УЗИ, МРТ при пентаде Кантрелла у плода
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Неполная пентада Кантрелла: клиническое наблюдение и обзор литературы
Пентада Кантрелла (ПК) — редкий врожденный порок, характеризующийся пятью составляющими дефектами: передней брюшной стенки, нижней части грудины, передней части диафрагмы, перикарда в диафрагмальной части и врожденным пороком сердца. Классическая форма пентады Кантрелла, включающая все пять признаков, встречается достаточно редко. Модифицированная классификация пентады Кантрелла разделяет порок на полный, частичный и неполный. Ограниченное число работ описывает успешное лечение новорожденных с пентадой Кантрелла, при этом публикаций о неполном пороке еще меньше.У мальчиков ПК встречается несколько чаще, чем у девочек, в соотношении 1,35:1. При анализе исходов у 153 детей с ПК, несмотря на хирургическое вмешательство, сообщается о летальности, доходящей до 52 % . Достаточно ограниченное число работ описывает успешное лечение новорожденных с ПК, при этом публикаций о неполном пороке еще меньше.
В данной работе авторы описали клиническое наблюдение успешного хирургического лечения новорожденного мальчика с неполной пентадой Кантрелла.
Описание клинического наблюдения
При обследовании по месту жительства в июле 2020 г. у женщины 36 лет на первом ультразвуковом скрининге по поводу беременности в 12-13 нед. обнаружено омфалоцеле больших размеров у плода, содержащее печень. От предложенной биопсии хориона женщина отказалась, решила сохранить беременность до результатов второго ультразвукового исследования (УЗИ). При дальнейшем наблюдении в 20 и 33 нед. по данным магниторезонансной томографии (МРТ) диагностированы диафрагмальная грыжа, омфалоцеле с дислокацией практически всей печени у плода мужского пола, заподозрен врожденный порок сердца (гипоплазия дуги аорты), плацентоцентез — 46, XY. Несмотря на заключения консилиумов двух федеральных учреждений на разных сроках о сомнительном прогнозе для жизни новорожденного, женщина приняла решение сохранить беременность.
Данному пациенту первым этапом была устранена диафрагмальная грыжа с формированием временного вместилища для грыжи пупочного канатика путем подшивания к краям дефекта передней брюшной стенки силастикового мешка с целью последующего постепенного погружения содержимого грыжи в брюшную полость, что позволило снизить внутрибрюшное и соответственно внутригрудное давление, обеспечить благоприятные условия для заживления диафрагмы и таким образом стабилизировать у ребенка дыхательную и сердечно-сосудистую систему, провести обследование сердечно-сосудистой системы в более благоприятных условиях и исключить внутрисердечные пороки.
При этом выбранная тактика позволила безопасно выполнить второй этап хирургической коррекции — радикальную пластику передней брюшной стенки на 14-е сутки жизни с полным восстановлением нормальных анатомо-физиологических соотношений, к этому времени диафрагма и средостение заняли свое правильное топографическое расположение.
Заключение
Источник:
Каганцов И.М., Баиров В.Г., Сухоцкая А.А., и др. Неполная пентада Кантрелла: клиническое наблюдение и обзор литературы.
Российский вестник детской хирургии, анестезиологии и реаниматологии. — 2021. — Т. 11. — №3. — C. 375-386.
Пентада Кантрелла: клиническое наблюдение
Актульность. Пентада Кантрелла - редкий комбинированный порок развития, представленный пятью признаками: торакоабдоминальным дефектом передней брюшной стенки, дефектами нижней части грудины, передней части диафрагмы и диафрагмального перикарда, а также врожденными аномалиями сердца. Прогноз неблагоприятный. Заболевание может быть диагностировано уже в первом триместре беременности. Исходы и врачебная тактика определяются характером кардиального порока и тяжестью внесердечных аномалий.
Описание. Мы представляем случай трехмесячной выживаемости ребенка из двойни с полной формой пентады Кантрелла. У ребенка при осмотре визуализировался большой дефект передней брюшной стенки, прикрытый тонкими оболочками. Супраумбиликально определялись сокращения эктопированного сердца. Имели место челюстно-лицевые деформации по типу «заячьей губы» и «волчьей пасти». Выражена общая мышечная гипотония. Наличие у ребенка сложного порока сердца явилось противопоказанием к выполнению хирургической коррекции омфалоцеле. В связи с этим была предпринята попытка частичного погружения эвентрированных органов в брюшную полость терапевтическими методами - омфалоцеле с рождения фиксировано за пуповину вертикально (рис. 1 см. на вклейке). В 3 месяца ребенок выписан домой при стабильных показателях гемодинамики с применением кислородного концентратора.
Заключение. В случае рождения ребенка с пентадой Кантрелла тактика и стратегия должны основываться на стабилизации гемодинамических показателей и подготовке к дальнейшему оперативному лечению с использованием современных принципов лечения и выхаживания.
Ключевые слова
Пентада Кантрелла - редкая врожденная сочетанная патология, характеризующаяся пятью пороками развития: центральным, супраумбиликальным торакоабдоминальным дефектом передней брюшной стенки; дефектом дистального сегмента грудины; эктопией сердца с отсутствием диафрагмального сегмента перикарда; диафрагмальной грыжей; врожденными внутрисердечными аномалиями.
Первое описание этого порока было сделано J.R. Cantrell с соавт. в 1958 году [1], в котором сообщалось о пяти случаях сочетания вышеперечисленных аномалий развития. Полная пентада Кантреллa - тяжелый и редкий, обычно фатальный синдром; чаще встречаются неполные формы, представляющие собой комбинацию двух или трех пороков [2].
В литературе имеется небольшое количество публикаций о данной врожденной патологии. Распространенность варьирует от 5,5 до 7,9 на 1 млн живорожденных [3].
По мнению W.M. Toyama, пентаду Кантрелла делят на 3 класса [4]: 1-й класс (определенный диагноз) - имеются все 5 врожденных пороков, характерных для пентады Кантрелла; 2-й класс (вероятный диагноз) - представлены 4 врожденных дефекта, в том числе пороки сердца и аномалии развития передней брюшной стенки; 3-й класс - неполный вариант, представленный различными комбинациями врожденных дефектов, включающий аномалию грудины.
Этиология пентады Кантрелла неизвестна. Описаны спорадические случаи возникновения заболевания. Чаще страдают мальчики (2,7:1). В литературе описаны единичные случаи сочетания с сиреномелией (синдром русалки), анэнцефалией, синдромом амниотических перетяжек [5]. Описаны семейные случаи изолированной эктопии сердца и пентады Кантрелла [6].
При патологоанатомическом исследовании встречаются определенные виды дефектов: дефект передней брюшной стенки: в 63% случаев - омфалоцеле, у ряда больных описан гастрошизис; стернальные дефекты: расщепление грудины (26%), отсутствие мечевидного отростка (10%), отсутствие нижних двух третей грудины (9%); вентральный ретростернальный дефект диафрагмы в 91% случаев [7]; внутрисердечные аномалии - являются постоянным и обязательным симптомом пентады Кантрелла: большие вентрикулярные септальные дефекты в 100% случаев, атриальные септальные дефекты в 52%, стеноз легочной артерии в 33%, тетрада Фалло в 20% случаев [8]; отсутствие перикарда - в 75% случаев.
Экстракардиальные аномалии включают в себя челюстно-лицевые дефекты, такие как «волчья пасть» и «заячья губа», пороки развития центральной нервной системы: гидроцефалия, анэнцефалия, мальформации скелета: косолапость, отсутствие большеберцовой или лучевой костей, абдоминальные аномалии: полиспления, агенезия желчного пузыря, незавершенный поворот толстой кишки [8, 9].
Наряду с пороками развития описаны хромосомные аберрации [10]. Особое внимание уделяется проведению пренатальной диагностики. Пренатально пентада Кантрелла может быть выявлена при ультразвуковом исследовании уже с 10-й недели беременности. Ультразвуковая диагностика в 2D-реконструкции в первом триместре и дополнительное использование 3D-режима улучшает визуализацию аномалий плода практически в любом его положении [11]. Если по данным ультразвуковой диагностики выставлен диагноз пентада Кантрелла, целесообразно проведение хромосомного анализа, в связи с указаниями на возможную ассоциацию с трисомией, а также с синдромом Тернера [9]. Кроме ультразвуковой диагностики проводится магнитно-резонансная визуализация, которая в сочетании с эхокардиографией плода позволяет провести оптимальную оценку комбинации пороков при данной врожденной патологии. Прогноз определяется тяжестью поражения сердца и других сочетанных с ним аномалий.
Описание
Мы представляем пациента с полной формой пентады Кантрелла, находившегося на лечении в отделении реанимации и интенсивной терапии новорожденных и недоношенных детей «Областного перинатального центра» города Ярославля, продолжительность жизни которого составила 3 месяца.
Недоношенный мальчик, массой тела 2650 г, длиной 50 см, окружность головы 34 см, окружность груди 30 см, у женщины 34 лет от 1-й беременности из монохориальной биамниотической двойни, наступившей самопроизвольно. Роды оперативные в сроке 35-36 недель гестации путем экстренного кесарева сечения в связи с плацентарной недостаточностью и страданием 1-го плода. Пренатально у 2-го плода выявлена пентада Кантрелла. В анамнезе хронический пиелонефрит, первичное бесплодие, фетоплацентарная недостаточность 1-го плода. Оценка по шкале Апгар второго ребенка составила 4/5 баллов.
У 1-го ребенка из двойни, мальчика, масса тела которого при рождении составила 1440 г, длина тела 44 см, окружность головы 29,5 см, окружность груди 26 см, оценка по шкале Апгар 6/8 баллов, на фоне задержки внутриутробного развития по гипотрофическому типу постнатально диагностирован врожденный порок сердца - перимембранозный дефект межжелудочковой перегородки 5 мм, два дефекта межпредсердной перегородки 5 и 2 мм, не потребовавшие оперативного лечения. Ребенок выписан из стационара в удовлетворительном состоянии в возрасте одного месяца пяти дней жизни из отделения патологии новорожденных.
Состояние 2-го ребенка из двойни с диагнозом пентада Кантрелла после рождения тяжелое за счет выраженной дыхательной недостаточности, потребовавшей проведения искусственной вентиляции легких в родильном блоке с рождения. Ребенок переведен в отделение реанимации и интенсивной терапии новорожденных для дальнейшего обследования и лечения.
У ребенка при осмотре визуализировался большой дефект передней брюшной стенки, прикрытый тонкими оболочками. Супраумбиликально определялись сокращения эктопированного сердца. Имели место челюстно-лицевые деформации по типу «заячьей губы» и «волчьей пасти». Выражена общая мышечная гипотония.
Результаты лабораторных исследований указывали на отсутствие изменений в гемограмме, умеренное повышение трансаминаз, отрицательные маркеры воспаления. Согласно ультразвуковому исследованию брюшной полости омфалоцеле содержало левую долю печени, поджелудочную железу, кишечные петли, желудок и сердце. Данные компьютерной томографии не дали дополнительной информации.
На рентгенограмме органов грудной клетки отмечалось сужение средостения, образованное сосудистым пучком. Легочные поля не изменены. При риноскопии выявлено значительное искривление носовой перегородки, с фактически полной обтурацией правого носового хода.
Нейросонография указывала на умеренное диффузное повышение эхогенности вещества головного мозга, небольшое расширение ликворо содержащих пространств, высокий индекс резистентности передней и средней мозговых артерий. Данные изменения при ультразвуковом мониторинге с положительной динамикой по показателям мозгового кровотока.
Гастроптоз подтвержден рентгеноконтрастным исследованием. Со стороны органов зрения пороков развития не обнаружено. Проведенное кариотипирование хромосомных аномалий не выявило.
С целью уточнения тактики ведения ребенок был консультирован специалистами ФГБУ Научный центр акушерства, гинекологии и перинатологии им. академика В.И. Кулакова Минздрава России.
Ведущим симптомом в клинической картине являлась дыхательная недостаточность, требовавшая респираторной поддержки в жестких режимах традиционной искусственной вентиляции легких (ИВЛ), с переводом в дальнейшем на осцилляторную высокочастотную вентиляцию в течение двух дней. После чего отмечалась положительная динамика в виде перевода на традиционную ИВЛ с постепенным снижением параметров вентиляции, сменой на неинвазивные методы дыхательной поддержки (CPAP), масочную подачу кислорода в возрасте одного месяца 15 дней жизни. За время нахождения в отделении ребенок перенес аспирационную пневмонию.
Ребенку со сложным пороком сердца потребовалось назначение вазопростана с первых часов жизни для поддержания гемодинамики. Гемодинамические показатели стабилизировались с применением вазопрессорной терапии к 7-м суткам жизни.
Проводилось комбинированное питание: энтеральное - через назогастральный зонд; парентеральное питание через центральный венозный катетер с расчетом всех компонентов - белков, жиров и углеводов по физиологической потребности для данного гестационного возраста. Особое внимание уделялось профилактике дискинезии желудочно-кишечного тракта в связи с наличием пороков развития грудины, диафрагмы и передней брюшной стенки.
Наличие у ребенка сложного порока сердца стало противопоказанием к выполнению хирургической коррекции омфалоцеле. В связи с этим была предпринята попытка частичного погружения эвентрированных органов в брюшную полость терапевтическими методами - омфалоцеле с рождения фиксировано за пуповину вертикально (рис. 1 см. на вклейке). В динамике отмечалось сокращение диаметра дефекта с образованием плотной корки и активная эпителизация по периметру (рис. 2 см. на вклейке).
В возрасте 2 месяцев 12 дней мальчик переведен для дальнейшего лечения в кардиологическое отделение.
Прибавка массы составила 1500 г за три месяца жизни, увеличение длины тела на 5 см за этот период жизни. В неврологическом статусе - с возраста одного месяца жизни фиксировал взгляд, в 2 месяца следил за погремушкой с поворотом головы. В 3 месяца ребенок выписан домой при стабильных показателях гемодинамики с применением кислородного концентратора.
На четвертые сутки пребывания в домашних условиях отмечалось резкое ухудшение состояния за счет нарастания симптомов дыхательной недостаточности. Ребенку вызвана скорая помощь, к моменту приезда которой диагностирована остановка сердца. Проводимые реанимационные мероприятия успеха не имели. Констатирована смерть в возрасте трех месяцев 4 суток жизни.
Пентада Кантрелла представляет собой крайне редкий комбинированный порок развития с неблагоприятным прогнозом. При полной форме данной сочетанной врожденной патологии, особенно при сочетании с аномальным кариотипом рекомендуется прерывание беременности. В случае пренатально диагностированной пентады Кантрелла и при настойчивом желании родителей сохранить ребенка возможно вынашивание беременности с дальнейшей междисциплинарной оценкой консилиумом способа родоразрешения, тактики ведения ребенка в раннем неонатальном периоде, объемом и последовательностью постнатальных вмешательств.
В нашем случае решение о пролонгировании беременности у женщины с длительным бесплодием было продиктовано наличием второго полноценного плода.
Лечебная тактика определяется размерами дефекта передней брюшной стенки, типом эктопии сердца и сочетанными аномалиями [9]. После рождения хирургическое лечение омфалоцеле должно быть незамедлительным. Одновременно должна осуществляться попытка устранения диафрагмального и перикардиального дефектов [12, 13].
Сложности хирургической коррекции у детей с данной патологией зачастую связаны с дефицитом торакоабдоминальных тканей и невозможностью погружения эктопированного сердца в грудную клетку из-за анатомических особенностей. У некоторых пациентов имеется выраженная дыхательная недостаточность, обусловленная гипоплазией легких, осложняющая проведение анестезиологического пособия, течение послеоперационного периода.
Летальность больных со сложной врожденной мальформацией, прооперированных в первые дни жизни превышает 50% [14]. B.S. Norma с соавт. сообщили о благоприятных исходах хирургической коррекции пороков сердца у 22 пациентов с пентадой Кантрелла без его эктопии [13]. L.K. Hornberger с соавт. опубликовали результаты наблюдения и лечения 13 больных пентадой Кантрелла с эктопированным сердцем (4 торакальных и 9 торакоабдоминальных эктопий). Трое из пациентов, с выраженной экстракардиальной патологией, погибли до хирургического вмешательства. Десять больных прооперировано в неонатальном периоде. Продолжительность жизни в пяти случаях составила от 3,5 до 9,5 года. Авторы пришли к выводу, что новорожденные дети с торакальной и торакоабдоминальной эктопией в сочетании с врожденным пороком сердца, при отсутствии клинически значимых экстракардиальных аномалий, имели высокий шанс успешно перенести операцию [15].
В 2008 J.H. Van Hoorn с соавт. представили 58 новорожденных, из которых 33 имели полную и 23 - неполную формы пентады Кантрелла. Двух пациентов классифицировать окончательно не удалось. Четырнадцать больных имели эктопированное сердце без грубых структурных аномалий, 16 - типичное расположение его с внутрисердечным дефектом и 23 - как эктопию, так и врожденный порок сердца. У 29 пациентов присутствовали и другие аномалии, характерные для пентады Кантрелла. 27 из 58 новорожденных умерли в первые сутки после рождения. Смертность была выше в группе детей с полной формой пентады Кантрелла и ассоциированными аномалиями [16].
Заключение
Знание такого редкого заболевания, как пентада Кантрелла специалистами ультразвуковой диагностики, акушерами-гинекологами, неонатологами и детскими хирургами позволяет проводить квалифицированное консультирование женщины с ранних сроков беременности о целесообразности пролонгирования беременности в декретированные сроки, решать вопросы пренатального мониторинга лабораторных и ультразвуковых показателей в условиях многопрофильной акушерской клиники.
Исход и врачебная тактика ведения ребенка с пентадой Кантрелла во многом определяются не только его формой, но и более ранней пренатальной диагностикой аномалии, которая позволяет применять высокотехнологичную медицинскую помощь на всех этапах лечения.
Случай трехмесячной продолжительности жизни ребенка с полной формой пентады Кантрелла, описанный нами, с абдоминальной эктопией сердца, сопутствующими внесердечными аномалиями при отсутствии хирургической коррекции врожденного порока - явление уникальное. Причина смерти нашего больного, вероятнее всего, носит кардиальный характер. Травма, тампонада сердца, внезапная смерть, эндокардит, периферическая эмболия, сердечная недостаточность, аритмия, как осложнения и причина смерти детей с пентадой Кантрелла описаны в литературе [14].
По нашему мнению, значимым фактором, приводящим к летальному исходу в домашних условиях, является прогрессирующая дискинезия желудочно-кишечного тракта (срыгивания, рвота) на фоне диспноэ у детей с врожденными пороками сердца, которая приводит к развитию аспирационной пневмонии, декомпенсации дыхательной и сердечно-сосудистой систем.
В случае рождения ребенка с пентадой Кантрелла тактика и стратегия должны основываться на стабилизации гемодинамических показателей и подготовке к дальнейшему оперативному лечению с использованием современных принципов лечения и выхаживания.
Список литературы
Об авторах / Для корреспонденции
Пентада Кантрелла у плода
Пентада Кантрелла - это врожденный порок внутриутробного развития, затрагивающий сердце и близлежащие структуры. Это редкий синдром, встречающийся у 1 из 200000 живорожденных. Множественные аномалии затрагивают разные органы, у плода сердце смещается относительно нормального положения и выпирает через дефект в нижнем секторе грудной клетки или верхней части брюшной стенки. Шансы на выживание ребенка с таким диагнозом крайне малы.

Что это такое
Заболевание относится к множественным врожденным аномалиям и имеет код по МКБ 10 Q89.7. Характеризуется множественными аномалиями, затрагивающими:
- дистальную часть грудины;
- диафрагму;
- сердце;
- нижний отдел перикарда;
- переднюю брюшную стенку.
У плода сердце смещается относительно нормального положения и выпирает через дефект в нижнем секторе грудной клетки или верхней части брюшной стенки. Встречаются случаи, когда через абдоминальную расщелину выпадают органы живота - желудок, петли кишечника.
Причины заболевания
Почему возникает патология, учеными до сих пор не выяснено. Известно лишь, что дело в неправильном строении мезодермы, которая образуется на 14-18 день внутриутробного развития. Происходит сбой, и ее сегмент формируется с нарушениями. Из-за этого ткани брюшной стенки срастаются неправильно. Предполагается связь с хромосомными аномалиями.
Риск развития пентады Кантрелла возрастает, если женщина во время беременности подвергалась воздействию неблагоприятных факторов:
- радиации, токсинов и тяжелых металлов;
- употребляла наркотики или алкоголь;
- перенесла инфекционные заболевания;
- принимала тератогенные лекарства.
Признаки и диагностика
Новорожденные с синдромом пентады Кантрелла появляются на свет с явным дефектом грудины и брюшной стенки. Порок сердца приводит к недостаточному снабжению органов и тканей кислородом. Из-за этого возникает одышка, а кожа и слизистые приобретают синюшный оттенок. Наблюдается втяжение межреберных промежутков и нижней части грудной клетки. Кроме типичных дефектов, возможны сочетанные аномалии лица и черепа, гидроцефалия.
Диагностировать патологию можно уже в середине второго триместра при ультразвуковом исследовании. У плода определяется расщепление грудины, дефекты брюшной стенки, отсутствие мечевидного отростка. Сердце сокращается за пределами грудной клетки, а органы живота выходят наружу в составе грыжевого мешка. Слышны интракардиальные шумы, а левый и правый желудочек сердца соединяются отверстием. Для подтверждения диагноза также проводят рентгенографию или компьютерную томографию, исключить хромосомные аномалии позволяет кариотипирование.
Лечение

Малыша сразу подключают к аппарату искусственной вентиляции легких. Необходимы препараты для поддержания сердечной деятельности и артериального давления. Аномалии развития устраняют только хирургическим путем. Тактика и объем операции зависят от типа и величины дефектов, сопутствующих состояний. В тяжелых случаях врачи рекомендуют проводить поэтапную коррекцию.
Прогноз
Более 80% детей с пентадой Кантрелла погибают в первые дни после рождения. Сохранить жизнь ребенка удается только при легких формах и раннем хирургическом вмешательстве. Но даже успешная операция не приводит к полному выздоровлению, она лишь облегчает состояние малыша. В большинстве случаев прогноз остается неблагоприятным - дети с таким синдромом имеют крайне низкую выживаемость.
Пентада Кантрелла - это сочетание тяжелых внутриутробных аномалий, с которыми дети обычно не выживают. Так как причины заболевания неизвестны, не существует и мер профилактики. При выявлении патологии на ранних сроках рекомендуют прерывание беременности.
Видео

* Представленная информация не может быть использована для самостоятельной постановки диагноза, определения лечения и не заменяет обращение к врачу!
Пентада Кантрелла
Пентада Кантрелла - это редкое врожденное заболевание, которое характеризуется комбинацией из пяти пороков развития: дефектами передней брюшной стенки и дистального отдела грудины, эктопией сердца, диафрагмальной грыжей, внутрисердечными аномалиями. Патологию связывают с некоторыми генетическими аномалиями, однако точные причины ее развития пока не установлены. Диагностика пентады Кантрелла осуществляется в антенатальном периоде с помощью УЗИ, эхокардиографии плода, магнитно-резонансной томографии. Тактика ведения беременности, родоразрешения и постнатального лечения определяется тяжестью порока сердца и степенью жизнеспособности плода.
МКБ-10



Общие сведения
Пентада получила название в честь американского педиатра Джеймса Кантрелла, который в 1958 году совместно с коллегами описал характерный синдром у 5 пациентов. Частота встречаемости заболевания - 5,5-7,9 случаев на 1 млн. новорожденных. Соотношение мальчиков и девочек среди заболевших составляет 2,7:1. Патология имеет крайне неблагоприятное течение, может быть ассоциирована с сиреномиелией (синдромом русалки), анэнцефалией, синдромом амниотических перетяжек. Ввиду тяжести течения пентады Кантрелла в практической неонатологии есть ряд сложностей при проведении адекватного лечения пациентов.

Причины
Пентада Кантрелла не относится к наследственным и хромосомным заболеваниям. Большинство случаев патологии имеют спорадический характер, возникают в семьях без отягощенного анамнеза. К возможным факторам риска относят тератогенные влияния на ранних сроках беременности: употребление алкоголя и наркотиков, прием антидепрессантов, длительный контакт с хлорсодержащими веществами.
Патогенез
Множественные грубые пороки развития связаны с нарушениями дифференцировки эмбриональной мезодермы. Патология возникает на 14-18 день эмбрионального периода и затрагивает большое количество тканей организма. При детальном изучении механизма формирования пентады установлено, что поражение диафрагмы и перикарда обусловлено аномальным развитием поперечных перетяжек. Дефекты мышечно-кожной стенки живота вызваны задержкой миграции структур мезодермы.
Классификация
Ранее синдром Кантрелла подразделяли на полный, когда у ребенка одновременно присутствовали все пять патологий, и неполный, когда имелись разнообразные комбинации из 2-4 аномалий, входящих в пентаду. В практической медицине наибольшее значение получила классификация Toyama, согласно которой все случаи заболевания подразделяются на 3 класса:
- Первый класс. Представляет собой полную пентаду Кантрелла и позволяет установить определенный диагноз этой патологии. Болезнь носит фатальный характер, возможности лечения живорожденных детей в основном ограничиваются паллиативной помощью.
- Второй класс. Характеризуется наличием 4-х врожденных аномалий из классической пентады, причем обязательно присутствуют пороки сердца и дефекты стенки живота. В таком случае пациентам устанавливают вероятный диагноз.
- Третий класс. Проявляется при неполной экспрессии мутантных генов. Имеет наиболее благоприятное течение среди всех вариантов данного заболевания. У детей отмечаются аномалии нижнего отрезка грудины в сочетании с одним или несколькими другими составляющими пентады.
Симптомы пентады Кантрелла
Первым признаком, который виден при рождении ребенка, является отсутствие центрального отдела брюшной стенки. Чаще всего поражение происходит по типу омфалоцеле, когда внутренние органы выпячиваются (эвентрируются) наружу через дефекты в области пупочного кольца. Реже встречается гастрошизис. Дефекты грудной стенки представлены отсутствием мечевидного отростка и расщеплением грудины. У 91% детей обнаруживается ретростернальный дефект диафрагмы.
Внутрисердечные аномалии являются постоянным признаком пентады Кантрелла. У всех новорожденных определяются дефекты межпредсердной и межжелудочковой перегородок, в 75% случаев отсутствует перикард. Более трети детей имеют стеноз легочной артерии, у каждого пятого пациента обнаруживается тетрада Фалло. Пентада клинически проявляется критическими расстройствами гемодинамики, которые требуют постоянной медикаментозной и/или аппаратной поддержки.
Для пациентов с пентадой Кантрелла характерны сопутствующие аномалии челюстно-лицевой зоны (волчья пасть и заячья губа), пороки развития головного мозга (гидроцефалия, анэнцефалия), множественные скелетные аномалии (косолапость, отсутствие трубчатых костей). Дефекты брюшной стенки нередко сочетаются с аномалиями внутренних органов: полиспленией, агенезией желчного пузыря, незавершенным поворотом кишечника.
Осложнения
При полной форме пентады возможно самопроизвольное прерывание беременности на ранних сроках. Если ребенок сохраняет жизнеспособность, существует риск преждевременных родов, грубой задержки внутриутробного развития, мертворождения. Кардиальные пороки, которые манифестируют сразу после рождения, вызывают застойные явления в легких, тяжелую гипоксемию и гипоксию, сердечно-легочную недостаточность - основную причину смерти в раннем постнатальном периоде.
К другим факторам неонатальной смертности при пентаде Кантрелла относят тампонаду сердца, аритмии, периферическую эмболию. Одним из важнейших осложнений при неполной форме порока является дискинезия пищеварительного тракта. Частые рвоты и срыгивания у младенцев становятся причиной асфиксии и летального исхода. Сочетание срыгиваний и диспноэ приводит к развитию аспирационной пневмонии и дыхательной недостаточности.
Диагностика
Постановка диагноза пентады Кантрелла проводится во внутриутробном периоде жизни ребенка благодаря современным методам визуализации. Заподозрить патологию удается при плановом посещении акушера-гинеколога и обязательном скрининге первого триместра беременности. При выявлении аномалий по данным УЗИ беременной женщине назначается расширенная программа обследования. Для диагностики применяются следующие методы:
- УЗИ беременности. С 10-й недели гестации возможна визуализация пороков сердца, недоразвития тканей брюшной стенки и других аномалий, которые входят в классическую пентаду Кантрелла. Исследование проводится в 3D-режиме для точной диагностики независимо от положения эмбриона в полости матки.
- Эхокардиография плода. Диагностированный порок сердца - абсолютное показание к пренатальной ЭхоКГ, с помощью которой определяют степень анатомо-функциональных аномалий развития сердечной области.
- МРТ плода. МР-томография выполняется после 17-18 недели беременности, чтобы максимально детально визуализировать все пороки пентады Кантрелла. Исследование необходимо для выбора программы лечения, определения прогноза для ребенка.
- Хромосомный анализ. Исследование назначается для диагностики трисомии с синдрома Тернера, которые зачастую ассоциированы с неполной формой пентады. Материал для генетической диагностики получают методом кордоцентеза или амниоцентеза.
Постнатальная диагностика проводится живорожденным младенцам для уточнения степени тяжести пороков. С этой целью назначают рентгенографию органов грудной клетки, УЗИ брюшной полости, рентгенографию ЖКТ с контрастированием. Чтобы исключить сопутствующие пороки ЦНС, проводится нейросонография. Программа обследования дополняется лабораторным мониторингом: клинический и биохимический анализы крови, исследование газов крови, анализы на острофазовые показатели.
Дифференциальная диагностика
При пентаде Кантрелла первого-второго класса постановка диагноза не затруднена, поскольку при первом осмотре ребенка выявляются патогномоничные признака патологии. Сложности могут возникать при вариантах порока с неполной экспрессией, которые нужно дифференцировать с изолированной торакальной эктопией сердца, синдромом Беквита-Видемана, хромосомными аномалиями плода и последствиями внутриутробных инфекций.
Лечение пентады Кантрелла
При полной форме заболевания и нежизнеспособности ребенка рекомендовано прерывание беременности по медицинским показаниям. В остальных случаях показано пролонгирование беременности с постоянным наблюдением у акушера-гинеколога и мониторингом состояния плода. Родоразрешение проводится планово путем кесарева сечения, после чего ребенка переводят в неонатальную реанимацию для поддержки витальных функций и определения плана дальнейших действий.
Детям с пренатально выявленными пороками сердца и относительно благоприятным прогнозом по возможности проводят антенатальное кардиохирургическое лечение. Внутриутробная коррекция кардиальных аномалий стабилизирует состояние ребенка, повышает шансы на успешное вынашивание беременности и живорождение. В остальных случаях операции проводят в неонатальном периоде при отсутствии медицинских противопоказаний.
При невозможности радикальной коррекции пентады проводится частичное погружение органов в брюшной полость, их фиксация консервативными способами. Для нормализации гемодинамики детям назначается вентиляция легких: ИВЛ, неинвазивная дыхательная поддержка CPAP, респираторная поддержка на портативном дыхательном концентраторе. Самостоятельное сосание невозможно, поэтому питание проводится через назогастральный зонд или через центральный венозный катетер.
Прогноз и профилактика
При полной пентаде Кантрелла гибель ребенка зачастую происходит еще во внутриутробном периоде либо в первые дни после рождения. Пациенты с неполными формами патологии, соответствующими второму и третьему классу по Toyama, имеют больше шансов на выживание в неонатальном периоде, однако прогноз для жизни в целом неблагоприятный. Меры профилактики заключаются в медико-генетическом консультировании пар, которые имеют отягощенную наследственность.
1. Неполная пентада Кантрелла: клиническое наблюдение и обзор литературы// И.М. Каганцов// Российский вестник детской хирургии, анестезиологии и реаниматологии. - 2021. - №3.
2. Ранняя диагностика пентады Кантрелла у новорожденного ребенка/ Н.А. Харитонова// Российский педиатрический журнал. - 2020. - №3.
4. Пентада Кантрелла: клиническое наблюдение/ Д.В. Ашерова-Юшкова// Акушерство и гинекология. - 2015. - №4.
Лучевая диагностика врожденных пороков сердца у детей при экстрастернальной эктопии сердца (Пентада Кантрелла)

Цель публикации: наглядная презентация возможностей современной лучевой диагностики врожденных пороков сердца (ВПС) в случае крайне редко встречающейся врожденной аномалии развития — пентады Кантрелла, сочетающей в себе наличие интракардиальной патологии, торакоабдоминального дефекта передней грудной и брюшной стенок, дефекта нижней трети грудины, отсутствие диафрагмального сегмента перикарда и диафрагмальной грыжи. Представлено клиническое наблюдение из собственной практики. Наиболее часто встречающиеся врожденные пороки сердца при аномалии Кантрелла — это тетрада Фалло, аномалия Эбштейна, сочетанные с отсутствием перикарда. Возможности современной лучевой диагностики позволяют пренатально определить наличие пентады Кантрелла у плода уже на сроке гестации 10-11 недель методом ультразвукового исследования, при необходимости детальной визуализации типов поражения высокой информативностью обладает магнитно-резонансная томография плода (МРТ). Постнатально для постановки точного диагноза вида ВПС используется эхокардиографическое исследование (ЭХО-КГ) и мультиспиральная компьютерная томография (МСКТ). МСКТ позволяет получить наглядные трехмерные изображения анатомии порока сердца и спрогнозировать тактику хирургического лечения. Стратегия лечения и прогноз напрямую зависят от сочетания аномалий развития в рамках пентады Кантрелла. В работе представлено клиническое наблюдение новорожденного с пентадой Кантрелла, включающей ВПС-атрезию легочной атерии (АЛА) с септальными дефектами и сопутствующими пороками.
Полный текст
Врожденные пороки сердца занимают второе место по частоте встречаемости после врожденных пороков развития нервной системы.
Пентада Кантрелла — редчайший врожденный порок развития плода, включающий в себя аномалию развития сердца. Причиной развития пентады Кантрелла считается нарушение закладки эмбриологического развития в возрасте 14-18 дней после зачатия в виде отсутствия латерального сегмента мезодермы [3]. Встречаемость порока крайне редка — 5,5 на 1 млн новорожденных. Впервые упоминание о заболевании было в 1958 году [3], когда в синдром пентады Кантрелла были включены 5 признаков нарушения эмбрионального развития [3]: верхнепупочный торакоабдоминальный дефект передней брюшной стенки, дефект дистального сегмента грудины, отсутствие диафрагмального сегмента перикарда, диафрагмальная грыжа, врожденные сердечные аномалии. В рамках международной классификации Тояма в зависимости от сочетания вышеописанных дефектов между собой принято выделять 3 класса порока [6]: 1-й класс — наличие всех пяти аномалий развития, что является истинным проявлением пентады Кантрелла, 2-й класс — наличие 4 дефектов, одним из которых является врожденная аномалия сердца, предполагает постановку возможного диагноза, 3-й класс — наличие различной комбинации дефектов, включающих в себя дефект грудины. Послеродовой и дородовой прогнозы зависят от тяжести внутрисердечных пороков развития и связанных с ними нарушений [4]. Наиболее частой внутрисердечной патологией врожденной пентады Кантрелла являются тетрада Фалло, двойное отхождение сосудов от левого желудочка (ДОС), атрезия трикуспидального клапана, аномалия Эбштейна, тотальный аномальный дренаж легочных вен (ТАДЛВ), атрезия легочной артерии (АЛА), коарктация аорты (КоАо), транспозиция магистральных сосудов (ТМС), атрезия митрального клапана, функционально единственный желудочек (ЕЖ) [3]. Среди экстракардиальных аномалий наиболее часто встречаются омфалоцеле и краниофациальные дефекты [3]. Известны случаи успешного хирургического лечения с приемлемой среднесрочной и долгосрочной перспективой [2]. По типу эктопии порок классифицируют на цервикальный, цервикоторакальный, торакальный, торакоабдоминальный.
Частота встречаемости ПК среди новорожденных составляет от 5,5 до 7,9 на 1 млн живорожденных. Данная аномалия развития не имеет приоритета к гендерной принадлежности, однако в некоторых источниках приведены случаи наиболее часто встречающейся ПК среди мальчиков [5]. В 2008 году в PubMed были опубликованы результаты частоты встречаемости ПК среди новорожденных. Были зафиксированы 58 случаев рождения, среди них 33 случая имели 1-й класс ПК по Тояма и 23 случая 3-й класс ПК по Тояма [5]. 29 новорожденных имели сочетанные аномалии развития. 37 из 58 новорожденных с ПК имели летальный исход в течение первых дней жизни, посмертно было заключено, что все случаи полных форм ПК сочетались с сопутствующими экстракардиальными аномалиями [5].
Необходимым объемом диагностических методов исследования в рамках определения типа порока являются рутинные анализы крови, мочи, электроэнцефалограмма. В рамках лучевых методов исследования для постановки диагноза обязательным является выполнение обзорной рентгенограммы грудной клетки, эхокардиографического исследования сердца [1].
Представляем клинический случай доношенного новорожденного с редчайшим пороком развития — пентадой Кантрелла, включающей в себя аномалию развития сердца.
Случай из практики
Доношенная девочка переведена в отделение анестезиологии и реанимации кардиохирургии перинатального центра ГБОУ ВО «СПбГПМУ» на 2-е сутки после рождения в Демьянской ЦРБ. В анамнезе: ребенок от вторых срочных самостоятельных родов, ручное отделение последа, седьмой беременности, во время беременности мать на учете не состояла, простудные заболевания в анамнезе. Масса ребенка при рождении 2215 г, рост 44 см. Апгар 7/8 баллов, состояние с рождения тяжелое, неврологическая симптоматика проявлялась гипотонией, гипорефлексией, акроцианозом. С 1-х суток жизни ребенок находился на искусственной вентиляции легких (ИВЛ), с жесткими параметрами, показатели сатурации 95 %. В легких дыхание проводилось с обеих сторон с наличием единичных хрипов проводного характера. Тоны сердца ритмичные. ЧСС — 130/мин. АД 52/25 мм рт. ст. Живот не вздут, мягкий, диурез сохранен. Локально определялся дефект передней грудной и брюшной стенок, в просвете дефекта зияло сердце с омфалоцеле (содержащим, предположительно, желудок), непокрытые кожно-апоневротическим компонентом. Дефект был прикрыт марлевой повязкой, смоченной в подогретом растворе фурацилина. В Демьянской ЦРБ получала лечение: антибиотики ампициллин 50мг/кг 2 раза/сутки внутривенно, инфузионная терапия (глюкоза 10 %, аминовен 10 %) в V = ФП, дицинон, обезболивающие, инотропная поддержка (дофамин 0,5 % 2мкг/кг × мин).
При осмотре новорожденной на момент поступления в кардиохирургического отделения перинатального центра ГБОУ ВО СПбГПМУ выявлены все 5 признаков синдрома, в том числе врожденный порок сердца, заболевание классифицировано как 1-й класс по Тояма (рис. 1).

Рис. 1. Новорожденная девочка, 2-х суток жизни, поступившая с диагнозом врожденная эктопия сердца, омфалоцеле
Ребенок был осмотрен коллегией специалистов: неонатологом, кардиологом, кардиохирургом, торакоабдоминальным хирургом, неврологом. Состояние ребенка расценено как стабильное, тяжелое, сознание на момент осмотра медикаментозно угнетено. При поступлении пациентке выполнена обзорная рентгенограмма грудной и брюшной полостей, визуализирующая сердечную тень в проекции эпигастрия, что дало представление о эвентрации сердца на переднюю брюшную стенку, однако не отражало анатомию внутрисердечных структур. Верхняя доля правого легкого ателектазирована, в дистальных отделах обоих легких выявлены признаки интерстициального отека легочной паренхимы. Контуры диафрагмы четкие, ровные (рис. 2). Учитывая отсутствие возможности проведения трансторакального ЭХО-КГ, в силу отсутствия кожно-апоневротического компонента, покрывающего сердце, в рамках предоперационного планирования принято решение о выполнении мультиспиральной компьютерной томографии (МСКТ) области грудной клетки с целью определения анатомии внутрисердечных структур и определения типа врожденного порока сердца.
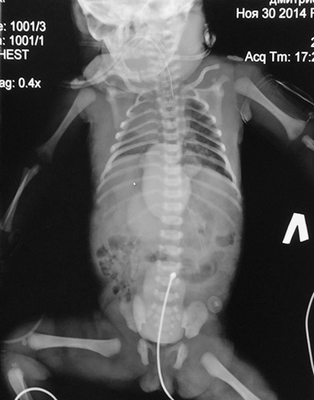
Рис. 2. Обзорный ренгтеновский снимок, показывающий атипичную локализацию сердечной тени, асимметричное положение диафрагмы, неравномерную пневматизацию легочной ткани
Девочке 2-х суток жизни выполнена МСКТ в условиях медикаментозной седации с проведением проспективной ЭКГ-синхронизации, болюсного внутривенного контрастирования (в/в) препаратом Визипак 270 в объеме 6 мл в периферическую вену правой нижней конечности. Сканирование осуществлено на аппарате Philips Ingenity 128, kv 80, mas 350, протяженность сканирования 250 мм. Выполнено обзорное сканирование грудной клетки и брюшной полости до контрастного усиления, по результатам которого определяется диффузная неравномерность пневматизации легочной паренхимы, с наличием участков ателектазированной легочной ткани в верхних и нижних долях легких с обеих сторон, в обеих плевральных полостях определяется умеренное количество выпота (рис. 3). Сердце визуализировано вне грудной полости (рис. 4). В грыжевом мешке определяется фрагмент паренхиматозного органа брюшной полости, по нативным денситометрических показателям идентичный паренхиме печени (50HU), как показано на рисунке 5.
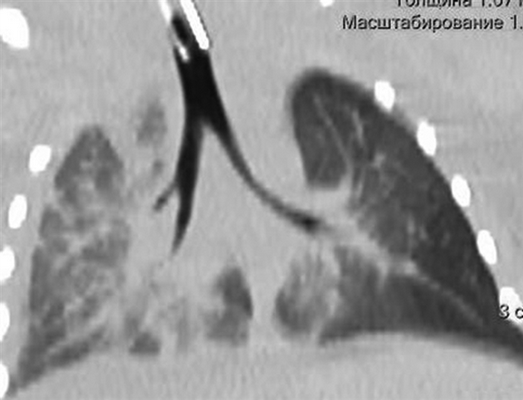
Рис. 3. Мультиспиральная компьютерная томография, мультипланарная реконструкция (MPR) легких в корональной проекции, в просвете трахеи интубационная трубка, пневматизация легких диффузно неравномерная с наличием участков ателектазированной легочной ткани
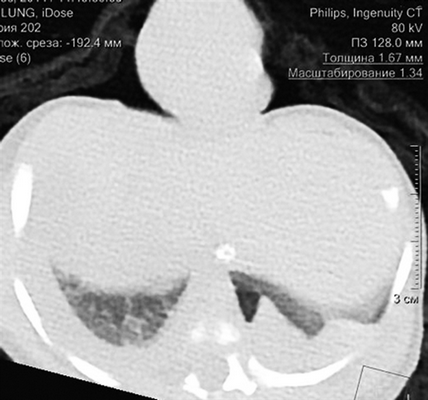
Рис. 4. Мультиспиральная компьютерная томография (МСКТ) легких, аксиальная проекция, патологический выпот в обеих плевральных полостях, эктопия сердца на переднюю грудную стенку
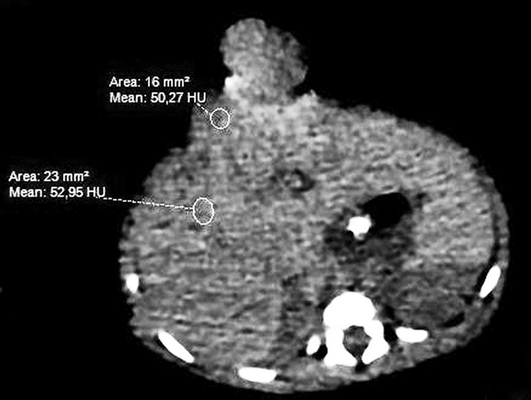
Рис. 5. МСКТ брюшной полости, аксиальная проекция, визуализирован грыжевой мешок, содержащий правую долю печени
После в/в контрастрования сканирование произведено в двух направлениях — каудокраниальном и краниокаудальном с целью визуализации анатомии сердца, определения сопутствующих пороков. В первую очередь требовалось уточнение анатомии камер сердца. По результатам исследования визуализированы полости правого и левого желудочков (ПЖ и ЛЖ), разделенные межжелудочковой перегородкой, с наличием дефекта в подклапанном отделе.
Правосторонняя предсердно-желудочковая конкордатность сохранена, анатомия правого предсердия определена наличием впадающего в него устья нижней полой вены. Левосторонняя внутрисердечная конкордантность оставалась под сомнением, так как ЛЖ сообщался с полостью, не имеющей в своей анатомии устьев легочных вен (рис. 6). Анатомия магистральных артерий также не являлась типичной. На (рис. 7, а) отчетливо визуализируется сформированный выходной тракт ЛЖ, корень аорты (Ао) с отходящими от лицевых синусов коронарными артериями, восходящий отдел Ао, дуга Ао с отходящими от нее брахиоцефальными сосудами, нисходящий отдел грудной Ао. Размеры всех вышеуказанных отделов магистральной артерии соответствовали росто-весовым показателям новорожденной (рис. 7, б).
Рис. 6. МСКТ, МPR. Четырехкамерная позиция, правосторонняя предсердно-желудочковая конкордантность сохранена, левосторонняя сомнительна
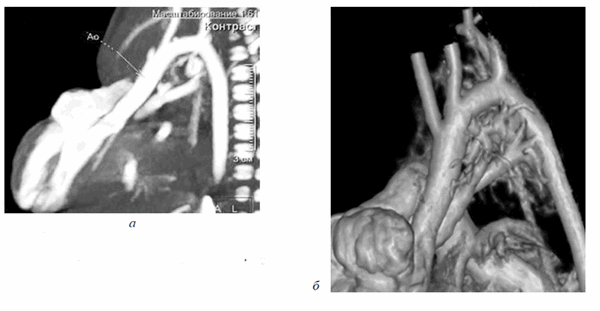
Рис. 7. МСКТ, сагиттальная проекция. (а) Визуализирован выходной тракт ЛЖ, сформированный корень Ао, восходящий отдел Ао, дуга Ао, нисходящий отдел Ао; МСКТ, объемная реконструкция (3D-volume rendering) сердца (б)
Источник кровоснабжения малого круга кровообращения в виде наличия легочной артерии (ЛА) отсутствовал, выходной отдел ПЖ не сформирован, правосторонняя предсердно-артериальная конкордантность нарушена. При детальной оценке полученных изображений визуализированы артериальные сосуды, отходящие от дуги Ао и направляющиеся к легким с обеих сторон. Общее количество больших аортолегочных коллатералей (БАЛК) — 2 (рис 8, а, б).
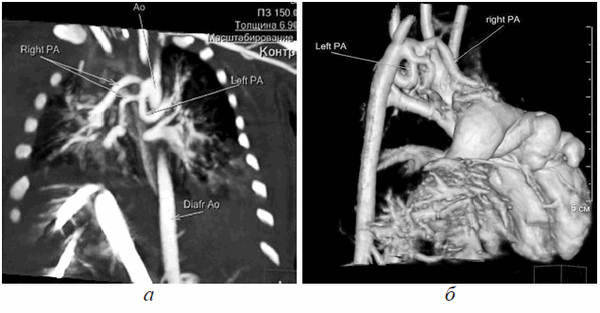
Рис. 8. МСКТ, MPR, определяются БАЛКи к правому и левому легким (а); МСКТ, 3D volume rendering, визуализация БАЛК к легким (б)
К правому легкому определено наличие двух ветвей БАЛК, одна из которых самостоятельно отходит от дуги Ао, вторая является ветвью БАЛКи, идущей также от дуги Ао, но имеющей деление на правую и левую ветви к одноименным легким соответственно. При анализе анатомии левого предсердия (ЛП) типичного расположения легочных вен (ЛВ) не отмечалось. Полость, сообщающаяся с ЛЖ, вбирала в себя единый венозный коллектор ЛВ, в который дренировались ЛВ обоих легких. Таким образом, был сделан вывод о том, что полость сообщающаяся с ЛЖ и включающая в себя единый венозный коллектор ЛВ, является ЛП (рис. 9). Анализируя структуры МПП и МЖП, отмечались множественные дефекты перегородок.
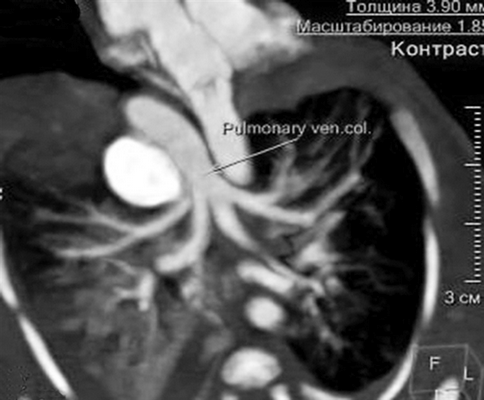
Рис. 9. МСКТ, аксиальная проекция, показан единый коллектор легочных вен
По результатам проведенного исследования было сделано заключение о наличии у пациента врожденного порока сердца — атрезия ЛА IV типа, БАЛКи ДМПП, ДМЖП, единый коллектор ЛВ в условиях эктопии сердца.
По совокупности результатов проведенного обследования собран повторный врачебный консилиум с последующим решением о необходимости экстренного хирургического вмешательства, однако учитывая тяжесть состояния пациента, проведение одноэтапной радикальной коррекции врожденных пороков невозможно, необходимо проведение хирургического лечения в 2 этапа. Первично выполнена частичная коррекция пентады Кантрелла с резекцией VI, VII, VIII ребер с формированием ложа в левой плевральной полости и низведением в нее сердца, формирование диафрагмы синтетической заплатой. Второй этап хирургического лечения с целью коррекции врожденного порока сердца решено отложить на 3-15 дней после завершения первичного вмешательства.
В раннем послеоперационном периоде состояние ребенка тяжелое, относительно стабильное, обусловлено дыхательной недостаточностью, сердечной недостаточностью, в том числе по причине врожденного порока сердца, объемом и травматичностью хирургического вмешательства. По завершении операции выполнена обзорная рентгенограмма грудной и брюшной полостей, на которой тень сердца расположена в левой половине грудной клетки (рис. 10), в верхних сегментах правого легкого сохраняется гиповентиляция, в остальных отделах легких признаки отека, контуры диафрагмы не четкие, в грудной и брюшной полостях определяются дренажи, в просвете трахеи интубационная трубка.
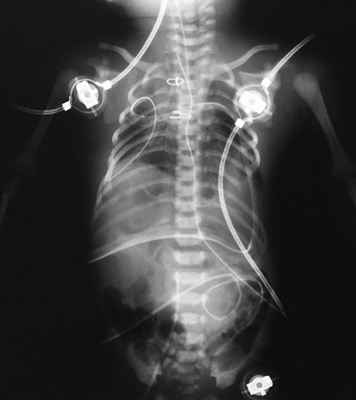
Рис. 10. Обзорный рентгеновский снимок, 1-е сутки после операции, сердце низведено в левую половину грудной клетки, положение диафрагмы асимметричное, пневматизация легких неравномерная
На фоне инфузии адреналина, милринона, метазона гемодинамика пациента оставалась относительно стабильной. Лабораторно отмечались признаки ацидоза, гиперкапнии. За время наблюдения состояние ребенка ухудшалось преимущественно за счет дыхательной недостаточности, отмечалось нарастание сердечной и печеночной недостаточностей, наблюдалась анурия, несмотря на проводимую стимуляцию лазиксом. Учитывая прогрессивное ухудшение состояния, ребенок переведен на высокочастотную ИВЛ (ВЧ ИВЛ), начат перитонеальный диализ. На фоне ВЧ ИВЛ выполнена обзорная рентгенограмма (рис. 11), пневматизация обоих легких улучшилась, сохраняются признаки отека легочной паренхимы. На 2-е сутки после операции состояние ребенка продолжало прогрессивно ухудшаться, несмотря на продолжающуюся терапию, и расценено как крайне тяжелое. На фоне высокодозной инотропной поддержки гемодинамика продолжала оставаться неустойчивой, отмечалась брадикардия, гипотония. После неоднократно проведенного непрямого массажа сердца, повторного перитонеального диализа на фоне введения максимальных доз иноторопных препаратов наблюдалась асистолия, констатирована клиническая и биологическая смерть пациента.
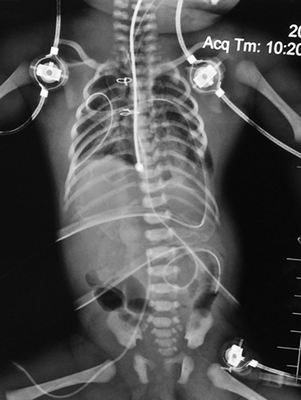
Рис. 11. Обзорный рентгеновский снимок после проведения ВЧ ИВЛ, пневматизация легочной паренхимы более равномерная
По результатам патологоанатомического вскры тия у ребенка зафиксированы множественные врожденные пороки развития — пентада Кантрелла (омфалоцеле, расщепление нижней 1/3 грудины, дефект передней части диафрагмы, отсутствие диафрагмального отдела перикарда, торакоабдоминальная эктопия сердца). Врожденный комбинированный порок сердца расценен как общий артериальный ствол с гипоплазией легочных артерий, рассыпной тип ветвления Ао, открытое овальное окно, ДМЖП, ДМПП, гипертрофия миокарда. Причина смерти — острая сердечно-сосудистая недостаточность, дыхательная недостаточность IV степени, почечная недостаточность, ДВС-синдром.
Современная пренатальная диагностика позволяет уже в 1-м триместре беременности выявлять данную патологию методом ультразвукового исследования. Дообследование плода с выявленной аномалий — пентада Кантрелла возможно при проведении МРТ плода [1]. Исходы и врачебная тактика определяются характером кардиального порока и тяжестью внесердечных аномалий [4]. В случае рождения ребенка с пентадой Кантрелла тактика и стратегия должны основываться на стабилизации гемодинамических показателей и подготовке к дальнейшему оперативному лечению с учетом современных принципов лечения и выхаживания. При полной форме пентады Кантрелла одноэтапная коррекции порока — устранение эвентрации, низведение сердца и гемодинамическая коррекция ВПС — приводит чаще всего к летальному исходу, связанному с большим травматизмом оперативного вмешательства, а также неизбежной гипергидротацией легочной ткани, приводящей к дыхательной недостаточности, связанной с гипоплазией легочной ткани и невозможностью адекватного газообмена для отключения аппарата ИВЛ [2]. При двухэтапной коррекции результат негативен по причине гипоплазии легочной ткани на микроскопичесом уровне, но отсрочен по времени.
Возможности современной МСКТ позволяют достоверно ответить на вопрос анатомии врожденного порока сердца и сочетанных аномалий развития [1].
Читайте также: