УЗИ, МРТ при синдроме Меккеля-Грубера у плода
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Поскольку непосредственные этиологические факторы аномалии развития не известны, в работе выделены основные группы причин, способствующих формированию пороков развития головного мозга. Большое значение отводится ранней диагностике пороков развития, котора
Clinical case of semilobar holoprosencephaly / A. V. Serezhkina*, **, 1, I. G. Khmelevskaya*, **, N. S. Razinkova*, **, T. A. Minenkova*, **, I. I. Zhiznevskaya*, **, A. S. Plekhanova* / * Federal State Budget Educational Establishment of Higher Education Kursk State Medical University Ministry of Health Care of Russian Federation, Kursk, Russia / ** Regional public health institution Regional Children's Clinical Hospital Committee of Health Kursk region, Kursk, Russia
Резюме. Поскольку непосредственные этиологические факторы аномалии развития не известны, в работе выделены основные группы причин, способствующих формированию пороков развития головного мозга. Большое значение отводится ранней диагностике пороков развития, которая позволяет своевременно решить вопрос о возможности пролонгирования беременности, что определяется видом порока, совместимостью с жизнью и прогнозом в отношении постнатального развития. В исследовании рассматриваемого порока развития большую роль играют такие современные методы, как пренатальная ультразвуковая диагностика, нейросонография, рентгеновская компьютерная и магнитно-резонансная томография головного мозга, имеющие достаточно высокую информативность. Указаны сроки гестации, позволяющие выявить структурные дефекты головного мозга. Медико-генетическое консультирование помогает выявить риск появления больного потомства. Проведена дифференциальная диагностика семилобарной с другими формами голопрозэнцефалии. Также отмечены возможные клинические проявления рассматриваемой нозологии. В данной статье представлен клинический случай семилобарной голопрозэнцефалии, диагностированной у мальчика в возрасте 1 месяц. При поступлении мать предъявляла жалобы на срыгивания и периодическое беспокойство сына. Объем и результаты обследования ребенка изложены ниже. Выявлена сопутствующая патология в виде пупочной грыжи, врожденной аномалии развития мочевой системы: подковообразная почка; водянки яичек и головчатой формы гипоспадии. После проведения курса поддерживающей терапии пациент был выписан в стабильном состоянии. В настоящее время специфическое лечение голопрозэнцефалии отсутствует. Оперативные вмешательства на головном мозге проводятся редко ввиду тяжести состояния больных, в связи с чем лечение данной патологии возможно только с помощью хирургической коррекции симптомов. Длительная дыхательная и кардиоваскулярная дисфункция предопределяет летальный исход заболевания.
Семилобарная голопрозэнцефалия считается умеренной формой голопрозэнцефалии. Так же, как и другие типы, этот можно диагностировать внутриутробно по отсутствию межполушарной щели (кроме задней части мозга).
Непосредственные этиологические факторы аномалии развития не известны. Многие авторы выделяют две группы причин развития порока: наследственные и экологические. Наследственные представлены хромосомными аномалиями, ведущими к анеуплоидии: трисомия 13 (синдром Патау), трисомия 18 (синдром Эдвардса), трисомия 21 (синдром Дауна), синдром триплоидии. Нередко встречающиеся мутации, связанные с голопрозэнцефалией: синдром 13 q, синдром Генуя, хвостовой дисгенез, синдром Айкарди, псевдотрисомия 13, синдром Меккеля - Грубера.
К экологическим причинам относятся сахарный диабет (СД) у матери, прием беременной салицилатов, ретиноевой кислоты, статинов, мизопростола, метотрексата, дифенилгидантоина, употребление алкогольных напитков. Доказана роль ионизирующего излучения в I триместре беременности. В основе патогенетических механизмов развития голопрозэнцефалии лежит нарушение формирования головного мозга по срединной линии. Зачастую эти процессы происходят на 5-10 неделях беременности [6].
Голопрозэнцефалия - порок, который может быть результатом заболеваний, характерных для Х-сцепленного, аутосомно-рецессивного и аутосомно-доминантного типа наследования, а также сочетания голопрозэнцефалии с инсулинзависимым СД [2].
Семилобарная голопрозэнцефалия характеризуется слиянием лобных долей при наличии незначительной перегородки в задней части с присутствием серпа и межполушарной щели. Клинически проявляться данная форма аномалии может по-разному: отсутствием носовой перегородки, анофтальмией, близким расположением глаз, аномалиями радужной оболочки и сетчатки, предчелюстными агенезиями, срединной расщелиной нёба и губы. Возможны дефекты других систем организма, в том числе врожденные пороки сердца, дисплазии яичек и половых органов, кистозность почек, кишечные мальротации, пупочные грыжи, почечные дисплазии, водянка плода и т. п. [8].
Голопрозэнцефалию можно диагностировать начиная с 13-14 недели гестации с помощью ультразвукового исследования (УЗИ). В большинстве случаев диагноз ставят на 20-24 неделях.
Пренатальная диагностика голопрозэнцефалии основана на обнаружении сочетанных аномалий лицевого черепа, лица и головного мозга. Определение аномалий лица заставляет заподозрить и начать более подробный поиск интракраниальных изменений, таких как единственный желудочек мозга, сращение таламусов, отсутствие межполушарной щели, микроцефалия. По данным УЗИ невозможно уверенно дифференцировать алобарную и семилобарную голопрозэнцефалию. Диагностика лобарной голопрозэнцефалии связана с большими трудностями [9].
Использование трансвагинальной эхографии способствовало накоплению опыта ультразвуковой визуализации структур мозга плода на ранних этапах развития, что дало возможность диагностики голопрозэнцефалии уже в конце I триместра беременности [5].
В пренатальном периоде ведущий метод диагностики - сонография. Алобарную и семилобарную форму голопрозэнцефалии необходимо диагностировать с помощью УЗИ в течение первой половины беременности, поэтому прибегать к другим технологиям нет необходимости. Диагностика лобарной формы голопрозэнцефалии возможна только с помощью магнитно-резонансной томографии (МРТ). В постнатальном периоде МРТ служит методом выбора.
Семилобарная форма голопрозэнцефалии - результат частичного недоразделения мозга на левую и правую полусферы. Существуют определенные критерии для дифференциальной диагностики этой формы с алобарной и лобарной формами порока. При семилобарной голопрозэнцефалии два полушария мозга частично разделены в задней части, имеется один общий желудочек с рудиментарными задними рогами. Алобарная и семилобарная формы часто сочетаются с микроцефалией и реже с макроцефалией. При алобарной и семилобарной голопрозэнцефалии всегда отсутствует мозолистое тело [2].
Дифференциальная диагностика аномалии развития головного мозга с помощью МРТ основана на следующих признаках: при алобарной форме мозг малых размеров и содержит одну единую полость с дорзальным саком вместо третьего и боковых желудочков, таламусы соединены вместе, нет обонятельных луковиц и обонятельных трактов; мальформации лица - наиболее неблагоприятный вариант. При семилобарной голопрозэнцефалии мозг также маленький, с рудиментами затылочных долей. Межполушарная щель имеется в переднем или заднем отделах. Поскольку мозолистое тело отсутствует частично или полностью, то дорзальный сак поднимается высоко с формированием межполушарной ликворной кисты. Самая легкая форма голопрозэнцефалии - лобарная. При ней гемисферы большого мозга отделены друг от друга, кроме передних отделов, боковые желудочки соединены между собой за счет агенезии прозрачной перегородки. Мозолистое тело отсутствует [1].
В качестве примера приведен клинический случай семилобарной голопрозэнцефалии, диагностированной у ребенка 1 месяца. Даниил Р. поступил в Курскую детскую клиническую больницу с жалобами на срыгивания и периодическое беспокойство.
При сборе анамнеза стало известно, что ребенок от 7-й беременности, протекавшей на фоне приема регулона, цикличных менструаций, четвертых преждевременных домашних родов, мать на учете в женской консультации не состояла. При рождении вес мальчика составил 1700 г, рост - 42 см. Ребенок осмотрен участковым педиатром на 10-е сутки. Нейросонография по месту жительства показала: межполушарная щель смещена влево, визуализируется большое количество жидкостного элемента. Мальчик был госпитализирован в Курскую ОДКБ в отделение № 3 для обследования и лечения.
Аллергологический и наследственный анамнез не отягощен.
Объективные данные при поступлении: общее состояние ребенка средней степени тяжести. Малыш на грудном вскармливании. Вес - 3600 г, рост - 51 см. Слизистые чистые, влажные. Склеры - субиктеричные. Дыхание ритмичное, с частотой 36 в минуту, хрипов нет. Тоны сердца ритмичные. Частота сердечных сокращений - 134 в минуту, артериальное давление (АД) - 85/55 мм рт. ст. Пупочная грыжа небольших размеров. Наружные половые органы сформированы по мужскому типу, обе половины мошонки увеличены в размерах. Головка полового члена открыта, отверстие уретры смещено книзу.
В неврологическом статусе: сознание ребенка ясное, улыбается. Окружность головы - 37,5 см, большой родничок - 2,0 × 2,0 см на уровне костей черепа. Голова гидроцефальной формы. Менингиальные симптомы отсутствуют. Двигательная активность сохранена. Мышечный тонус умеренно повышен по флексорному типу в сгибателях конечности. Определяются рефлексы Бабинского, Моро, ползания, опоры и автоматической ходьбы, при тракции за руки голову выводит. В положении на животе голову выводит, опора на предплечья.
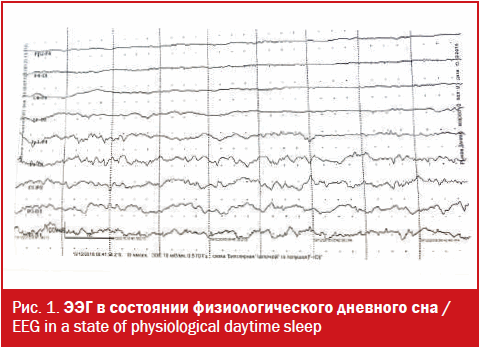
Даниилу Р. была проведена ЭЭГ в состоянии физиологического дневного сна (20 минут), на которой достоверных изменений эпилептиформного характера не было выявлено (рис. 1).
При УЗИ головного мозга было обнаружено, что структуры мозга сформированы неправильно, правое полушарие практически полностью отсутствует. Ядра таламуса и структуры задней черепной ямки сохранены, но правое ядро таламуса гипоплазировано. Эхогенность левого полушария средняя. Рисунок извилин и борозд отчетливый слева. Межполушарная щель в сечении через тела боковых желудочков - 1,7 (норма - до 4 мм). Субдуральное пространство - 0 мм (норма - 2 мм).Субарахноидальное пространство справа - 1,7 мм; слева - 1,8 (норма - 2 мм). Правый боковой желудочек отсутствует. Передний рог - 4,0 мм, латеральный рог - 3,0 мм, задний рог - 7,0 мм. Третий желудочек в сечении через тела боковых желудочков - 3,0 мм (норма - 3 мм), форма желудочка неправильная. Четвертый желудочек в сагиттальном сечении - 3,0 мм (норма - 4 мм). Контуры сосудистых сплетений ровные, толщина левого - 7,0 мм, структура однородная. Мозжечок, таламус, подкорковые ядра - эхогенность повышена справа, слева без особенностей, эхоструктура однородная. Заключение: врожденная аномалия развития головного мозга, характерная для алобарной формы голопрозэнцефалии.
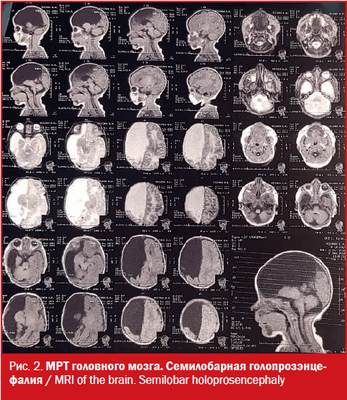
МРТ: на серии Т1- и Т2-взвешенных томограмм в сагиттальной и аксиальной проекции получено изображение супратенториальных структур головного мозга. Правые лобная, теменная и частично височная доли (кроме гиппокампа), мозолистое тело и прозрачная перегородка отсутствуют, нижний червь мозжечка гипопластичный. Правый зрительный бугор увеличен в размерах, визуализируется частично правая затылочная доля. Левое полушарие большого мозга, полушария мозжечка и ствол сформированы правильно. Ретенции желудочковой системы нет. Признаков объемного воздействия не отмечено. Базальные цистерны открыты. В заключении отмечено, что полученные данные могут соответствовать МР-картине семилобарной голопрозэнцефалии (рис. 2).
Из сопутствующей патологии отмечено наличие пупочной грыжи и врожденной аномалии развития мочевой системы (подковообразная почка, водянка яичек и головчатая форма гипоспадии).
Во время пребывания в стационаре ребенок находился на грудном вскармливании, получал ноотропную поддержку - курс церебролизина, метаболическую (витамин D3) и симптоматическую терапию (Урсофальк).
Пациент выписан в стабильном состоянии. Вес мальчика при выписке составил 3960 г (+360 г). Неврологический статус - без динамики.
Согласно данным C. Olsen и соавт. [4], среди 78 родившихся детей общая смертность составила 32% в течение первых 48 часов и 38,5% — в течение 1-й недели соответственно. При синдромальных случаях смертность в течение первых 48 часов жизни составила 57%, у больных с алобарной формой выживаемость на 1-й неделе — 50%. До 12 месяцев с изолированной формой голопроз-энцефалии дожили 54%, при синдромальных случаях — 14%, несиндромальных в сочетании с другими пороками развития головного мозга — 25%. Высокая смертность, по-видимому, связана с дисфункцией ствола или гипоталамических структур головного мозга в сочетании с полиорганной патологией. Длительная дыхательная и кардио-васкулярная дисфункция предопределяет летальный исход заболевания [3]. В настоящее время специфическое лечение голопрозэнцефалии отсутствует. Оперативные вмешательства на головном мозге проводятся редко ввиду тяжести состояния больных, в связи с чем лечение данной патологии возможно только с помощью хирургической коррекции симптомов (аномалий развития лица). При тяжелых формах заболевания отмечаются также трудности вскармливания новорожденных, связанные со слабостью сосания, невозможностью проглатывания пищи, поперхиванием, рвотой с риском аспирации, эзофагии и др. [7]. В связи с этим в некоторых случаях применяется гастротомия (при исключении центрального нарушения питания) [9]. При наличии декомпенсированной гидроцефалии рекомендовано нейрохирургическое лечение. При наличии эпилептических приступов проводится подбор противосудорожной терапии с использованием антиконвульсантов под контролем обычной ЭЭГ и видео-ЭЭГ-мониторинга. M. Barr и соавт. [1] доказали, что для восстановления нарушенных физиологических ритмов сна можно использовать внешний шум (например, радио).
В заключение отмечаем, что голопрозэнцефалия может входить в структуру хромосомных или моногенных заболеваний, а также встречаться как изолированная мальформация. Этиология голопрозэнцефалии остается недостаточно изученной. В диагностике рассматриваемого порока развития большую роль играют такие современные методы, как пренатальная ультразвуковая диагностика, нейросонография, рентгеновская компьютерная и магнитно-резонансная томография головного мозга, имеющие достаточно высокую информативность. Медико-генетическое консультирование помогает выявить риск появления больного потомства. Установлено, что молодые пары, дающие жизнь ребенку со стандартной трисомией, имеют неспецифический (около 1%) риск рекуррентной трисомии 13-й хромосомы.
КОНФЛИКТ ИНТЕРЕСОВ. Авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить.
CONFLICT OF INTERESTS. Not declared.
Литература/References
- Barr M. Jr., Cohen M. M. Jr. Autosomal recessive alobar holoprosencephaly with essentially normal facies // Am J Med Genet. 2002; 112: 28.
- Barr M. Jr., Cohen M. M. Jr. Holoprosencephaly survival and performance // Am J Med Genet. 1999; 89: 116.
- Cohen M. M. Jr. Problems in the definition of holoprosencephaly // Am J Med Genet. 2001; 103: 183.
- Olsen C. L., Hughes J. P., Youngblood L. G. et al. Epidemiology of holoprosencephaly and phenotypic characteristics of affected children. New York state 1984-1989 // Am J Med Genet. 1997; 73: 217.
- Барашнев Ю. И. Перинатальные повреждения нервной системы у новорожденных. Руководство по безопасному материнству / Под ред. Ю. И. Барашнева. М.: Триада-Х, 1998. С. 373-432. [Barashnev Yu. I. Perinatal damage to the nervous system in newborns. Safe Motherhood Guide / Pod red. Yu. I. Barashneva. M.: Triada-KH, 1998. Pp. 373-432.]
- Аминофф М. Дж., Гринберг Д. А., Саймон Р. П. Клиническая неврология. М.: МЕДпресс-информ, 2009. 480 c. [Aminoff M. Dzh., Grinberg D. A., Saymon R. P. Clinical neurology. M.: MEDpress-inform, 2009. P. 480.]
- Неонатология: Национальное руководство. Краткое издание / Под ред. Н. Н. Володина. М.: ГЭОТАР-Медиа, 2014. [Neonatology: National Guidelines. Short edition / Pod red. N. N. Volodina. M.: GEOTAR-Media, 2014.]
- Никифоров А. С., Гусев Е. И. Общая неврология. 2-е изд., испр. и доп. М.: ГЭОТАР-Медиа, 2015. [Nikiforov A. S., Gusev Ye. I. General neurology. 2nd ed., Rev. and add. M.: GEOTAR-Media, 2015.]
- Петрухин А. С. Детская неврология. В 2 т. Том 1. М.: ГЭОТАР-Медиа, 2009. 272 c. [Petrukhin A. S. Pediatric neurology. In 2 volumes, Volume 1. M.: GEOTAR-Media, 2009. P. 272.]
А. В. Серёжкина* , ** , 1
И. Г. Хмелевская* , **, доктор медицинских наук, профессор
Н. С. Разинькова* , **, кандидат медицинских наук
Т. А. Миненкова* , **
И. И. Жизневская* , **, кандидат медицинских наук
А. С. Плеханова*
* ФГБОУ ВО КГМУ Минздрава России, Курск, Россия
** ОБУЗ ОДКБ, Курск, Россия
Плод - сердце, B-режим
[EN] Эхограмма №413: Сердце плода в B-режиме.
Изображение получено с помощью УЗ сканера SonoAce-X4 (снят с производства).
Публикации по теме
Коарктация аорты у плода - ультразвуковая диагностика врожденных пороков сердца. Известно, что диагноз данного порока сердца опирается на прямой признак - визуализацию места сужения аорты, и, возможно, расширение проксимального отдела аорты. Однако четко визуализировать участок сужения аорты у плода достаточно трудно и удается только в единичных наблюдениях. Порок можно увидеть лишь тогда, когда имеется уменьшение диаметра перешейка аорты более чем на 1/3 по сравнению с нормой для каждого срока беременности. Ключом к пренатальной диагностике коарктации аорты является комплексный учет данных, получаемых как при изучении четырехкамерного среза сердца (дилатация правого желудочка, гипоплазия левого желудочка), так и при оценке самих главных артерий.
Пренатальная диагностика синдрома Меккеля-Грубера. Синдром Меккеля-Грубера (дизэнцефалия спланхнокистозная) - комплекс множественных летальных врожденных пороков развития с аутосомнорецессивным типом наследования. В связи с большой редкостью этого синдрома, единичными публикациями в отечественной литературе, посвященными его диагностике, представляем собственный опыт пренатальной диагностики 3 случаев синдрома Меккеля-Грубера.
Плод - позвоночник, B-режим
[EN] Эхограмма №414: Позвоночник плода в B-режиме.
УЗИ, МРТ при синдроме Меккеля-Грубера у плода
КГБУЗ «Красноярский межрайонный родильный дом №4», Красноярск, Россия
Лечебно-диагностический центр Международного института биологических систем им. С.М. Березина, Красноярск, Россия
Случай мальформации Денди—Уокера с доношенной беременностью и родами
Синдром Денди—Уокера — редкий вид патологии ЦНС, представляющий собой врожденный порок развития каудального отдела ствола и червя мозжечка, ведущий к неполному раскрытию срединной (Мажанди) и латеральных (Лушка) апертур IV желудочка мозга. Пациенток с мальформацией Денди—Уокера относят к высокой группе риска по развитию осложнений в течении беременности, и их ведение требует высокой квалификации врачей неврологов и акушеров-гинекологов. Представлено клиническое наблюдение случая доношенной беременности у пациентки с синдромом Денди—Уокера.
Пороки развития нервной системы занимают ведущее место в структуре аномалий развития, причем около 80% из них представлены гидроцефалией различного генеза. Согласно представленным результатам анализа данных мониторинга врожденных пороков развития в 31 регионе РФ за период 2006—2012 гг., их частота составила 23,04 на 1000 рождений [1].
Синдром Денди—Уокера (Dandy—Walker Malformation) — врожденный порок развития каудального отдела ствола и червя мозжечка, ведущий к неполному раскрытию срединной (Мажанди) и латеральных (Лушка) апертур IV желудочка мозга [2].
Синдром Денди—Уокера — редкий вид патологии ЦНС (порок развития задней черепной ямки) с частотой встречаемости среди живорожденных детей 1 случай на 25 000—35 000, однако среди детей с врожденной гидроцефалией он диагностируется значительно чаще — от 3,5 до 12% случаев [3, 4].
Впервые описание мальформации было представлено в 1914 г. американским нейрохирургом W. Dandy в соавторстве с К. Blackfun [5] в статье, посвященной изучению разных форм гидроцефалии. В 1942 г. J. Taggart и A. Walker предложили вариант хирургической коррекции описанной патологии, а в 1954 г. С. Benda объединил в названии одного синдрома фамилии двух ученых — W. Dandy и A. Walker [6].
Этиология и патогенез болезни до настоящего времени остаются неясными. В большинстве случаев заболевание носит спорадический характер, но встречаются и семейные случаи. В 29—55% случаев синдром Денди—Уокера сочетается с другими мальформациями (синдромы Меккеля, Меккеля—Грубера, Варбурга, Тернера и другие хромосомные аберрации), а также с поражениями отдельных органов и систем (поли-, синдактилия, врожденные пороки сердца, поликистоз почек, расщелины неба и др.) [3, 7, 8]. Предположительно на развитие этой патологии влияют перенесенная в ранние сроки беременности цитомегаловирусная инфекция, коревая краснуха, не исключена связь с наличием сахарного диабета, осложненного диабетической фетопатией, приемом варфарина при беременности [8, 9].
В классической форме синдром включает следующие проявления: внутреннюю гидроцефалию различной степени; расширение III желудочка до формирования кисты задней черепной ямки из-за ее увеличения, связанного со смещением вверх латеральных синусов и мозжечкового намета; гипоплазию червя мозжечка (и полушарий) [2].
Для синдрома Денди—Уокера характерна вариабельность клинических признаков [3, 10]. У некоторых больных выявляют гипоплазию червя мозжечка или небольшие кисты при нормальной заднечерепной ямке (вплоть до полного отсутствия червя мозжечка), с недостаточно развитым стволом мозга. Церебральные аномалии могут включать явления дисплазии (нейрональная гетеротопия, шизэнцефалия, дисгенезия мозолистого тела и голопрозэнцефалия). Степень гидроцефалии варьирует от небольшой дилатации боковых и III желудочков до выраженной окклюзионной водянки. Уровень обструкции сильвиева водопровода и препятствие току цереброспинальной жидкости (ЦСЖ) находятся в месте выходных отверстий IV желудочка.
Выделяют полную и неполную формы синдрома Денди—Уокера. Полная форма характеризуется агенезией червя мозжечка и наличием явной коммуникации между IV желудочком и кистой в области большой цистерны. Неполная форма — это частичная агенезия нижней части червя мозжечка, в связи с чем коммуникация IV желудочка с кистой большой цистерны прослеживается не на всем протяжении червя [3].
Клиническими проявлениями синдрома Денди—Уокера у младенцев являются увеличенный размер головы, истончение и выпячивание кости в затылочной части черепа, гидроцефалия (не всегда), мягкость черепных костей и слишком большие роднички, беспокойное поведение, мышечные спазмы и судороги, слабость крика, нистагм, спонтанные движения рук и ног, спастический тетрапарез, замедление моторного развития. У взрослых — тошнота, рвота, раздражительность, судороги, проблемы со зрением, нарушение координации движений, среди которых размашистость, нечеткость и шаткость походки (вплоть до невозможности передвигаться самостоятельно), интеллектуальное отставание [3, 11].
Диагностика синдрома Денди—Уокера возможна в пренатальный период при проведении скрининговой трансвагинальной эхографии в 13—14 нед беременности. После рождения ребенка постановка диагноза осуществляется на основе сбора анамнеза, неврологического осмотра, нейросонографии и проведения магнитно-резонансной томографии (МРТ) головного мозга с сагиттальным и аксиллярным обзором IV желудочка (с целью выявления аномалии строения мозга и гидроцефалии после закрытия родничков), офтальмологического обследования, консультации нейрохирурга [7, 12—15].
Прогноз для жизни и здоровья при синдроме Денди—Уокера зависит от наличия сочетанных аномалий развития и срока диагностики. По данным литературы [3], показатели постнатальной заболеваемости и смертности выше в случаях, когда синдром диагностирован в пренатальном периоде, а не у новорожденного.
Лечение синдрома Денди—Уокера оперативное. Для снижения внутричерепного давления (ВЧД) за счет оттока ЦСЖ проводится шунтирование IV мозгового желудочка с последующей симптоматической терапией (диуретики в сочетании с препаратами калия, лечебная гимнастика, массаж). В настоящее время широкое применение получило вентрикулоперитонеальное шунтирование, позволившее улучшить прогноз и качество жизни таких пациентов, в том числе девочек, достигающих детородного возраста [16].
При нарастании гидроцефалии и/или нарушении функции шунта необходимо произвести его ревизию: в I и II триместрах предпочтительным является вентрикулоплевральный шунт, в III — для предотвращения преждевременных родов используются вентрикулоатриальный или вентрикулоплевральный шунты. При отсутствии эффекта от проводимой терапии рекомендуется досрочное родоразрешение с предварительным проведением профилактики респираторного дистресс-синдрома плода дексаметазоном. За 48 ч до родоразрешения профилактически назначаются антибиотики для уменьшения частоты инфицирования шунта [16].
Способ родоразрешения напрямую зависит от состояния пациентки. При отсутствии клинической симптоматики родоразрешение через естественные родовые пути не противопоказано, а процент акушерских осложнений не выше, чем в общей популяции. Положительным является низкий риск возникновения спаек или инфицирования дистального конца шунта. Во втором периоде родов происходит повышение давления ЦСЖ, увеличение ВЧД, поэтому при ведении потужного периода предпочтительно укорочение потуг путем проведения рассечения промежности. Во время родов профилактика антибиотиками должна быть продолжена [16, 18].
Выбор оперативного родоразрешения обусловливается наличием неисправности в работе шунта и повышением ВЧД, приводящим к наличию клинической симптоматики и возможности провоцирования внутричерепного кровоизлияния. После стабилизации состояния больной производится кесарево сечение. По мнению некоторых авторов, более предпочтительно общее обезболивание, так как регионарная анестезия противопоказана при повышенном ВЧД. Теоретически, при регионарной анестезии какое-то количество анестетика вместе с ЦСЖ может оказаться в брюшной полости, являясь причиной неадекватного обезболивания. Однако имеются публикации [20] об успешном применении регионарной анестезии в родах у таких больных. Родоразрешение проводится в условиях многопрофильного стационара, имеющего в структуре отделение нейрореанимации.
Представляем собственное клиническое наблюдение случая доношенной беременности у пациентки с синдромом Денди—Уокера.
Больная И., 33 лет, встала на учет по беременности в женскую консультацию на сроке 7—8 нед. Беременность первая, желанная, наступила спонтанно.
Пациентка имеет II группу инвалидности. Закончила педагогический институт, работала психологом в школе, но в связи с прогрессирующим снижением зрения с работы уволилась. В поликлинике по месту жительства наблюдалась только офтальмологом, неврологом не осматривалась в течение последних 20 лет.
Больная обратилась за помощью в период беременности; прегравидарная подготовка не проводилась.
При первичном обращении жалобы на периодическую головную боль, не сопровождающуюся тошнотой, временами шаткость при ходьбе, ухудшение памяти. В неврологическом статусе: когнитивные функции сохранные, соответствуют уровню образования. Глазные щели D>S, зрачки неправильной формы, несимметричные, слабо реагируют на свет, движения глазных яблок не ограничены, функции черепных нервов сохранны; парезов нет, рефлексы с рук и ног высокие, симметричные; патологические рефлексы отсутствуют, чувствительность не нарушена, пальценосовую пробу выполняет с легкой интенцией с обеих сторон, в пробе Ромберга легкое пошатывание, менингеальных симптомов нет. Передвигается самостоятельно, без посторонней помощи.
Беременная отнесена к высокой группе по оценке пренатальных факторов риска. Был проведен консилиум с участием нейрохирургов, неврологов, офтальмологов, акушеров-гинекологов с диагнозом: «беременность 10 нед. Синдром Денди—Уокера, тетравентрикулярная сообщающаяся шунтзависимая выраженная гидроцефалия. Хронический цефалгический синдром, легкий вестибулоатактический синдром. Вторичная оперированная глаукома II «С» — OD, IV «С» — OS, частичная осложненная катаракта обоих глаз, частичное помутнение роговицы». Решение консилиума: на основании действующих регламентирующих документов — вынашивание беременности не противопоказано 1 .
Пациентка поставлена в известность о том, что во время беременности физиологически увеличивается объем циркулирующей крови, повышаются ВЧД и внутрибрюшное давление, которые могут привести к нарушению работы шунта и декомпенсации заболевания. Был разработан план ведения пациентки в соответствии с порядком оказания медицинской помощи женщинам в периоды беременности, родов и послеродовом с учетом беременности высокой группы риска 2 . Рекомендовано проведение МРТ головного мозга после 20 нед беременности.
Дополнительно отмечаются истончение мозолистого тела, атрофические изменения перивентрикулярного белого вещества.
Заключение: МР-картина варианта мальформации Денди—Уокера. Внутренняя тетравентрикулярная гидроцефалия. Послеоперационные изменения в теменно-затылочных областях (см. рисунок).
Рекомендации по ведению беременной были выполнены в полном объеме, пациентка осматривалась неврологом и офтальмологом в динамике — 1 раз в месяц до 30 нед и 1 раз в 2 нед до родоразрешения. Беременность протекала без осложнений.
Данные протоколов ультразвукового исследования плода в I, II и III триместрах беременности в пределах нормы. Гемодинамические нарушения в системе мать—плацента—плод не выявлены. Кардиотокография плода на сроках 32 и 36 нед беременности: базальный ритм 134—139 ударов в 1 мин, нормотип.
При катамнестическом наблюдении в течение 3 лет состояние пациентки стабильное. В настоящее время оперативное лечение не требуется.
Комплексный подход, обсуждение диагноза с привлечением специалистов разного профиля (невролог, нейрохирург, нейрофизиолог, радиолог, акушер-гинеколог, терапевт), оптимальный объем обследования и динамическое наблюдение за течением беременности позволили пролонгировать и благополучно родоразрешить пациентку путем операции кесарева сечения при доношенном сроке.
Энцефалоцеле
Энцефалоцеле - это порок развития черепа и головного мозга, при котором часть мозгового вещества оказывается вне черепной коробки вследствие дефекта костной ткани. Проявляется видимым выпячиванием на стыке костей, сопровождается разнообразными симптомами в зависимости от размеров и локализации. Возможны судороги, гидроцефалия, ликворея, зрительные расстройства, признаки умственной отсталости. Диагноз энцефалоцеле основывается на клинической картине аномалии развития при подтверждении результатами КТ и МРТ. Лечение оперативное, проводится удаление грыжевого мешка и закрытие костного дефекта.
МКБ-10
Общие сведения
Энцефалоцеле - врожденное заболевание, которое исторически относили к группе мозговых грыж, включающих также менингомиелоцеле и энцефалоцистоцеле. Однако позже грыжевое происхождение патологии было опровергнуто, поскольку сама по себе грыжа предполагает выпячивание правильно развитых тканей, а в случае энцефалоцеле мозговое вещество имеет структурные дефекты. Аномалия встречается с частотой 1 случай на 4000-5000 новорожденных, то есть достаточно редко. Прогноз при энцефалоцеле зависит от тяжести патологии. Порок часто является множественным, в этом случае даже успешное оперативное лечение не позволяет компенсировать возникшие нарушения по причине умственной отсталости или стойкой гидроцефалии. Терапия заболевания продолжает разрабатываться.
Причины энцефалоцеле
Энцефалоцеле представляет собой аномалию развития головного мозга, причиной которой становится воздействие тератогенных факторов на ранних сроках беременности. Нарушать правильную закладку мозга и его оболочек могут:
- внутриутробные инфекции;
- прямое токсическое действие на плод (употребление алкоголя и наркотиков, курение, различные медикаменты);
- хроническая гипоксия.
Определенную роль играет наследственность. Особенно это касается врожденных синдромов, частью клинической картины которых является энцефалоцеле. К числу таких патологий относится, например, синдром Меккеля-Грубера, включающий также микроцефалию, микрофтальм и другие пороки.
Классификация
Возможна передняя и задняя локализация. Передние энцефалоцеле располагаются между лобной и носовой костью, иногда выпячивание проходит через внутренний край глазницы. Задние образования, в свою очередь, делятся на верхние и нижние по отношению к затылочному бугру. Также существуют базальные энцефалоцеле. Частота их не превышает 1,5%, при этом они представляют наибольшую опасность в связи с невыраженной клиникой и сложностями диагностики. В практической педиатрии диагноз, как правило, включает указание конкретных костей, между которыми располагается выпячивание, например, височно-сфеноидальное, носо-орбитальное энцефалоцеле и т. д.
Симптомы энцефалоцеле
Клинические проявления крайне разнообразны ввиду разных размеров и локализации. Образование представлено выпячиванием округлой формы и мягкоэластической консистенции, всегда расположенным по средней линии на стыке костей черепа. Кожа над ним бывает неизмененной, но чаще можно заметить рубцовую деформацию либо истончение и цианотичность. Большие энцефалоцеле иногда флюктуируют и пульсируют, что могут обнаружить как педиатр, так и родители малыша. Иногда пульсация и изменение окраски проявляются только при напряжении и плаче, но отсутствуют в состоянии покоя.
При передних энцефалоцеле возможно затруднение носового дыхания, ликворея. Всегда присутствует истинный гипертелоризм, то есть увеличение расстояния между глазницами, которое часто сопровождается зрительными расстройствами. Носовая перегородка расширена и уплощена, заметно ее смещение в здоровую сторону. Для затылочных образований более характерна неврологическая симптоматика, судороги и отставание в развитии, поскольку при такой локализации выпячивание может достигать больших размеров и в нем задействуется больший объем мозгового вещества. По этой же причине задние мозговые грыжи часто сочетаются с микроцефалией.
Диагностика
Первичная диагностика возможна уже в первые дни и часы жизни ребенка, поскольку образование визуализируется сразу после рождения. Любое опухолеподобное выпячивание изначально расценивается как энцефалоцеле с последующей дифференциальной диагностикой. Чаще всего приходится исключать полипы и ангиомы.
Диагноз уточняется на основании данных КТ и МРТ, при этом проводится серия снимков, подтверждающих наличие костного дефекта и присутствие мозговых тканей в выпячивании. Просвечивание (трансиллюминация) позволяет заподозрить наличие в образовании нервных тканей. Кроме того, даже легкое надавливание вызывает беспокойство пациента в связи с резким повышением внутричерепного давления.
Лечение энцефалоцеле
Лечение только оперативное. Срочность операции определяется состоянием ребенка. Чаще всего вмешательство по удалению энцефалоцеле планируется в возрасте от 3 лет. Такой срок необходим для того, чтобы понаблюдать за развитием головного мозга, в том числе за участком в составе выпячивания. Кроме того, ребенок этому времени уже достаточно окрепнет, чтобы благополучно перенести такую сложную операцию. Противопоказаниями являются повышенное внутричерепное давление, выраженные психические и неврологические расстройства. Истончение кожи над энцефалоцеле, наоборот, является показанием к срочному хирургическому вмешательству.
Операция осуществляется в два этапа. Сначала выполняется закрытие костного дефекта и отсечение ножки грыжи. В раннем возрасте отверстие в черепе можно закрыть лоскутом надкостницы, при более поздних операциях (в возрасте более 3 лет) применяется костный трансплантат.
Второй этап операции - косметическое иссечение поверхностно расположенной оставшейся части грыжевого мешка. Проводится через 15-20 дней после первого этапа (3-6 месяцев в случае интракраниального доступа). Тактику операции в каждом конкретном случае определяют в зависимости от степени вовлеченности мозговых тканей в энцефалоцеле.
Прогноз и профилактика
Исход более благоприятный в случае небольшого размера грыж, особенно передних. Затылочные грыжи, включающие значительный объем мозгового вещества, а также труднооперабельные базальные грыжи значительно ухудшают прогноз. Двухэтапная операция в раннем возрасте приводит к излечению, тем не менее, практически 100% детей после вмешательства имеют отставания в нервно-психическом развитии. Возможна умственная отсталость различной степени выраженности.
Основными послеоперационными осложнениями являются ликворея и нарастающая внутричерепная гипертензия. В дальнейшем это может привести к развитию инфекционных процессов. Наименее благоприятный прогноз - при синдромальных формах, включающих множественные пороки развития, часто несовместимые с жизнью. Профилактика энцефалоцеле возможна только в антенатальном периоде.
Читайте также: