Задний эмбриотоксон и аномалия Аксенфельда-Ригера у ребенка
Добавил пользователь Дмитрий К. Обновлено: 22.01.2026
Синдром Аксенфельда-Ригера (Axenfeld-Rieger syndrome) является генетически гетерогенной группой нарушений морфогенеза, связанной с неправильным развитием переднего отдела глаза, зубов и брюшной области.
Синдром Аксенфельда-Ригера включает в себя:
- аномалия Аксенфельда, характеризующаяся только наличием заднего эмбриотоксона в сочетании с иридогониодисгенезом,
- аномалия Ригера, к предыдущим симптомам присоединяется еще ряд симптомов со стороны переднего сегмента глаза,
- синдром Ригера, глазные проявления сочетаются с целым рядом общих симптомов: гипоплазия срединной линии лица, телекантус с широким и плоским корнем носа, аномалии развития зубов (отсутствие верхнечелюстных резцов, микродентизм, анодонтия), пупочная грыжа, врожденные пороки сердца, конструктивная тугоухость, задержка умственного развития, и др.). В 50% случаев встречается вторичная глаукома.
Синдром Аксенфельда-Ригера наследуется по аутосомно- доминантному типу.
Табл.1. Типы синдрома Аксенфельда-Ригера (синим цветом выделены гены, анализ которых проводится в «Центре Молекулярной Генетики»).
| Тип | OMIM | Локус | Ген | Белок | Тип наследования |
| RIEG1 | 180500 | 4q25 | PITX2 | фактор транскрипции | АД |
| RIEG2 | 601499 | 13q14 | - | - | АД |
| RIEG3 | 601090 | 6p25 | FOXC1 | фактор транскрипции | АД |
Ген PITX2 кодирует белок, действующий как транскрипционный фактор и регулирующий экспрессию гена проколлаген-лизил-гидроксилазы. Этот белок играет важную роль в развитии глаз, зубов и органов брюшной полости, а также, участвующий в базальной и гормон-регулируемой активности пролактина. Мутации в этом гене связаны с сидромом Аксенфельда-Ригера тип 1, аномалией Петерса, дермоидной кистой роговицы и с гипоплазией радужки с глаукомой тип 2. Ген находится на хромосоме 4, в локусе 4q25, и состоит из 7 экзонов. Описано около 30 мутаций.
Ген FOXC1 кодирует белок, действующий также как фактор транскрипции. Белок FOXC1 играет важную роль в раннем развитии, особенно в формировании структур в передней части глаза, также участвует в нормальном развитии других частей тела, включая сердце, почки и мозг. Мутации в этом гене связаны с сидромом Аксенфельда-Ригера тип 3, гипоплазией радужки с глаукомой тип 1. Ген находится на хромосоме 6, в локусе 6p25, и состоит из 1 экзона. Описано около 45 мутаций.
В Центре Молекулярной Генетики проводится прямая диагностика синдрома Аксенфельда-Ригера тип 1 и тип 3 путем прямого секвенирования кодирующей области генов PITX2 и FOXC1.
Редкие болезни: синдром Аксенфельда-Ригера

Синдром Аксенфельда-Ригера — редкое генетическое заболевание, поражающее преимущественно глаза, зубы и пищеварительный тракт. Приблизительно 7000 человек в России и 35 000 человек во всем мире страдают от этой болезни. Как она проявляется и можно ли ее вылечить, рассказывает MedAboutMe.
Во всем виноваты гены
Синдром Аксенфельда-Ригера — это генетическое заболевание. Виной всему поломка двух генов — PITX2 и FOXC1. По данным медицинской литературы, не исключено и поражение других генов, но точных данных по этому вопросу пока нет.
Ген PITX2 кодирует одноименный белок, участвующий в развитии глаз, зубов и многих органов брюшной полости, а также регулирует выработку одного из гормонов гипофиза — пролактина. Существует около тридцати мутаций этого гена.
Ген FOXC1 также кодирует одноименный белок, который влияет на эмбриональное развитие органа зрения. Он также оказывает влияние на формирование других структур — в частности, головного мозга, почек и сердца. Описано около 45 мутаций этого гена.
Синдром Аксенфельда-Ригера наследуется по аутосомно-доминантному типу. Это означает, что для развития болезни достаточно одного «сломанного гена», полученного от матери или отца. При этом ген не содержится в половых хромосомах — Х или Y. Мальчики и девочки болеют одинаково часто. Вероятность передачи болезни составляет 50%.
Что затрагивает болезнь

При синдроме Аксенфельда-Ригера прежде всего поражаются глаза. В медицинской литературе описаны различные проявления этой болезни, но чаще всего при обследовании выявляются такие нарушения:
- Аномалия Аксенфельда, при которой в переднюю камеру глаза выступает задний эмбриотоксон — молочно-белая утолщенная линия Швальбе, видимая по периферии роговицы.
- Аномалия Ригера, при которой наблюдаются изменения в переднем сегменте глаза.
У большинства людей развивается гипоплазия стромы радужки — цветной части глаза. Она меняется, но как именно — предсказать невозможно. У одних людей она деформируется — принимает необычную форму и, например, зрачок становится щелевидным. У других появляется несколько зрачков или один необычно большого размера. Нередки случаи, когда зрачок смещается в сторону и сопровождается выворотом века.
Примерно у половины людей, страдающих этим недугом, нарушается внутриглазное давление, и развивается опасное состояние — глаукома. Повышенное давление приводит к пережатию зрительного нерва и проявляется прогрессирующим снижением зрения, нередко — сильной болью. Без лечения глаукома может привести к слепоте.
Для синдрома Аксенфельда-Ригера характерно двухстороннее поражение глаз, однако изменения обычно развиваются асимметрично. Как правило, первые признаки болезни замечают родители еще на первом году жизни ребёнка. Поначалу кажется, что это только косметический дефект. Долгие годы болезнь может оставаться стабильной и не приводит к ухудшению зрения. И только при развитии глаукомы и поражении нерва зрение начинает резко ухудшаться вплоть до полной слепоты.
Синдрома Аксенфельда-Ригера нередко сопровождается и другими аномалиями развития. А.В. Петраевский в своей работе описывает возможное появление таких изменений:
- недоразвитие верхней челюсти;
- уменьшение или отсутствие отдельных зубов — чаще всего на верхней челюсти;
- аномалии строения носа;
- выворот верхней губы; ;
- аномалии строения паховой области;
- гипоспадия — смещение отверстия мочеиспускательного канала у мальчиков;
- нарушения слуха;
- врожденные пороки сердца;
- задержка умственного развития.
Как диагностируют

Заподозрить болезнь может офтальмолог на первом профилактическом приеме через месяц после рождения ребёнка. При выраженных изменениях зрачка специалист может осмотреть новорожденного уже в родильном доме. Как правило, болезнь диагностируют в первые месяцы жизни младенца. Реже — спустя полгода и более, и только в том случае, если изменения органа зрения слабо выражены — например, наблюдается незначительная деформация зрачка.
Уточнить, насколько сильно поражен орган зрения, можно с помощью обследования. Обязательно проводится гониоскопия — осмотр передней камеры глаза. Врач измеряет внутриглазное давление, изучает сетчатку, оценивает остроту зрения и проводит компьютерную периметрию. По результатам — принимает решение о дальнейшей терапии.
Подтвердить диагноз и найти «сломанный»» ген можно с помощью молекулярно-генетического анализа. Для этого нужно сдать кровь. По анализу крови можно обнаружить дефектные гены и убедиться, что аномалии развития глаза действительно связаны с синдромом Аксенфельда-Ригера.
Женщины и мужчины, страдающие этим недугом, могут передать болезнь своим детям. Диагностировать синдром Аксенфельда-Ригера можно и внутриутробно — если взять на анализ материнскую кровь, амниотическую жидкость или ворсины хориона. Это инвазивное обследование — нужно будет сделать прокол тканей, поэтому оно проводится только по показаниям при подтвержденном генетическом заболевании у родителей. Исследование делают во втором триместре беременности.
Как лечат
Терапия при синдроме Аксенфельда-Ригера всегда симптоматическая. Если повышается внутриглазное давление, и развивается глаукома, назначаются гипотензивные препараты. Они снижают давление и препятствуют сдавлению зрительного нерва. Если медикаментозное лечение не помогает, проводится операция. Лечение всегда подбирается индивидуально с учетом того, какие органы затронуты изменениями и как недуг влияет на качество жизни.
Существуют также и другие необычные болезни. Об одной из них можно прочитать в статье «Синдром Урбаха-Вите: болезнь бесстрашия».
Синдром Беквита-Видеманна: укоротить язык, чтобы выжить

«Укоротить язык»: оказывается, так может звучать не только совет утомленных родителей маленьким болтунам, но и самое настоящее врачебное назначение. Макроглоссия, при которой язык вырастает так, что во рту не помещается, — один из симптомов синдрома Беквита-Видеманна, генетической аномалии, которая возникает без особых причин. Как ее диагностируют, что грозит малышам с большим языком, и можно ли предупредить заболевание, рассказывает MedAboutMe.
У 9 из 10 детей с синдромом причины заболевания неизвестны

Синдром гигантизма с пупочной грыжей, он же синдром Беквита-Видеманна — заболевание редкое. В среднем оно проявляется у одного из 13 000 младенцев. В 9 из 10 случаев никаких предпосылок к аномалии нет, дети с синдромом рождаются у здоровых родителей. Еще в 10% отмечают влияние наследственности: если у близких родственников есть такое же заболевание, шансы у ребёнка родиться с особенностями несколько выше. Если же синдром есть у матери или отца, то вероятность наследования — 50%.

Пока неясно, почему это происходит, но уже есть статистические данные: процедура ИКСИ, инъекционное оплодотворение сперматозоидами яйцеклетки, также повышает вероятность развития синдрома Беквита-Видеманна у ребёнка на фоне генетического здоровья родителей или доноров. Объяснения явлению еще нет, но у детей, родившихся после зачатия методом ИКСИ, синдром гигантизма с пупочной грыжей встречается несколько чаще, чем у остальных малышей.
У 8 из 10 детей с синдромом Беквита-Видеманна при генетическом анализе отмечают мутацию гена IGF-2 в 11-й паре хромосом. Этот ген называют инсулиноподобным фактором роста, а его изменение означает, что различные органы и ткани вдруг начинают расти неравномерно, опережая остальные по сроку развития. Среди вероятных мутаций — наличие только отцовского гена в обеих хромосомах или дублирование генов. А в 20% случаев мутацию вообще невозможно выявить, и диагноз ставят по симптомокомплексу болезни без подтверждения генетиком.
Гигантизм и пуповинная грыжа
Среди первых симптомов болезни, обнаруживаемых еще во время УЗИ, отмечают повышенные показатели веса и роста ребёнка, превышающие срок беременности. Иногда можно заметить, что показатели ассиметричны, например, отмечают увеличение внутренних органов: печени, селезенки, почек, нередко — языка.

Осиа Уорни, малышка с синдромом Беквита-Видеманна, показывала язычок окружающим еще во время ультразвукового исследования. Доктор и медсестра, наблюдавшие эту картину, нашли ситуацию «необыкновенно милой», а вот мама девочки встревожилась.
Осиа родилась некрупной, она — одна из пары близнецов, и у ее сестры Индиго никаких особенностей не отмечалось. После появления на свет девочке пришлось 7 дней провести в реанимации: помимо увеличенного язычка у нее выявили пониженный уровень сахара в крови, а это тоже один из первых симптомов заболевания.
Через 7 месяцев малышке провели первую операцию — пластику языка. Увеличенная мышца не помещалась во рту, мешала дышать, есть и учиться разговаривать. Сегодня Осиа полтора года, и она не отличается по здоровью от родной сестры, хотя каждые 6 месяцев ей приходится проходить обследование на онкофакторы: у людей с синдромом Беквита-Видеманна очень велики риски появления раковых опухолей.
Во время беременности макроглоссию, увеличенный язык, замечают довольно редко, да и сам этот симптом встречается не при каждом случае заболевания. Перинатальные признаки болезни — размеры плода, увеличенная пуповина, плацента, повышенное количество околоплодных вод.
После рождения, как правило, преждевременного, первым сигналом для наблюдения за ребёнком становятся показатели роста и веса. Крупный и недоношенный малыш — в группе риска. Гигантизм может быть, и чаще всего так и происходит, не полным. Могут быть увеличены внутренние органы, ушные раковины, язык. Однако все это в первые месяцы может проходить незамеченным или списываться на индивидуальные особенности ребёнка.
Тревожные сигналы — это низкий уровень глюкозы в крови, а также пуповинная грыжа, которая встречается настолько часто, что даже включена в название синдрома. Из-за порока развития брюшной стенки грыжа может быть достаточно большой: в пупочное кольцо и расходящийся диастаз выпадает часть кишечника, порой даже печень и другие органы пищеварения.
Среди дополнительных частых симптомов — гидронефроз, почечные кисты и почечная недостаточность, возможно наличие эмбриональных опухолей, микроцефалии, недоразвития лицевых мускулов и увеличенных глазных яблок.
Шансы на здоровье

Хотя в раннем детстве детишки с синдромом Беквита-Видеманна могут сильно отличаться от сверстников, выделяясь несоответствием размеров частей тела друг другу и в особенности высоким ростом, то к подростковому возрасту, как правило, эти особенности исчезают. Однако с рождения и как минимум до 8 лет дети переживают критический период развития — это специфика болезни.
Для коррекции размеров внутренних органов и вправления грыжи малышам нередко приходится проходить через ряд операций. Уменьшение размера языка тоже может происходить поэтапно.

Оливия Джиллис, маленькая подданная Великобритании, сегодня готовится в первый класс. Как и все британцы, она пойдет туда в 4 года. Девочка уже может нормально есть и говорить, хотя для этой возможности ей пришлось несколько раз побывать в операционной.
Оливия — один из ярких примеров малышей с макроглоссией, развившейся из-за синдрома Беквита-Видеманна. Сразу после рождения стало ясно, что операции не избежать. Язычок занимал все пространство во рту ребёнка, и кормить грудью малышку мама не смогла. Девочка получала молоко через трубку.
К полугоду ее язык не просто не помещался во рту: он полностью закрывал подбородок! В 6 месяцев ей провели первую операцию по коррекции формы, и ребёнок начал свободно дышать. Однако потребовалось еще две операции, чтобы вернуть языку малышки естественные пропорции.
Дополнительные опасности, которые приносит синдром Беквита-Видеманна, — это гипогликемия, пониженный уровень глюкозы, гипокальциемия, сниженный иммунитет и частые респираторные заболевания. А еще у детей очень высок риск развития онкологических опухолей, так, 7,5% случаев сопровождаются раком почек. Причем болезнь развивается быстро и агрессивно, поэтому все малыши и взрослые с синдромом должны регулярно проходить исследования.
Несмотря на сложности, взрослые пациенты отзываются о своем здоровье вполне позитивно. Они ведут полноценную жизнь, при которой основным напоминанием о заболевании является только необходимость несколько чаще посещать врачей для ранней диагностики возможных проблем.

Ребёнок проявляет беспокойство и жалуется на страшные сны? Не может сосредоточиться на занятии, волнуется и часто жалуется на больной живот? Наш тест поможет определить уровень тревожности у ребёнка и подскажет дальнейшую тактику поведения.
Аномалия Кимерли
Аномалия Кимерли — наличие в структуре первого шейного позвонка дополнительной костной дужки, ограничивающей движения позвоночной артерии и вызывающей синдром ее сдавления. Аномалия Кимерли характеризуется головокружением, шумом в ушах, шаткостью походки и расстройством координации, «мушками» и потемнением в глазах, приступами потери сознания и внезапной мышечной слабости. Возможны двигательные и чувствительные расстройства, возникновение ТИА и ишемического инсульта. Диагностируется аномалия Кимерли при рентгенографическом исследовании краниовертебрального перехода, проведении магнитно-резонансной ангиографии, дуплексного сканирования и УЗДГ сосудов головы и шеи. Сосудистые нарушения, которыми сопровождается аномалия Кимерли, подлежат комплексному консервативному лечению. Операция по резекции аномальной дуги производится лишь в тяжелых случаях.
МКБ-10


Общие сведения
Наряду с аномалией Киари, платибазией и ассимиляцией атланта аномалия Кимерли относится к так называемым краниовертебральным мальформациям — врожденным нарушениям строения области сочленения черепа с первыми шейными позвонками. По некоторым данным аномалия Кимерли встречается у 12-30% людей. Вызывая сдавление позвоночной артерии, аномалия Кимерли сопровождается хронической ишемией в задних отделах мозга. Однако такая ситуация возникает далеко не всегда. Сама по себе аномалия Кимерли не является заболеванием и ее наличие не говорит о том, что именно она вызывает сосудистые нарушения в бассейне позвоночной артерии. При обследовании пациентов, у которых имеется синдром позвоночной артерии и аномалия Кимерли, лишь у 25% обнаруживается причинно-следственная связь между наличием аномалии и развитием синдрома.
Патогенез
Правая и левая позвоночные артерии отходят от соответствующих подключичных артерий. Каждая позвоночная артерия проходит вдоль шейного отдела позвоночника, находясь в канале, образованном отверстиями поперечных отростков его позвонков. Затем она входит в большое затылочное отверстие, попадая таким образом в полость черепа. Позвоночные артерии и их ветви образуют так называемый вертебро-базилярный бассейн, кровоснабжающий часть спинного мозга в шейном отделе позвоночника, мозжечок и ствол мозга. Выходя из шейного канала позвоночная артерия огибает шейный позвонок и горизонтально проходит в широкой костной борозде, где она может свободно перемещаться при движениях головы. Костная дужка, наличием которой характеризуется аномалия Кимерли, расположена над костной бороздой и ограничивает движения позвоночной артерии в этом месте.
Аномалия Кимерли может приводить к развитию синдрома позвоночной артерии двумя путями: за счет срабатывания периваскулярных вегетативно-ирритативных механизмов симпатической иннервации и за счет уменьшенного поступления крови в вертебро-базилярный бассейн из-за механического сдавления позвоночной артерии. Факторами, приводящими к тому, что аномалия Кимерли становиться клинически значимой, являются атеросклероз, поражение сосудистой стенки при васкулитах, шейный спондилоартроз, остеохондроз шейного отдела позвоночника, артериальная гипертензия, наличие других краниовертебральных мальформаций, рубцовый процесс, черепно-мозговая травма или травма позвоночника с повреждениями в области краниовертебрального перехода. К возникновению клинической картины синдрома позвоночной артерии у пациентов с аномалией Кимерли могут приводить травмы плеча, вызывающие повреждение ограниченной костной дужкой позвоночной артерии по хлыстовому механизму.
Классификация
В неврологии выделяют 2 вида аномалии Кимерли. Первая характеризуется наличием костной дужки, соединяющей суставной отросток атланта с его задней дугой. Во втором варианте аномалия Кимерли представлена костной дужкой между суставным отростком атланта и его поперечным отростком.
Аномалия Кимерли может иметь односторонний характер и ли наблюдаться с обоих сторон первого шейного позвонка. Кроме того, аномалия Кимерли может быть полной и неполной. Полная аномальная костная дужка имеет вид полукольца, неполная костная дужка представляет собой дугообразный вырост.
Симптомы аномалии Кимерли
Клинические проявления, которыми сопровождается аномалия Кимерли, обусловлены уменьшенным притоком крови к задним отделам головного мозга. В результате пациенты испытывают шум в ухе или обоих ушах (свист, звон, гул, шипение), мелькание «мушек» или мерцание «звездочек» перед глазами, внезапное преходящее потемнение в глазах. Указанные симптомы усиливаются при поворотах головы. Поскольку аномалия Кимерли сопровождается нарушением кровоснабжения мозжечка, то возникают головокружение и шаткость походки, которые также могут усугубляться при поворотах головой. На фоне некомфортного положения головы или перенапряжения мышц шеи при аномалии Кимерли у пациентов могут наблюдаться приступы потери сознания. Возможна внезапно возникающая мышечная слабость, приводящая к падению больного без потери сознания.
В случаях более тяжелого течения аномалия Кимерли может сопровождаться головной болью, тремором рук и ног, нистагмом, нарушениями координации, гипестезией и/или мышечной слабостью части лица или туловища, чувствительными и двигательными расстройствами одной или нескольких конечностей. Могут наблюдаться транзиторные ишемические атаки в вертебро-базилярном бассейне. Особо тяжелым осложнением наличия аномалии Кимерли является ишемический инсульт.
Диагностика
При обращении пациента с симптомами недостаточности кровообращения в вертебро-базилярном бассейне головного мозга в первую очередь производят рентгенографию черепа и рентгенографию позвоночника в шейном отделе. Аномалия Кимерли, как правило, достаточно четко визуализируется на боковых рентгенограммах области краниовертебрального перехода. При наличие ушного шума для исключения лор-патологии (кохлеарный неврит, хронический средний отит, лабиринтит) может потребоваться консультация отоларинголога, проведение аудиометрии и других исследований слуха. Производится также исследование вестибулярного анализатора (вестибулометрии, электронистагмографии, стабилографии).
Рентгенография ШОП (боковая проекция). Дополнительное костное кольцо в области задней дужки С1 (аномалия Кимерли)
Поскольку выявленная аномалия Кимерли может не являться причиной синдрома позвоночной артерии, неврологу необходимо исключить другие возможные причины вертебро-базилярной недостаточности. Выявить тромбоз, артерио-венозную мальформацию или аневризму сосудов головного мозга, сдавление сосуда объемным образованием (опухоль, киста или абсцесс головного мозга) способна контрастная ангиография. Определить насколько клинически значима аномалия Кимерли, т. е. степень ее влияния на кровообращение в вертебро-базилярном бассейне, позволяет применение целого ряда гемодинамических исследований: УЗДГ экстракраниальных сосудов, транскраниальной допплерографии, дуплексного сканирования и магнитно-резонансной ангиографии сосудов головного мозга. С их помощью при аномалии Кимерли возможно выявить локализацию сдавления позвоночной артерии и ее зависимость от положения головы и шеи.
Лечение аномалии Кимерли
Аномалия Кимерли требует лечения в случае наличия клинических и гемодинамических признаков нарушения кровообращения в вертебро-базилярном бассейне, связанного именно с данной патологией. Пациенты, у которых имеется аномалия Кимерли, должны соблюдать некоторые меры предосторожности в рамках охранительного режима. При аномалии Кимерли следует избегать форсированных физических нагрузок, резких околозапредельных поворотов головой, стоек на голове, кувырков, спортивных занятий и игр, связанных с ударами головой (борьба, футбол, спортивная гимнастика и пр.). При прохождении массажа или мануальной терапии шейного отдела позвоночника пациенту необходимо предупреждать массажиста и мануального терапевта о том, что у него аномалия Кимерли. Ухудшение состояния пациента является поводом к незамедлительному обращению к врачу.
В большинстве случаев аномалия Кимерли, приводящая к клиническим проявлениям сосудистой недостаточности, подлежит консервативному лечению. Проводится сосудистая терапия направленная на улучшение мозгового кровотока (ницерголин, винпоцетин, винкамин, циннаризин). По показаниям под контролем коагулограммы при аномалии Кимерли применяются препараты, улучшающие реалогические свойства крови (пентоксифиллин). В комплексную терапию включают также антиоксиданты, ноотропы, нейропротекторы и метаболические препараты (пирацетам, препараты гинкго билоба, никотиноил-гамма-аминобутировая кислота, мельдоний).
Аномалия Кимерли на сегодняшний день не является показанием для проведения хирургического лечения. Необходимость в оперативном лечении может возникнуть при декомпенсированном течении синдрома позвоночной артерии, приводящем к выраженной недостаточности кровообращения в вертебро-базилярном бассейне при отсутствии достаточного коллатерального кровоснабжения. Операция при аномалии Кимерли заключается в резекции аномальной дуги и мобилизации позвоночной артерии. В послеоперационном периоде пациентам необходимо ношение воротника Шанца сроком от 2 до 4 недель.
Задний эмбриотоксон и аномалия Аксенфельда-Ригера у ребенка
ГБОУ ВПО "Волгоградский государственный медицинский университет" Минздравсоцразвития России
Семейно-наследственный случай аномалии Аксенфельда в сочетании с дистрофией Штаргардта (клиническое наблюдение)
Приведен клинический случай аномалии Аксенфельда (задний эмбриотоксон), отмеченной в трех поколениях одной семьи. При этом два члена семьи (отец, мать) не состоят в кровном родстве. У сына с наиболее выраженными проявлениями указанной аномалии имеется, кроме того, типичная дистрофия Штаргардта обоих глаз.
Одной из разновидностей семейно-наследственного иридокорнеального дисгенеза является синдром Аксенфельда—Ригера. Последний включает такие составляющие, как аномалия Аксенфельда, аномалия Ригера, синдром Ригера [2—4, 6]. Если первая аномалия характеризуется только наличием заднего эмбриотоксона в сочетании с иридогониодисгенезом, а при второй к этим признакам присоединяется еще ряд симптомов со стороны переднего сегмента глаза, то при синдроме Ригера глазные проявления сочетаются с целым рядом общих симптомов, причем достаточно часто встречается вторичная глаукома [5]. У членов одной семьи степень выраженности тех или иных симптомов может значительно варьировать. Патология наследуется по аутосомно-доминантному типу, но может иметь и спорадическое происхождение [6].
Клинический случай. Больной К., 34 года. Жалобы на снижение зрения, которое стал отмечать 14 лет назад. Очки не помогают. С его слов, до 20-летнего возраста острота зрения обоих глаз была 1,0. Диагноз дистрофия Штаргардта обоих глаз был поставлен 10 лет назад. Другой офтальмопатологии не обнаруживалось. Острота зрения снижалась постепенно (в среднем на 0,1 каждые 3 года). Снижение зрения, ощутимо влияющее на качество жизни, отмечает с 30-летнего возраста. Периодически получает курсы медикаментозной дедистрофической терапии. У кровных родственников заболеваний глаз не отмечает.
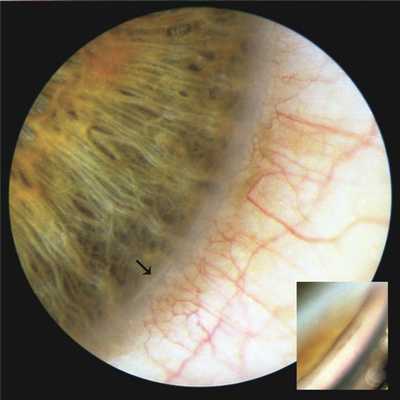
Офтальмостатус. Правый глаз: Vis OD=0,4, не корригируется, ROD M 0,5 дптр 100° — М 1,5 дптр 10°, внутриглазное давление (ВГД) 21 мм рт.ст. Придаточный аппарат без особенностей. Биомикроскопически отмечается небольшой по протяженности фрагмент заднего эмбриотоксона, расположенный в нижневнутреннем отделе окружности лимба. Гониоскопия: угол передней камеры открыт, переднее пограничное кольцо Швальбе в месте проекции заднего эмбриотоксона белой окраски, на его поверхности имеются остатки мезодермальной ткани радужки (рис. 1). Рисунок 1. Задний эмбриотоксон правого глаза больного К. (указан стрелкой). На вставке гониоскопическая картина этого участка. Пояснения в тексте. Других изменений переднего отрезка и оптических сред не отмечено.
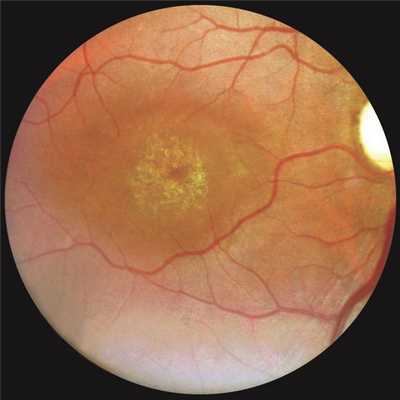
Глазное дно: в центральной области очаговые изменения, характерные для дистрофии Штаргардта, поверхность их покрыта золотистым световым рефлексом, описанным А.М. Водовозовым [1] (рис. 2). Рисунок 2. Глазное дно правого глаза больного К. Пояснения в тексте. Явных элементов желтопятнистой дистрофии в окружности основного очага не отмечено. Периферия глазного дна без особенностей.
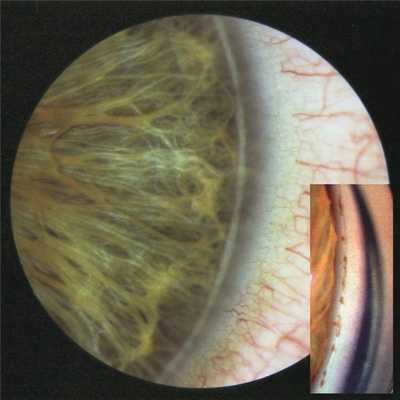
Левый глаз: Vis OS=0,3, не корригируется, ROS M 0,5 дптр 95° — H 0,5 дптр 5°, ВГД 20 мм рт.ст. Придаточный аппарат без особенностей. Биомикроскопически отмечается протяженный задний эмбриотоксон, практически занимающий 2 /3 окружности лимба (наружный, верхний и нижний отделы). Гониоскопия: угол передней камеры в целом открыт, однако отмечаются отдельные гониосинехии, экранизирующие шлеммов канал. Переднее пограничное кольцо Швальбе в области заднего эмбриотоксона более интенсивного белого цвета, чем на правом глазу. На поверхности кольца скопления остатков мезодермальной ткани радужки в большем количестве, чем в правом глазу (рис. 3). Рисунок 3. Задний эмбриотоксон левого глаза больного К. На вставке гониоскопическая картина этого участка. Пояснения в тексте. Других изменений переднего отрезка и оптических сред не отмечено.
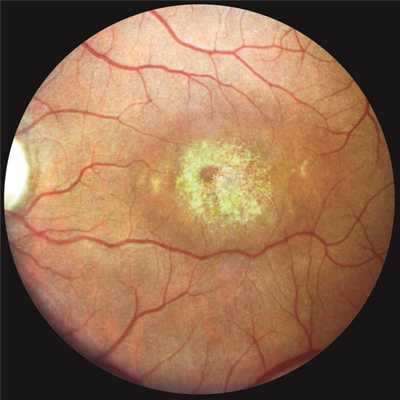
Глазное дно: в центральной области изменения, характерные для дистрофии Штаргардта, аналогичные таковым на правом глазу, но более выраженные (рис. 4), Рисунок 4. Глазное дно левого глаза больного К. Пояснения в тексте. в частности, значительно ярче золотистый стационарный световой рефлекс. Вокруг основного очага разбросаны 5—6 желтопятнистых очажков, которые отсутствуют на дне правого глаза.
Проведен ряд дополнительных исследований.
Статическая периметрия. В центральной части поля зрения обоих глаз чередование относительных и абсолютных скотом. Последние в большем количестве присутствуют в поле зрения левого глаза.
Флюоресцентная ангиография обоих глаз. Имеется типичная для дистрофии Штаргардта картина «бычьего глаза», идентичная в обоих глазах (рис. 5, 6). Рисунок 5. Флюоресцентная ангиограмма правого глаза больного К. Пояснения в тексте. Рисунок 6. Флюоресцентная ангиограмма левого глаза больного К. Пояснения в тексте.
Оптическая когерентная томография (ОКТ) сетчатки обоих глаз. Выраженное истончение нейроэпителия в макуле, деструкция фоторецепторного слоя, расширение контура фовеа, эксцентричная фиксация (смещена кверху от макулы). Изменения более выражены в левом глазу (рис. 7, 8). Рисунок 7. ОК-томограмма макулярной области правого глаза больного К. Пояснения в тексте. Рисунок 8. ОК-томограмма макулярной области левого глаза больного К. Пояснения в тексте.
По данным стандартной электроретинографии, имеются умеренные изменения наружных слоев центральных отделов сетчатки, более выраженные в левом глазу. Тонографические показатели обоих глаз в пределах нормы.
Учитывая возможный семейно-наследственный характер имеющейся у больного К. патологии, провели обследование его кровных родственников: 54-летней матери, 57-летнего отца, 12-летней дочери.
Существенной офтальмопатологии у них не отмечено. Однако у всех выявлен задний эмбриотоксон обоих глаз. Наиболее выражен он у дочери больного К. (рис. 9). Рисунок 9. Задний эмбриотоксон в наружном отделе левого глаза дочери больного К. У отца и матери пациента К. эмбриотоксон носит фрагментарный, в целом незначительно выраженный характер (рис. 10, 11). Рисунок 10. Задний эмбриотоксон (указан стрелками) в наружном отделе правого глаза отца больного К. Рисунок 11. Задний эмбриотоксон в наружном отделе левого глаза матери больного К. Вместе с тем у матери эмбриотоксон более выражен. При этом ни у одного из членов семьи гониоскопически не зафиксированы изменения в области угла передней камеры, свидетельствующие о спаянии мезодермальной ткани радужки с передним пограничным кольцом Швальбе, имевшем место у пациента К. У членов семьи пациента К. не отмечено и признаков дистрофии Штаргардта, а также других существенных изменений глазного дна.
Приведенное клиническое наблюдение представляет интерес по нескольким причинам.
Прежде всего удалось проследить наследование аномалии Аксенфельда в виде заднего эмбриотоксона в трех поколениях одной семьи. При этом данная аномалия имелась у обоих представителей старшего поколения (родителей больного К.), не состоящих в кровном родстве, что подтверждает сведения о достаточной распространенности этой аномалии [6].
Аномалия Аксенфельда в обследуемой семье наименее выражена у родителей больного К., тогда как у него самого протяженность заднего эмбриотоксона и интенсивность его белой окраски значительны, особенно в левом глазу. Кроме того, отмеченные у больного К. остатки мезодермальной ткани радужки на переднем пограничном кольце Швальбе свидетельствуют о возможности формирования у пациента синдрома Ригера как следующей за аномалией Аксенфельда клинической формы. Видимо, своевременно разобщившиеся спайки между радужкой и передним пограничным кольцом Швальбе «разорвали» последующую патогенетическую цепь, состоящую из таких звеньев, как корэктопия, атрофия радужки, формирование в ней отверстий, вторичная глаукома.
У дочери больного К. имеется задний эмбриотоксон, несколько менее выраженный, чем у отца, и нет признаков перехода в аномалию Ригера, как у него.
Представляет интерес сочетание у больного К. двух семейно-наследственных патологических состояний с локализацией в разных отделах глазного яблока: аномалии Аксенфельда и дистрофии Штаргардта. У других членов его семьи дистрофических изменений глазного дна не отмечено. Не исключая случайный характер такого сочетания, все же следует вспомнить приведенное выше предположение A. Spallone [7] о том, что синдром Аксенфельда—Ригера является заболеванием не только переднего, но и заднего отрезка глазного яблока. В приведенном случае, кроме того, обращает на себя внимание наличие у больного К. дистрофического процесса в структурах глазного дна, более выраженного именно в левом глазу, в котором и аномалия Аксенфельда имеет значительно более яркие проявления, чем в правом.
Данный случай показателен тем, что у пациента с дистрофией Штаргардта имеются признаки другой врожденной семейно-наследственной патологии — аномалии Аксенфельда (в ее переходной — в аномалию Ригера — форме). Эти признаки были обнаружены лишь в 34-летнем возрасте, хотя они существовали практически с рождения в отличие от дистрофии Штаргардта, клинически проявившейся значительно позже. Несомненно, благоприятным для пациента моментом является отсутствие у него вторичной глаукомы, осложняющей иридокорнеальный дисгенез.
Мы далеки от мысли проводить генетические параллели между аномалией Аксенфельда и дистрофией Штаргардта. Известно, что первая имеет аутосомно-доминантный тип наследования с высокой степенью пенетрантности, а для второго заболевания характерен аутосомно-рецессивный вариант передачи. Вместе с тем общеизвестен факт сочетания различной врожденно-наследственной патологии у одного человека. Вероятнее всего, это имело место и в случае, описанном в литературе ранее [7]. Вывод может быть один: при любой офтальмологической семейно-наследственной патологии, локализующейся в одном отделе глаза, следует помнить о возможности ее сочетания с иной патологией в другом отделе глаза.
Читайте также: