УЗИ, МРТ при лобарной голопрозэнцефалии у плода
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Поскольку непосредственные этиологические факторы аномалии развития не известны, в работе выделены основные группы причин, способствующих формированию пороков развития головного мозга. Большое значение отводится ранней диагностике пороков развития, котора
Clinical case of semilobar holoprosencephaly / A. V. Serezhkina*, **, 1, I. G. Khmelevskaya*, **, N. S. Razinkova*, **, T. A. Minenkova*, **, I. I. Zhiznevskaya*, **, A. S. Plekhanova* / * Federal State Budget Educational Establishment of Higher Education Kursk State Medical University Ministry of Health Care of Russian Federation, Kursk, Russia / ** Regional public health institution Regional Children's Clinical Hospital Committee of Health Kursk region, Kursk, Russia
Резюме. Поскольку непосредственные этиологические факторы аномалии развития не известны, в работе выделены основные группы причин, способствующих формированию пороков развития головного мозга. Большое значение отводится ранней диагностике пороков развития, которая позволяет своевременно решить вопрос о возможности пролонгирования беременности, что определяется видом порока, совместимостью с жизнью и прогнозом в отношении постнатального развития. В исследовании рассматриваемого порока развития большую роль играют такие современные методы, как пренатальная ультразвуковая диагностика, нейросонография, рентгеновская компьютерная и магнитно-резонансная томография головного мозга, имеющие достаточно высокую информативность. Указаны сроки гестации, позволяющие выявить структурные дефекты головного мозга. Медико-генетическое консультирование помогает выявить риск появления больного потомства. Проведена дифференциальная диагностика семилобарной с другими формами голопрозэнцефалии. Также отмечены возможные клинические проявления рассматриваемой нозологии. В данной статье представлен клинический случай семилобарной голопрозэнцефалии, диагностированной у мальчика в возрасте 1 месяц. При поступлении мать предъявляла жалобы на срыгивания и периодическое беспокойство сына. Объем и результаты обследования ребенка изложены ниже. Выявлена сопутствующая патология в виде пупочной грыжи, врожденной аномалии развития мочевой системы: подковообразная почка; водянки яичек и головчатой формы гипоспадии. После проведения курса поддерживающей терапии пациент был выписан в стабильном состоянии. В настоящее время специфическое лечение голопрозэнцефалии отсутствует. Оперативные вмешательства на головном мозге проводятся редко ввиду тяжести состояния больных, в связи с чем лечение данной патологии возможно только с помощью хирургической коррекции симптомов. Длительная дыхательная и кардиоваскулярная дисфункция предопределяет летальный исход заболевания.
Семилобарная голопрозэнцефалия считается умеренной формой голопрозэнцефалии. Так же, как и другие типы, этот можно диагностировать внутриутробно по отсутствию межполушарной щели (кроме задней части мозга).
Непосредственные этиологические факторы аномалии развития не известны. Многие авторы выделяют две группы причин развития порока: наследственные и экологические. Наследственные представлены хромосомными аномалиями, ведущими к анеуплоидии: трисомия 13 (синдром Патау), трисомия 18 (синдром Эдвардса), трисомия 21 (синдром Дауна), синдром триплоидии. Нередко встречающиеся мутации, связанные с голопрозэнцефалией: синдром 13 q, синдром Генуя, хвостовой дисгенез, синдром Айкарди, псевдотрисомия 13, синдром Меккеля - Грубера.
К экологическим причинам относятся сахарный диабет (СД) у матери, прием беременной салицилатов, ретиноевой кислоты, статинов, мизопростола, метотрексата, дифенилгидантоина, употребление алкогольных напитков. Доказана роль ионизирующего излучения в I триместре беременности. В основе патогенетических механизмов развития голопрозэнцефалии лежит нарушение формирования головного мозга по срединной линии. Зачастую эти процессы происходят на 5-10 неделях беременности [6].
Голопрозэнцефалия - порок, который может быть результатом заболеваний, характерных для Х-сцепленного, аутосомно-рецессивного и аутосомно-доминантного типа наследования, а также сочетания голопрозэнцефалии с инсулинзависимым СД [2].
Семилобарная голопрозэнцефалия характеризуется слиянием лобных долей при наличии незначительной перегородки в задней части с присутствием серпа и межполушарной щели. Клинически проявляться данная форма аномалии может по-разному: отсутствием носовой перегородки, анофтальмией, близким расположением глаз, аномалиями радужной оболочки и сетчатки, предчелюстными агенезиями, срединной расщелиной нёба и губы. Возможны дефекты других систем организма, в том числе врожденные пороки сердца, дисплазии яичек и половых органов, кистозность почек, кишечные мальротации, пупочные грыжи, почечные дисплазии, водянка плода и т. п. [8].
Голопрозэнцефалию можно диагностировать начиная с 13-14 недели гестации с помощью ультразвукового исследования (УЗИ). В большинстве случаев диагноз ставят на 20-24 неделях.
Пренатальная диагностика голопрозэнцефалии основана на обнаружении сочетанных аномалий лицевого черепа, лица и головного мозга. Определение аномалий лица заставляет заподозрить и начать более подробный поиск интракраниальных изменений, таких как единственный желудочек мозга, сращение таламусов, отсутствие межполушарной щели, микроцефалия. По данным УЗИ невозможно уверенно дифференцировать алобарную и семилобарную голопрозэнцефалию. Диагностика лобарной голопрозэнцефалии связана с большими трудностями [9].
Использование трансвагинальной эхографии способствовало накоплению опыта ультразвуковой визуализации структур мозга плода на ранних этапах развития, что дало возможность диагностики голопрозэнцефалии уже в конце I триместра беременности [5].
В пренатальном периоде ведущий метод диагностики - сонография. Алобарную и семилобарную форму голопрозэнцефалии необходимо диагностировать с помощью УЗИ в течение первой половины беременности, поэтому прибегать к другим технологиям нет необходимости. Диагностика лобарной формы голопрозэнцефалии возможна только с помощью магнитно-резонансной томографии (МРТ). В постнатальном периоде МРТ служит методом выбора.
Семилобарная форма голопрозэнцефалии - результат частичного недоразделения мозга на левую и правую полусферы. Существуют определенные критерии для дифференциальной диагностики этой формы с алобарной и лобарной формами порока. При семилобарной голопрозэнцефалии два полушария мозга частично разделены в задней части, имеется один общий желудочек с рудиментарными задними рогами. Алобарная и семилобарная формы часто сочетаются с микроцефалией и реже с макроцефалией. При алобарной и семилобарной голопрозэнцефалии всегда отсутствует мозолистое тело [2].
Дифференциальная диагностика аномалии развития головного мозга с помощью МРТ основана на следующих признаках: при алобарной форме мозг малых размеров и содержит одну единую полость с дорзальным саком вместо третьего и боковых желудочков, таламусы соединены вместе, нет обонятельных луковиц и обонятельных трактов; мальформации лица - наиболее неблагоприятный вариант. При семилобарной голопрозэнцефалии мозг также маленький, с рудиментами затылочных долей. Межполушарная щель имеется в переднем или заднем отделах. Поскольку мозолистое тело отсутствует частично или полностью, то дорзальный сак поднимается высоко с формированием межполушарной ликворной кисты. Самая легкая форма голопрозэнцефалии - лобарная. При ней гемисферы большого мозга отделены друг от друга, кроме передних отделов, боковые желудочки соединены между собой за счет агенезии прозрачной перегородки. Мозолистое тело отсутствует [1].
В качестве примера приведен клинический случай семилобарной голопрозэнцефалии, диагностированной у ребенка 1 месяца. Даниил Р. поступил в Курскую детскую клиническую больницу с жалобами на срыгивания и периодическое беспокойство.
При сборе анамнеза стало известно, что ребенок от 7-й беременности, протекавшей на фоне приема регулона, цикличных менструаций, четвертых преждевременных домашних родов, мать на учете в женской консультации не состояла. При рождении вес мальчика составил 1700 г, рост - 42 см. Ребенок осмотрен участковым педиатром на 10-е сутки. Нейросонография по месту жительства показала: межполушарная щель смещена влево, визуализируется большое количество жидкостного элемента. Мальчик был госпитализирован в Курскую ОДКБ в отделение № 3 для обследования и лечения.
Аллергологический и наследственный анамнез не отягощен.
Объективные данные при поступлении: общее состояние ребенка средней степени тяжести. Малыш на грудном вскармливании. Вес - 3600 г, рост - 51 см. Слизистые чистые, влажные. Склеры - субиктеричные. Дыхание ритмичное, с частотой 36 в минуту, хрипов нет. Тоны сердца ритмичные. Частота сердечных сокращений - 134 в минуту, артериальное давление (АД) - 85/55 мм рт. ст. Пупочная грыжа небольших размеров. Наружные половые органы сформированы по мужскому типу, обе половины мошонки увеличены в размерах. Головка полового члена открыта, отверстие уретры смещено книзу.
В неврологическом статусе: сознание ребенка ясное, улыбается. Окружность головы - 37,5 см, большой родничок - 2,0 × 2,0 см на уровне костей черепа. Голова гидроцефальной формы. Менингиальные симптомы отсутствуют. Двигательная активность сохранена. Мышечный тонус умеренно повышен по флексорному типу в сгибателях конечности. Определяются рефлексы Бабинского, Моро, ползания, опоры и автоматической ходьбы, при тракции за руки голову выводит. В положении на животе голову выводит, опора на предплечья.
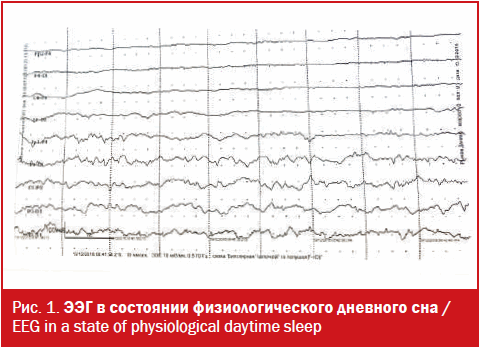
Даниилу Р. была проведена ЭЭГ в состоянии физиологического дневного сна (20 минут), на которой достоверных изменений эпилептиформного характера не было выявлено (рис. 1).
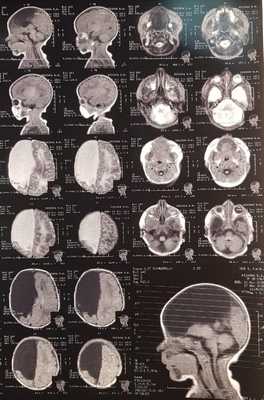
При УЗИ головного мозга было обнаружено, что структуры мозга сформированы неправильно, правое полушарие практически полностью отсутствует. Ядра таламуса и структуры задней черепной ямки сохранены, но правое ядро таламуса гипоплазировано. Эхогенность левого полушария средняя. Рисунок извилин и борозд отчетливый слева. Межполушарная щель в сечении через тела боковых желудочков - 1,7 (норма - до 4 мм). Субдуральное пространство - 0 мм (норма - 2 мм).Субарахноидальное пространство справа - 1,7 мм; слева - 1,8 (норма - 2 мм). Правый боковой желудочек отсутствует. Передний рог - 4,0 мм, латеральный рог - 3,0 мм, задний рог - 7,0 мм. Третий желудочек в сечении через тела боковых желудочков - 3,0 мм (норма - 3 мм), форма желудочка неправильная. Четвертый желудочек в сагиттальном сечении - 3,0 мм (норма - 4 мм). Контуры сосудистых сплетений ровные, толщина левого - 7,0 мм, структура однородная. Мозжечок, таламус, подкорковые ядра - эхогенность повышена справа, слева без особенностей, эхоструктура однородная. Заключение: врожденная аномалия развития головного мозга, характерная для алобарной формы голопрозэнцефалии.
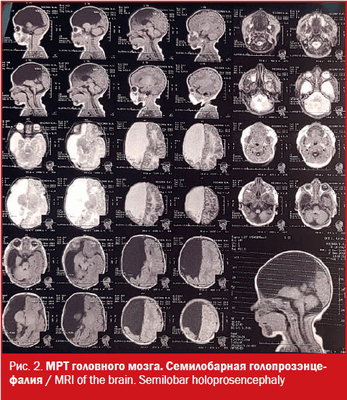
МРТ: на серии Т1- и Т2-взвешенных томограмм в сагиттальной и аксиальной проекции получено изображение супратенториальных структур головного мозга. Правые лобная, теменная и частично височная доли (кроме гиппокампа), мозолистое тело и прозрачная перегородка отсутствуют, нижний червь мозжечка гипопластичный. Правый зрительный бугор увеличен в размерах, визуализируется частично правая затылочная доля. Левое полушарие большого мозга, полушария мозжечка и ствол сформированы правильно. Ретенции желудочковой системы нет. Признаков объемного воздействия не отмечено. Базальные цистерны открыты. В заключении отмечено, что полученные данные могут соответствовать МР-картине семилобарной голопрозэнцефалии (рис. 2).
Из сопутствующей патологии отмечено наличие пупочной грыжи и врожденной аномалии развития мочевой системы (подковообразная почка, водянка яичек и головчатая форма гипоспадии).
Во время пребывания в стационаре ребенок находился на грудном вскармливании, получал ноотропную поддержку - курс церебролизина, метаболическую (витамин D3) и симптоматическую терапию (Урсофальк).
Пациент выписан в стабильном состоянии. Вес мальчика при выписке составил 3960 г (+360 г). Неврологический статус - без динамики.
Согласно данным C. Olsen и соавт. [4], среди 78 родившихся детей общая смертность составила 32% в течение первых 48 часов и 38,5% — в течение 1-й недели соответственно. При синдромальных случаях смертность в течение первых 48 часов жизни составила 57%, у больных с алобарной формой выживаемость на 1-й неделе — 50%. До 12 месяцев с изолированной формой голопроз-энцефалии дожили 54%, при синдромальных случаях — 14%, несиндромальных в сочетании с другими пороками развития головного мозга — 25%. Высокая смертность, по-видимому, связана с дисфункцией ствола или гипоталамических структур головного мозга в сочетании с полиорганной патологией. Длительная дыхательная и кардио-васкулярная дисфункция предопределяет летальный исход заболевания [3]. В настоящее время специфическое лечение голопрозэнцефалии отсутствует. Оперативные вмешательства на головном мозге проводятся редко ввиду тяжести состояния больных, в связи с чем лечение данной патологии возможно только с помощью хирургической коррекции симптомов (аномалий развития лица). При тяжелых формах заболевания отмечаются также трудности вскармливания новорожденных, связанные со слабостью сосания, невозможностью проглатывания пищи, поперхиванием, рвотой с риском аспирации, эзофагии и др. [7]. В связи с этим в некоторых случаях применяется гастротомия (при исключении центрального нарушения питания) [9]. При наличии декомпенсированной гидроцефалии рекомендовано нейрохирургическое лечение. При наличии эпилептических приступов проводится подбор противосудорожной терапии с использованием антиконвульсантов под контролем обычной ЭЭГ и видео-ЭЭГ-мониторинга. M. Barr и соавт. [1] доказали, что для восстановления нарушенных физиологических ритмов сна можно использовать внешний шум (например, радио).
В заключение отмечаем, что голопрозэнцефалия может входить в структуру хромосомных или моногенных заболеваний, а также встречаться как изолированная мальформация. Этиология голопрозэнцефалии остается недостаточно изученной. В диагностике рассматриваемого порока развития большую роль играют такие современные методы, как пренатальная ультразвуковая диагностика, нейросонография, рентгеновская компьютерная и магнитно-резонансная томография головного мозга, имеющие достаточно высокую информативность. Медико-генетическое консультирование помогает выявить риск появления больного потомства. Установлено, что молодые пары, дающие жизнь ребенку со стандартной трисомией, имеют неспецифический (около 1%) риск рекуррентной трисомии 13-й хромосомы.
КОНФЛИКТ ИНТЕРЕСОВ. Авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить.
CONFLICT OF INTERESTS. Not declared.
Литература/References
- Barr M. Jr., Cohen M. M. Jr. Autosomal recessive alobar holoprosencephaly with essentially normal facies // Am J Med Genet. 2002; 112: 28.
- Barr M. Jr., Cohen M. M. Jr. Holoprosencephaly survival and performance // Am J Med Genet. 1999; 89: 116.
- Cohen M. M. Jr. Problems in the definition of holoprosencephaly // Am J Med Genet. 2001; 103: 183.
- Olsen C. L., Hughes J. P., Youngblood L. G. et al. Epidemiology of holoprosencephaly and phenotypic characteristics of affected children. New York state 1984-1989 // Am J Med Genet. 1997; 73: 217.
- Барашнев Ю. И. Перинатальные повреждения нервной системы у новорожденных. Руководство по безопасному материнству / Под ред. Ю. И. Барашнева. М.: Триада-Х, 1998. С. 373-432. [Barashnev Yu. I. Perinatal damage to the nervous system in newborns. Safe Motherhood Guide / Pod red. Yu. I. Barashneva. M.: Triada-KH, 1998. Pp. 373-432.]
- Аминофф М. Дж., Гринберг Д. А., Саймон Р. П. Клиническая неврология. М.: МЕДпресс-информ, 2009. 480 c. [Aminoff M. Dzh., Grinberg D. A., Saymon R. P. Clinical neurology. M.: MEDpress-inform, 2009. P. 480.]
- Неонатология: Национальное руководство. Краткое издание / Под ред. Н. Н. Володина. М.: ГЭОТАР-Медиа, 2014. [Neonatology: National Guidelines. Short edition / Pod red. N. N. Volodina. M.: GEOTAR-Media, 2014.]
- Никифоров А. С., Гусев Е. И. Общая неврология. 2-е изд., испр. и доп. М.: ГЭОТАР-Медиа, 2015. [Nikiforov A. S., Gusev Ye. I. General neurology. 2nd ed., Rev. and add. M.: GEOTAR-Media, 2015.]
- Петрухин А. С. Детская неврология. В 2 т. Том 1. М.: ГЭОТАР-Медиа, 2009. 272 c. [Petrukhin A. S. Pediatric neurology. In 2 volumes, Volume 1. M.: GEOTAR-Media, 2009. P. 272.]
А. В. Серёжкина* , ** , 1
И. Г. Хмелевская* , **, доктор медицинских наук, профессор
Н. С. Разинькова* , **, кандидат медицинских наук
Т. А. Миненкова* , **
И. И. Жизневская* , **, кандидат медицинских наук
А. С. Плеханова*
* ФГБОУ ВО КГМУ Минздрава России, Курск, Россия
** ОБУЗ ОДКБ, Курск, Россия
Анэнцефалия
Анэнцефалия — это порок развития нервной системы, при котором у плода отсутствуют большие полушария мозга, недоразвиты кости свода черепа, мягкие ткани головы. Аномалия возникает под действием химических, биологических или физических тератогенных факторов. Большинство плодов с анэнцефалией погибают во внутриутробном периоде, и даже при живорождении срок жизни ребенка не превышает нескольких часов. Диагностика выполняется пренатально с помощью УЗИ, МРТ, амниоцентеза, биохимического скрининга. В такой ситуации показано прерывание беременности, при отказе родителей от аборта — оказание паллиативной помощи ребенку.
МКБ-10
Общие сведения
По данным ВОЗ, пороки развития ЦНС занимают первое место среди врожденных аномалий у плода (25%). Анэнцефалов стоит отличать от ацефалов (акефалов), у которых вовсе отсутствует головной мозг. Анэнцефалия встречается в США и России с частотой 1 на 10000 новорожденных, в Великобритании — 5:10000, в Китае — 8:10000. Заболевание чаще наблюдается у плодов женского пола. Риск повторения анэнцефалии при следующей беременности составляет до 5%, а при рождении женщиной двух детей с этим пороком ЦНС — возрастает до 10%. Вероятность аномалии у кровных родственников составляет не более 3%.
Причины
Порок формируется при нарушениях внутриутробной закладки нейроструктур в эмбриональном периоде формирования плода. Он считается мультифакториальным, точные причины такого состояния окончательно не установлены. Ученые предполагают, что появление анэнцефалии связано с воздействиями негативных факторов на ранних этапах беременности. Типичными тератогенными влияниями считаются:
К предрасполагающим факторам со стороны родителей относят генетические — наличие различных генных мутаций, частые случаи родственных браков и кровосмешения. Риск аномалий развития ЦНС у ребенка повышается, если в организме матери низкий уровень йода, фолатов и других витаминов группы В. Вероятность анэнцефалии повышается при тяжелом течении гестации, наличии у беременной женщины экстрагенитальных патологий: сахарного диабета, сердечной недостаточности, гипотиреоза.
Патогенез
Патоморфологически головной мозг плода с анэнцефалией представляет собой геморрагическую массу, в основном выполненную в виде недоразвитой сосудистой ткани, с элементами глии, единичных нервных клеток. Гистологически определяются аномальные сосуды, хориоидальные сплетения, беспорядочно расположенные фибробласты, кистозные полости с медуллярным эпителием. Ствол мозга и спинной мозг обычно имеют патологические структурные изменения, однако они выражены не так интенсивно.
Симптомы анэнцефалии
У живорожденных младенцев с анэнцефалией резко уменьшена мозговая часть черепа, отсутствуют плоские кости, недоразвиты мягкие ткани. На месте головного мозга находится аномальная сосудистая ткань, которая может быть покрыта кожей.
Такие дети пребывают в бессознательном состоянии, они слепые и глухие, не реагируют ни на какие внешние стимулы. Зачастую есть сопутствующие пороки: спинномозговые грыжи, патологии надпочечников, расщелины неба.
Хотя большинство живорожденных пациентов погибает практически сразу, в медицине описано по крайней мере три случая, когда дети смогли прожить более 2-х лет. Долгожителем среди анэнцефалов считается девочка Стефани Кин (более известная по прозвищу Бэби Кей), которая прожила 2,5 года и умерла от остановки сердца. У нее были проблемы с дыханием, которые требовали проведения ИВЛ. Двухлетний рубеж также преодолели Джексон Буэлл из Бостона, Виктория де Кристо из Бразилии.
Однако продление срока жизни анэнцефалов возможно только при постоянной кардиореспираторной поддержке, восполнении нутритивных потребностей. Ввиду отсутствия большей части мозга состояние младенцев остается стабильно тяжелым вплоть до смерти. Объем и необходимость паллиативной помощи для таких новорожденных поднимает важные вопросы биомедицинской этики.
Осложнения
Анэнцефалия признана 100% летальным пороком. В 75% случаев гибель плода происходит пренатально, что чревато замершей беременностью. Если это состояние не будет вовремя обнаружено, существует риск септических осложнений у женщины. К казуистическим на сегодня последствиям внутриутробной гибели плода относят литопедион, мумификацию плода. В остальных 25% случаев смерть младенца наблюдается в течение первых дней после родов из-за несовместимых с жизнью нарушений.
Диагностика
Аномалия обнаруживается антенатально во время плановых ультразвуковых скринингов беременности. Такой диагноз является абсолютным показанием к прерыванию беременности независимо от возраста плода, поэтому на акушеров-гинекологов ложится большая ответственность — необходимо как можно раньше установить диагноз, чтобы снизить риски для женщины. Для обнаружения порока применяются следующие методы:
- Пренатальное УЗИ. Ультразвуковое сканирование — основной способ диагностики, который при анэнцефалии демонстрирует неправильную форму головы, недоразвитие костно-мышечных структур черепа, отсутствие больших полушарий. Однако остальные структуры тела обычно развиты нормально. УЗИ позволяет выявить патологию с 11-12 недели гестационного срока.
- МРТ плода. Магнитно-резонансная томография — безопасный и наиболее информативный метод уточнения анатомических особенностей ЦНС, если результаты УЗИ дают сомнительную картину. Исследование проводится с 14-15 недель беременности.
- Амниоцентез. Инвазивная диагностика с забором околоплодных вод необходима, чтобы определить биохимические показатели, выполнить дифференциальную диагностику с врожденными синдромальными заболеваниями, вызванными генными или хромосомными мутациями.
- Биохимический скрининг. Заподозрить патологию удается по повышению уровня хорионического гонадотропина и альфа-фетопротеина, снижению количества протеина-А в первом триместре беременности.
Женщинам, которые планируют прервать беременность при выявленной анэнцефалии плода, требуется расширенная диагностика. Назначается стандартное гинекологическое исследование, а также набор анализов: клиническое исследование крови, анализы на вирусные гепатиты, ВИЧ-инфекцию. Впоследствии пациенткам рекомендовано медико-генетическое консультирование, чтобы оценить риски врожденных пороков при последующих беременностях.
Лечение анэнцефалии
Решение о прерывании беременности должны принимать родители. Единственным исключением является мертвый плод, когда женщине требуется экстренное хирургическое вмешательство. Живорожденным детям с анэнцефалией можно оказать только паллиативную помощь. Около 66% таких детей живут несколько часов после рождения, остальные погибают в первые минуты. Основные направления медицинской помощи при анэнцефалии:
- Респираторная поддержка. У детей случаются кризы в работе дыхательной системы, требующие назначения ИВЛ для адекватной оксигенации тканей организма.
- Нутритивная поддержка. Применяется парентеральное питание с использованием растворов глюкозы, специальных аминокислотных смесей для новорожденных.
Важной составляющей является психологическая помощь родителям на всех этапах от момента диагностики аномалии у плода, поскольку это становится тяжелым испытанием для семьи, у женщин зачастую возникают депрессии, апатии, суицидальные мысли. Помощь оказывают психологи, психиатры (в случае серьезных нарушений психоэмоционального состояния), верующим людям можно порекомендовать обратиться к представителям религии.
Прогноз и профилактика
Прогноз неблагоприятный, поскольку анэнцефалия заканчивается внутриутробной смертью плода или летальным исходом вскоре после того, как родился живой ребенок. Современные возможности медицины позволяют обеспечить такому младенцу достойный уход из жизни, если родители приняли решение не прерывать беременность.
Учитывая разнообразие предрасполагающих факторов, эффективные меры профилактики этого состояния пока не разработаны. Положительным для предупреждения тяжелых аномалий плода является прием беременной по назначению гинеколога витаминных добавок с солями фолиевой кислоты, дополнение рациона зелеными листовыми овощами, фасолью, апельсиновым соком, содержащими фолаты.
1. Особенности врожденных пороков головы и шеи/ В. А. Белая// Актуальная медицина: материалы I Студенческой научно-теоретической конференции, посвященной 120-летию со дня рождения С. И. Георгиевского. - 2018.
2. Эпидемиология анэнцефалии в крымском регионе/ Н. Н. Каладзе и др./ Таврический медико-биологический вестник. - 2018. - №1.
3. Анэнцефалия/ А.С. Николаева, И.С. Коршиков// VII Всероссийская (81-й Итоговая) студенческая научная конференция
Голопрозэнцефалия
Голопрозэнцефалия - тяжелое нарушение формирования мозга, при котором фиксируется полное или частичное отсутствие разделения на полушария в сочетании с другими пороками развития головы и лица. Симптомами данного состояния являются аномальное формирование лица с развитием циклопии, хоботкообразного носа (или отсутствия носа), заячьей губы и расщелины твердого неба. Наблюдаются генерализованные судороги и другие нарушения. Диагностика голопрозэнцефалии не представляет особых сложностей по причине выраженности расстройств, в ряде случаев возможно пренатальное выявление заболевания при помощи ультразвуковых или молекулярно-генетических методик. Лечение только симптоматическое. В зависимости от выраженности патологии больные погибают внутриутробно или в первые часы после рождения, реже живут несколько лет или доживают до взрослого возраста.
Голопрозэнцефалия - порок развития, характеризующийся различными по тяжести нарушениями разделения головного мозга на два полушария вплоть до формирования единого «мозгового пузыря». Эта аномалия является одной из наиболее распространенных нарушений формирования конечного мозга. Первое полное описание патологии было подготовлено в 1963-м году В. Де Маейром. Исследователь выделил три разновидности данного состояния: алобарную, семилобарную и лобарную. В 1993 году после дополнительного изучения специалисты выделили четвертый подтип голопрозэнцефалии - среднее межполушарное слияние, представляющего собой самый мягкий вариант этого заболевания. Встречаемость всех форм патологии по различным данным составляет 1 случай на 8000-16000 родов, особенно часто такие пороки выявляются у выходцев из Пакистана, Гавайев и Юго-Восточной Азии. У девочек голопрозэнцефалия диагностируется примерно в 2 раза чаще, чем у мальчиков.
Причины голопрозэнцефалии
Голопрозэнцефалия является полиэтиологичным состоянием, возникающим под влиянием разнообразных генетических и хромосомных дефектов. Пока не найдено четких указаний на существование взаимосвязи между определенными клиническими формами заболевания и типом нарушений генома больных. Одним из наиболее изученных генов, мутации которого приводят к голопрозэнцефалии, является SHH, расположенный на 7-й хромосоме. Продукт экспрессии гена представляет собой сигнальный белок, принимающий непосредственное участие в эмбриогенезе, в частности в процессах формирования центральной нервной системы. В зависимости от характера мутации SHH протеин имеет дефекты в своей структуре либо не выделяется вовсе. Это генетическое нарушение выявляется у 30-40% больных, механизм наследования аутосомно-доминантный с неполной пенетрантностью. В некоторых случаях вместо нарушений формирования мозга возникают такие микросимптомы как отсутствие одного резца, отсутствие уздечки языка и судорожный синдром.
Среди других генетических дефектов, способных приводить к появлению голопрозэнцефалии, выделяют мутации ZIC2, TGIF, SIX3 и некоторых других генов. Почти половина всех случаев заболевания связана не с генетическими, а хромосомными аномалиями - например, синдромом Патау, синдромом Эдвардса и триплоидией. Помимо этого голопрозэнцефалия выявляется при делециях отдельных участков 2-й, 7-й, 13-й, 21-й хромосом, что указывает на возможность поражения локализованных там генов. Подобные пороки развития головного мозга и лица также могут выступать как часть симптомокомплекса синдрома Меккеля-Грубера и ряда других состояний. Иногда конкретный механизм развития голопрозэнцефалии при перечисленных состояниях и других хромосомных патологиях остается неизвестным. Установлено, что начало развития дефектов центральной нервной системы и элементов черепа приходится на период между 1-м и 4-м месяцем вынашивания ребенка. Чем раньше возникают нарушения - тем тяжелее будут проявления заболевания.
Определенное влияние на развитие аномалий формирования мозга могут оказывать различные приобретенные факторы и особенности течения беременности. Так, при наличии у матери выраженного инсулинозависимого сахарного диабета вероятность родить ребенка с голопрозэнцефалией составляет 1%, что примерно в 200 раз выше, чем у здоровой женщины. Употребление некоторых лекарственных средств во время беременности также может приводить к такому врожденному нарушению - например, установлена взаимосвязь между заболеванием и приемом антихолестериновых препаратов (статинов). Определенную роль играют распространенные тератогены: токсины, этиловый спирт, элементы табачного дыма, ретиноевая кислота. Установление причин развития голопрозэнцефалии имеет значение для принятия решения о планировании следующих беременностей - в зависимости от характера и этиологии этого состояния вероятность повторного рождения ребенка с данной аномалией сильно различается.
Классификация голопрозэнцефалии
В настоящее время выделяют несколько клинических форм голопрозэнцефалии, которые различаются между собой тяжестью проявлений, выраженностью пороков развития и распространенностью в популяции. Врачам-генетикам не удалось выявить четкой взаимосвязи между отдельными типами заболевания и генетическими мутациями или хромосомными нарушениями, поэтому причины развития той, а не иной формы патологии остаются неизвестными. Предполагается, что это может зависеть от пенетрантности дефектного гена, того, какой именно участок хромосомы был подвержен повреждениям, условий протекания беременности и ряда других факторов. На сегодняшний день различают четыре основные формы голопрозэнцефалии:
- Алобарная форма - наиболее тяжелая форма, характеризующаяся грубейшими пороками развития головного мозга, лица и других органов. Диагностируется в 15-25% случаев заболевания.
- Семилобарная форма - классический вариант голопрозэнцефалии, проявляющийся тяжелыми, но менее выраженными аномалиями развития. Она также является самым распространенным типом заболевания, составляющим от 40 до 60% всех эпизодов патологии.
- Лобарная форма - более сглаженный вариант голопрозэнцефалии, при котором посредством хирургической коррекции и симптоматической терапии можно улучшить качество жизни больного и продлить ее до взрослого возраста. Эта разновидность составляет примерно 15-20% всех случаев заболевания.
- Вариант среднего межполушарного слияния - наиболее редкая форма голопрозэнцефалии, которая характеризуется смазанными симптомами и зачастую сильно отличается от классических типов патологии. Рассматривается как мягкая форма этого порока развития с 1993 года.
Некоторые специалисты в качестве отдельного варианта выделяют так называемые абортивные формы голопрозэнцефалии у фенотипически здоровых лиц. Характерные симптомы заболевания отсутствуют, однако у пациентов может наблюдаться отсутствие одного резца или уздечки языка, аномалии строения носовой полости. Изредка возникают судорожные припадки. Все перечисленные микросимптомы указывают на предположительное наличие генетических дефектов, которые могут передаться потомству и становиться причиной развития полноценной формы голопрозэнцефалии. Лицам с такими симптомами следует особенно тщательно подходить к вопросам пренатальной диагностики в период планирования беременности и развития плода.
Симптомы голопрозэнцефалии
Проявления голопрозэнцефалии сильно различаются в зависимости от формы патологии. Тем не менее, существуют общие симптомы, характерные практически для всех разновидностей заболевания. В список таких симптомов входят расщепление твердого нёба и верхней губы, выявляющиеся почти у всех пациентов (за исключением варианта среднего межполушарного слияния). Кроме того, у всех больных голопрозэнцефалией наблюдаются судорожные припадки, тяжелая умственная отсталость, нарушение рефлексов, патологии роговицы и сетчатки. При наиболее тяжелой алобарной форме заболевания выявляется циклопизм, отсутствие носа, резкое уменьшение размера головы и многочисленные пороки других органов. При этом типе голопрозэнцефалии в 70% случаев происходит самопроизвольный аборт или мертворождение, выжившие младенцы крайне редко доживают до 6-ти месяцев.
Семилобарная форма заболевания характеризуется более мягкими симптомами. Глаза больных близко расположены (гипотелоризм), голова несколько уменьшена в размерах, нередко имеются дефекты носа (недоразвитие одного носового хода). Голопрозэнцефалия этого типа, тем не менее, является достаточно тяжелым состоянием, больные умирают в первые два года жизни. Лобарная форма характеризуется еще более легкими проявлениями. При своевременной хирургической коррекции дефектов нёба и верхней губы возможно достижение пациентами подросткового и даже взрослого возраста. Летальность при этой разновидности голопрозэнцефалии во многом зависит от наличия или отсутствия сопутствующих заболеваний и пороков развития. Вариант среднего межполушарного слияния характеризуется отсутствием аномалий развития лица, однако умственная отсталость, судорожные припадки и другие неврологические проявления сохраняются.
Практически все симптомы голопрозэнцефалии выявляются сразу при рождении ребенка или еще на этапе внутриутробного развития. У пациентов могут обнаруживаться эндокринные расстройства, дисплазии почек, яичников, легких и других органов. Согласно данным медицинской статистики, именно эти нарушения в большинстве случаев становятся причиной летального исхода у больных голопрозэнцефалией. Иногда у пациентов также диагностируются иммунологические расстройства, врожденные пороки сердца, арефлексия и другие патологии. Из-за слабости мышц и наличия дефектов полости рта кормление таких детей нередко бывает затруднено, из-за чего они медленно набирают вес и отстают в физическом развитии.
В педиатрии и неонатологии диагностика голопрозэнцефалии не представляет особых затруднений, поскольку даже стертый вариант среднего межполушарного слияния достаточно легко выявить методами современной медицинской визуализации. Нередко это заболевание диагностируется еще на пренатальном этапе при проведении профилактических ультразвуковых исследований (иногда - уже на 12-й неделе беременности). На УЗИ можно отчетливо увидеть аномальное строение черепа и головного мозга плода. При алобарной форме мозг выглядит как наполненный жидкостью пузырь без каких-либо признаков разделения на полушария. Семилобарный тип голопрозэнцефалии характеризуется наличием борозды в задней части мозга, соответствующей неполному или начальному разделению на полушария. Лобарная форма заболевания несколько сложнее обнаруживается при помощи УЗИ, поскольку признаки нарушения разделения мозга фиксируются только в его глубоких слоях - мозолистом теле, таламусе и желудочках.
Определить наличие голопрозэнцефалии на пренатальном этапе развития можно при помощи методов молекулярной генетики. Материал для исследования в подозрительных случаях (неоднозначные результаты УЗИ, наличие аналогичных нарушений у родственников или при прошлых беременностях, микросимптомы у родителей, сахарный диабет у матери) берут методом амниоцентеза или биопсии ворсин хориона. Генетическая диагностика голопрозэнцефалии может включать в себя прямое секвенирование гена SHH для выявления мутаций, а также исследование кариотипа плода или ребенка для выявления хромосомных патологий. Примерно в 60% случаев заболевания изменения кариотипа не обнаруживаются, поэтому методика считается низкоспецифичной.
Лечение голопрозэнцефалии
Специфического лечения голопрозэнцефалии не существует. Осуществляют хирургическую коррекцию аномалий развития лица, проводят симптоматическую терапию. При алобарной и семилобарной формах заболевания к помощи нейрохирургов прибегают редко из-за тяжести состояния больного - ребенок попросту может не вынести такую операцию. При лобарной форме голопрозэнцефалии в первые 6 месяцев стараются произвести хирургическое вмешательство для устранения расщепления нёба и губы и формирования нормального носа. При всех типах патологии используют противосудорожную терапию. Коррекцию других нарушений осуществляют при наличии показаний. Пациентам, дожившим до детского или взрослого возраста, необходимо лечение у психиатра, дефектолога и других специалистов по причине выраженных отставаний в умственном развитии.
Прогноз при большинстве форм голопрозэнцефалии неблагоприятный - больные либо умирают в первые часы, дни, месяцы или годы жизни (алобарная и семилобарная формы), либо на всю жизнь остаются умственно отсталыми, страдают от судорожных припадков и других неврологических нарушений. Профилактика этого состояния сводится к ранней пренатальной диагностике методом ультразвукового исследования или генетических техник. При выявлении заболевания у плода ставится вопрос о прерывании беременности по медицинским показаниям. Пренатальная диагностика особенно актуальна, если кто-то из родителей имеет микросимптомы голопрозэнцефалии, если заболевание наблюдалось у близких родственников, если беременная страдает от сахарного диабета или подвергается воздействию тератогенов.
УЗИ, МРТ при лобарной голопрозэнцефалии у плода
Серёжкина А.В. 1, 2 Миненкова Т.А. 2 Разинькова Н.С. 2 Емельянова Т.А. 2 Матвиенко Е.В. 2 Тилипкина К.А. 2
Голопрозэнцефалия относится к порокам развития головного мозга, обусловленным неполным разделением эмбрионального переднего мозга. В норме передний мозг (прозэнцефалон) примерно на 32-е сутки гестации делится на две части: конечный мозг, или телэнцефалон (хвостатые ядра, скорлупу, гемисферы большого мозга), и промежуточный мозг (зрительные бугры, гипоталамусы, бледные шары). Результатом нарушения деления могут быть различные аномалии лица и/или головного мозга. Ключевые слова: голопрозэнцефалия, врожденные пороки развития, педиатрия.
1. Лучевая диагностика и терапия заболеваний головы и шеи : национальное руководство / гл. ред. тома Т. Н. Трофимова. - М. : ГЭОТАР-Медиа, 2013. - 888 с. -(Серия "Национальные руководства по лучевой диагностике и терапии" / гл. ред. серии С. К. Терновой).
2. Влияние различных факторов на плод/ Л.А. Озолиня, И.В. Бахарева, А.В. Тягунова - М. : ГЭОТАР-Медиа, 2017.
3. Неонатология: Национальное руководство. Краткое издание / Под ред. Н.Н. Володина. - М. : ГЭОТАР-Медиа, 2014.
4. The Merck Manual. Руководство по медицине. Диагностика и лечение / гл. ред. Марк Х. Бирс ; пер. с англ. под ред. А. Г. Чучалина. - 2-е изд. - М. : Литтерра, 2011. - 3744 с.
6. Общая неврология/ А. С. Никифоров, Е. И. Гусев. - 2-е изд., испр. и доп. - М. : ГЭОТАР-Медиа, 2015.
7. Патологическая анатомия : национальное руководство / гл. ред. М. А. Пальцев, Л. В. Кактурский, О. В. Зайратьянц. - М. : ГЭОТАР-Медиа, 2014. - 1264 с.
Семилобарная голопрозэнцефалия - серп мозга и межполушарная щель частично сформированы в задних отделах мозга. В передних отделах отмечают «слияние» гемисфер мозга, частичное «сращение» таламусов. Мозолистое тело как анатомическая структура отсутствует, но в области валика могут определять единичные волокна. Выделяют срединный межгемисферный вариант голопрозэнцефалии, когда межполушарная щель присутствует в лобных и затылочных отделах, а сращение (неразделение) происходит в задней лобной и теменной области. Данное состояние также называют синтелэнцефалией.
Этиологическими факторами в ее возникновении считают трисомию 13, 15 и 21 пары хромосом, синдром Дауна, другие хромосомные аберрации, воздействие ионизирующего излучения.
Голопрозэнцефалия - порок, который может быть результатом заболеваний, характерных для Х-сцепленного, аутосомно-рецессивного и аутосомно-доминантного типа наследования. Таким образом, для носителей аутосомно-доминантной голопрозэнцефалии процент рождения ребёнка со стёртой формой порока, составляет 14%, а с выраженной - 21%. Данный порок весьма разнороден, поэтому его проявление у одного из членов семьи может быть обусловлено основным заболеванием: например, если мать больна сахарным диабетом, то есть риск родить ребёнка с голопрозэнцефалией и он составляет 1 %. В случае, если у родителей присутствует синдромальная форма, то у детей может проявиться в случае повторения соответствующего синдрома. Цитогенетическая аномалия может иметь повторение в случае наличия порока у одного из потенциальных родителей. В случаях, если в роду не было ни одного случая проявления данного порока развития головного мозга, его появление может иметь место в 4-5% случаев. В некоторых из них возникновение аномалии объясняется дигенным наследованием. [5]
Эта аномалия является одной из наиболее распространенных нарушений формирования конечного мозга. Первое полное описание патологии было подготовлено в 1963-м году В. Де Маейром. Исследователь выделил три разновидности данного состояния: алобарную, семилобарную и лобарную. В 1993 году после дополнительного изучения специалисты выделили четвертый подтип голопрозэнцефалии - среднее межполушарное слияние, представляющего собой самый мягкий вариант этого заболевания. Встречаемость всех форм патологии по различным данным составляет 1 случай на 8000-16000 родов, особенно часто такие пороки выявляются у выходцев из Пакистана, Гавайев и Юго-Восточной Азии. У девочек голопрозэнцефалия диагностируется примерно в 2 раза чаще, чем у мальчиков. [2]
Проявления голопрозэнцефалии сильно различаются в зависимости от формы патологии. Тем не менее, существуют общие симптомы, характерные практически для всех разновидностей заболевания. В список таких симптомов входят расщепление твердого нёба и верхней губы, выявляющиеся почти у всех пациентов (за исключением варианта среднего межполушарного слияния). Кроме того, у всех больных голопрозэнцефалией наблюдаются судорожные припадки, тяжелая умственная отсталость, нарушение рефлексов, патологии роговицы и сетчатки. При наиболее тяжелой алобарной форме заболевания выявляется циклопизм, отсутствие носа, резкое уменьшение размера головы и многочисленные пороки других органов. [6] При этом типе голопрозэнцефалии в 70% случаев происходит самопроизвольный аборт или мертворождение, выжившие младенцы крайне редко доживают до 6-ти месяцев.
Семилобарная форма заболевания характеризуется более мягкими симптомами. Глаза больных близко расположены (гипотелоризм), голова несколько уменьшена в размерах, нередко имеются дефекты носа (недоразвитие одного носового хода). Голопрозэнцефалия этого типа, тем не менее, является достаточно тяжелым состоянием, больные умирают в первые два года жизни. Лобарная форма характеризуется еще более легкими проявлениями. При своевременной хирургической коррекции дефектов нёба и верхней губы возможно достижение пациентами подросткового и даже взрослого возраста. Летальность при этой разновидности голопрозэнцефалии во многом зависит от наличия или отсутствия сопутствующих заболеваний и пороков развития. Вариант среднего межполушарного слияния характеризуется отсутствием аномалий развития лица, однако умственная отсталость, судорожные припадки и другие неврологические проявления сохраняются. Семилобарная форма - классический вариант голопрозэнцефалии, проявляющийся тяжелыми, но менее выраженными аномалиями развития. Она также является самым распространенным типом заболевания, составляющим от 40 до 60% всех эпизодов патологии.
В педиатрии и неонатологии диагностика голопрозэнцефалии не представляет особых затруднений, поскольку даже стертый вариант среднего межполушарного слияния достаточно легко выявить методами современной медицинской визуализации. Нередко это заболевание диагностируется еще на пренатальном этапе при проведении профилактических ультразвуковых исследований (иногда - уже на 12-й неделе беременности). [3] На УЗИ можно отчетливо увидеть аномальное строение черепа и головного мозга плода. При алобарной форме мозг выглядит как наполненный жидкостью пузырь без каких-либо признаков разделения на полушария. Семилобарный тип голопрозэнцефалии характеризуется наличием борозды в задней части мозга, соответствующей неполному или начальному разделению на полушария. Лобарная форма заболевания несколько сложнее обнаруживается при помощи УЗИ, поскольку признаки нарушения разделения мозга фиксируются только в его глубоких слоях - мозолистом теле, таламусе и желудочках. [1]
Определить наличие голопрозэнцефалии на пренатальном этапе развития можно при помощи методов молекулярной генетики. Материал для исследования в подозрительных случаях (неоднозначные результаты УЗИ, наличие аналогичных нарушений у родственников или при прошлых беременностях, микросимптомы у родителей, сахарный диабет у матери) берут методом амниоцентеза или биопсии ворсин хориона. Генетическая диагностика голопрозэнцефалии может включать в себя прямое секвенирование гена SHH для выявления мутаций, а также исследование кариотипа плода или ребенка для выявления хромосомных патологий. Примерно в 60% случаев заболевания изменения кариотипа не обнаруживаются, поэтому методика считается низкоспецифичной. [4]
На МРТ при семилобарной голопрозэнцефалии межполушарную щель определяют между затылочными долями. Таламусы частично разделены, вследствие чего III желудочек небольшого размера. Лобные доли не разделены. Мозолистое тело не визуализируют.[1]
Голопрозэнцефалию возможно диагностировать, начиная с 13-14 нед гестации с помощбю ультразвукового исследования. В большинстве случаев диагноз ставят на 20-24-й неделе. [3] Пренатальная диагностика голопрозэнцефалии основана на обнаружении сочетанных аномалий лицевого черепа, лица и головного мозга. Определение аномалий лица заставляет заподозрить и начать более подробный поиск интракраниальных изменений, таких как единственный желудочек мозга, сращение таламусов, отсутствие межполушарной щели, микроцефалию. По данным УЗИ невозможно уверенно дифференцировать алобарную и семилобарную голопрозэнцефалию. Диагностика лобарной голопрозэнцефалии связана с большими трудностями.
В пренатальном периоде голопрозэнцефалию следует дифференцировать от вентрикуломегалии выраженной степени, гидранэнцефалии, энцефалоцеле, внутримозговых кист.
В качестве примера мы приводим клинический случай семилобарной голопрозэнцефалии, диагностированной у ребенка в возрасте 1 мес 4 дня в Курскую ОДКБ поступил ребенок Даниил Р., с жалобами на срыгивания, периодическое беспокойство.
Из анамнеза известно: ребенок от 7 беременности, на фоне приема регулона, цикличных менструаций, мать на учете не состояла, 4 преждевременных домашних родов. При рождении: вес 1700г, рост 42см. Ребенок осмотрен участковым педиатром на 10сутки. Нейросонография по м/ж межполушарная щель смещена влево, визуализируется большое количество жидкостного элемента. Госпитализирован в Курскую ОДКБ в отделение №3 для обследования и лечения.
Аллергоанамнез не отягощен. Наследственность не отягощена.
Объективные данные при поступлении: Общее состояние ребенка средней степени тяжести. На грудном вскармливании. Вес 3600г, рост 51см. Слизистые чистые, влажные. Склеры - субиктеричные. Дыхание ритмичное, хрипов нет. ЧД =36 в мин. Тоны сердца ритмичные. ЧСС =134 в мин, АД 85/55мм.рт.ст. Пупочная грыжа небольших размеров. Наружные половые органы сформированы по мужскому типу, обе половины мошонки увеличены в размерах. Головка полового члена открыта, отверстие уретры смещено книзу.
В неврологическом статусе: Сознание ясное. Улыбается. Окружность головы 37,5см, большой родничок 2,0х2,0см на уровне костей черепа. Голова гидроцефальной формы. Менингиальные симптомы отсутствуют. Двигательная активность сохранена. Мышечный тонус умеренно повышен по флексорному типу в сгибателях конечности. Рефлексы: Бабинский+, Моро+, ползания +, опора+, рефлекс автоматической ходьбы+, при тракции за руки голову выводит. В положении на животе голову выводит, опора на предплечья.
При УЗИ головного мозга: Структуры мозга сформированы неправильно, правое полушарие практически полностью отсутствует. Ядра таламуса и структуры задней черепной ямки сохранены, но правое ядро таламуса гипоплазировано. Эхогенность левого полушария средняя. Рисунок извилин и борозд отчетливый слева. Межполушарная щель в сечении через тела боковых желудочков :1,7 (N-до 4 мм). Субдуральное пространство : 0 мм (N-2 мм) Субарахноидальное пространство D=1.7 мм; S=1.8 (N-2 мм). Боковые желудочки: правый желудочек отсутствует. Передний рог s=4,0 мм, латеральный рог - s=3,0 мм, задний рог-s=7,0мм. Третий желудочек в сечении через тела боковых желудочков : 3,0 мм(N-3 мм), форма желудочка неправильная. Четвертый желудочек в сагиттальном сечении : 3,0 мм (N-4 мм). Сосудистые сплетения контуры ровные толщина:левое-7,0мм, структура однородная. Мозжечок, таламус, подкорковые ядра - эхогенность повышена справа повышена, слева без особенностей, эхоструктура однородная. Заключение: ВАР головного мозга, характерная для алобарной формы голопрозэнцефалии.
Так же пациенту Р. было проведено МРТ головного мозга: Заключение -полученные данные могут соответствовать МР-картине семилобарной голопрозэнцефалии.
Были проведены консультации специалистов и сделаны такие заключения:
- Консультация невролога: ВАР головного мозга, характерная для алобарной формы голопрозэнцефалии. Последствия перинатального гипоксически-ишемического поражения ЦНС. Синдром двигательных нарушений.
- Консультация генетика МГК: ВАР головного мозга: семилобарная голопрозэнцефалия. Перинатальная энцефалопатия. Синдром двигательных нарушений
За время пребывания в стационаре пациент Р. Находился на грудном вскармливании и получал ноотропную поддержку - курс церебролизина; метаболическую терапию - Витамин D3, левокаринитин ; симптоматическую терапию - Урсофальк.
Выписан в стабильном состоянии. Вес при выписке 3960г (+360г). Неврологический статус без динамики.
Специфического лечения голопрозэнцефалии не существует. Осуществляют хирургическую коррекцию аномалий развития лица, проводят симптоматическую терапию. При семилобарной форме заболевания к помощи нейрохирургов прибегают редко из-за тяжести состояния больного - ребенок попросту может не вынести такую операцию. Пациентам, дожившим до детского или взрослого возраста, необходимо лечение у психиатра, дефектолога. [5]
Прогноз при большинстве форм голопрозэнцефалии неблагоприятный - больные либо умирают в первые часы, дни, месяцы или годы жизни (алобарная и семилобарная формы), либо на всю жизнь остаются умственно отсталыми, страдают от судорожных припадков и других неврологических нарушений. У ребенка могут наблюдаться проблемы со зрением, обонятельные нарушения, эндокринологические расстройства, а также гидроцефалия. [7]
Читайте также:
- Классификация парезов и параличей лицевого нерва по House-Brackmann и Fisch
- Оценка структуры рельефа слизистой органа ЖКТ (желудка, кишечника) по рентгенограмме
- Плазмоцитома сосудистой оболочки глаза: признаки, гистология, лечение, прогноз
- Роль организма в развитии опухоли. Наследственность и опухолевый процесс.
- Бактерицидное действие света. Действие света на кожу