелудочной железы и печеночная недостаточность. В тяжелых случаях он живет не более одного года.
В старшем возрасте проявляются такие осложнения:
- трофические язвы на коже, вызванные локальными нарушениями кровообращения;
- частые переломы костей;
- повторные воспаления в легких;
- калькулезный холецистит;
- кардиосклероз;
- сепсис при контакте с инфекцией;
- нарушенное полового развитие.
Выделяют промежуточную форму β-талассемии с более доброкачественным течением. Она начинается у детей старшего возраста. Внешний вид ребенка не изменяется. Нет отставания в умственном и физическом развитии. Основные проявления вызываются увеличением селезенки и повышенной ломкостью костной ткани.
Клиника малой талассемии
Малая или гетерозиготная форма передается одним из родителей. Второй «здоровый» ген сглаживает повреждения. Симптомы талассемии могут длительно не проявляться или вызывать признаки, общие с другими заболеваниями:
- слабость, повышенную утомляемость;
- головные боли;
- головокружение.
При осмотре обращается внимание на следующее:
- бледность кожи с желтушным оттенком;
- возможно увеличение печени и селезенки.
У ребенка с малой формой резко увеличена восприимчивость к инфекциям, снижен иммунитет. Очень тяжело и часто протекают вирусные и кишечные инфекционные заболевания.
Во взрослом состоянии у женщины при беременности развивается водянка плода (повышение внутричерепного давления и скопление жидкости в желудочках мозга). Эта патология несовместима с жизнью ребенка, в случае нормальных родов ведет к тяжелым неврологическим и психическим отклонениям.
Минимальной β-талассемией называют синдром Сильвестрони — Бьянко. Эта форма заболевания протекает практически бессимптомно, обнаруживается случайно в семьях со случаями талассемии.
Диагностика талассемии должна начинаться с генетической консультации супругов перед зачатием ребенка. Во время беременности при необходимости проводят анализ амниотической жидкости. Уже на ранних сроках в эритроцитах плода можно обнаружить характерные изменения.
Порой внешний осмотр и выяснение семейного анамнеза позволяют заподозрить болезнь у ребенка без лабораторной диагностики.
В анализе крови выявляют:
- снижение уровня гемоглобина до 30-50 г/л при гомозиготном варианте и до 90 -110 г/л при гетерозиготном;
- низкий цветовой показатель (менее 0,5), который образуется малым насыщением клеток гемоглобином);
- рост ретикулоцитов (предшественников эритроцитов) до 4%.
При просмотре окрашенного мазка под микроскопом обращают внимание на следующее:
- наличие слабо окрашенных (гипохромных) эритроцитов;
- изменение размеров и формы красных клеток, эритроциты приобретают формы овала, серпа, шара.
Одна из характерных разновидностей серповидных клеток — дрепаноциты.
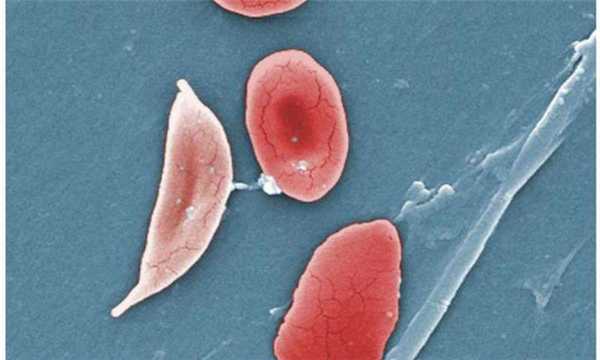
Дрепаноциты образуются при любых гемоглобинопатиях, содержат особую разновидность (S гемоглобин), который может в условиях низкой концентрации образовывать полимеры и изменять форму оболочки клетки
Биохимические тесты показывают нарушенный обмен железа, как при гемолитической анемии:
- высокий уровень свободного билирубина;
- повышенную концентрацию железа в сыворотке;
- сниженную способность к связыванию железа.
Исследование особенностей гемоглобина проводят с помощью ацетат-целлюлозной пленки. Подобный анализ позволяет определить количественный уровень фракций. Гомозиготная бета-талассемия отличается увеличенным уровнем фетального гемоглобина (в норме он содержится у плода, а у взрослого человека всего до 1%), неспособного переносить кислород.
Использование метода полимеразной цепной реакции позволяет изучить строение полипептидных цепочек гемоглобина.
Генетический анализ выявляет мутацию в одиннадцатой паре хромосом при β-талассемии, другие специфические изменения, типичные для прочих форм.
Исследование пунктата костного мозга проводится для выявления повышенного содержания незрелых эритроцитов в виде сидеробластов.
Рентгенологическое исследование способствует обнаружению дефектов костной ткани:
- участков остеопороза (сниженной плотности);
- увеличенной массы костей черепа;
- поперечной исчерченности на мелких костях кистей и стоп;
- на рентгенограмме черепа при большой форме β-талассемии видно типичное игольчатое поражение надкостницы, которое называется симптомом «волосатого черепа».
Ультразвуковое исследование подтверждает увеличение печени и селезенки, помогает обнаружить камни в желчевыводящей системе.
Лабораторные данные более отчетливо выражены при β-талассемии. Другие формы болезни не дают четкой картины.
Заболевания, с которыми необходимо проводить дифференциальную диагностику:
- железодефицитная анемия,
- серповидно-клеточная анемия,
- гемолитическая аутоиммунная анемия,
- наследственный микросфероцитоз.
Генетическая интерпретация видов бета-талассемии
Ученые-генетики выяснили интересную закономерность: люди имеют одинаковую мутацию генов, отвечающих за синтез гемоглобина, но клиника и степень выраженности заболевания у них отличаются.
Гены могут находиться в следующих состояниях:
- нормальные — характерны для здорового человека;
- поврежденные частично — «работает» неполно, из-за чего синтез полипептидных цепей недостаточен;
- разрушенные полностью — синтез останавливается.
По этому признаку виды талассемии делят на:
- минор — самая легкая форма, поврежден только один ген, внешне человек выглядит здоровым, по анализу крови можно предположить небольшую анемию;
- интермедиа — недостаток бета-цепей серьезно сказывается на синтезе, эритроциты недоразвиты, малокровие выражено с явными признаками, но организм еще может приспособиться, поэтому отсутствует необходимость в постоянных переливаниях крови;
- майор — мутации подверглись все гены, больному необходимы постоянные переливания крови по жизненным показаниям.
Генетические разновидности альфа-талассемии
По охвату мутацией генов, их локусов (определенных частей, отрезков), отвечающих за синтез альфа-цепочек полипептидов, предлагается выделение нескольких групп заболевания:
- мутация только в одном локусе — клинических проявлений нет;
- изменения двух (пары) локусов в одном или разных генах — анализ крови показывает снижение гемоглобина, уменьшение размера эритроцитов;
- поражение трех локусов — выражается в кислородной гипоксии органов, увеличении селезенки, возникновении Н гемоглобинопатии, образующийся гемоглобин нестоек, разлагаясь, вызывает гемолитическую анемию;
- мутация всех локусов — полностью прекращает синтез альфа-цепей, при подобной ситуации происходит внутриутробная гибель плода или ребенок умирает сразу после родов.
Генетические исследования подтвердили также неодинаковое значение пар генов, одна пара из двух является главной, а другая имеет второстепенную роль. Клинические проявления зависят от того, в какой паре произошла мутация.
Пересадка костного мозга считается наиболее результативным способом
Проблемы терапии
Лечение талассемии зависит от тяжести поражения эритропоэза, степени охвата генов процессами мутации. В настоящее время применяются следующие методы.
Диетический рацион направлен на снижение всасывания железа в кишечнике, рекомендуются орехи, какао, соя, чай.
Тяжелая форма требует регулярного переливания крови, эритроцитарной массы, размороженных и профильтрованных эритроцитов. Эффективность временная, возможны побочные эффекты, но главное — сохранить жизнь больному.
Терапия дополняется ежедневным устранением излишков железа с помощью введения (хелатов — специальных комплексов, повышающих действие лекарства). Для связывания железа назначается Десферал. Этот препарат предупреждает сидероз (патологическое состояние, вызванное отложением в тканях железа), но на уровень гемоглобина не влияет.
При возникновении резкого ухудшения состояния по аналогии с гемолитическими кризами показаны глюкокортикоиды в больших дозах.
Спленэктомия возможна при больших размерах селезенки детям после пятилетнего возраста. Наиболее оптимален возраст 8-10 лет. После удаления селезенки наступает период улучшения, но опасен риск присоединения инфекции.
Для трансплантации необходим донор, совпадающий по всем параметрам, лучше всего из близких родственников.
К симптоматическим средствам относятся препараты гепатопротекторного действия, большие дозы аскорбинки помогают вывести излишки железа из организма.
Все формы талассемии нуждаются в препаратах с фолиевой кислотой и витаминами группы В. На фоне присоединившейся инфекции, при беременности следует применять фолиевую кислоту в больших дозах, поскольку неэффективное кроветворения при талассемии значительно увеличивает ее потребление клетками.
Можно ли предупредить заболевание?
Талассемия считается до настоящего времени неизлечимым заболеванием. К мерам предупреждения относят дородовую диагностику для решения вопроса о наследовании признаков, своевременного прерывания беременности.

В родильном доме берется кровь из пяточки для экспресс-теста
Рожденный ребенок с легкой степенью болезни может прожить всю жизнь без тяжелых последствий. Но гарантировать именно эту форму патологии современная наука не может. Особенно следует подумать о целесообразности деторождения парам с гомозиготной наследственностью.
Методика исследования клеток плода во время беременности (забор осуществляется путем прокола матки через брюшную стенку) опасна возможным занесением инфекции и преждевременными родами. Поэтому лучше решать проблемы в медико-генетической консультации до плановой беременности.
Больных с талассемией ведут и наблюдают совместно гематологи, педиатры и терапевты.
Читайте также:
- Вдыхание холодной сжатой газовой смеси. Эффекты вдыхания холодной газовой смеси
- Удаление корней молочных зубов. Травмирование мягких тканей при удалении зубов
- Муж не обращает на меня внимание. В чем может быть причина?
- Субъективные оценки эффективности лечения. Отношение к лечению при наркомании
- УЗИ при внутриматочной спирали