
Гемолитический стрептококк и анемия
Гемолитические анемии – это группа анемий, которые характеризуются снижением продолжительности жизни эритроцитов и их ускоренным разрушением (гемолиз, эритроцитолиз) либо внутри кровеносных сосудов, либо в костном мозге, печени или селезенке.
В норме средняя продолжительность жизни эритроцитов составляет 110–120 суток. При гемолитической анемии жизненный цикл красных клеток крови укорачивается в несколько раз и составляет 15–20 суток. Процессы разрушения эритроцитов преобладают над процессами их созревания (эритропоэза), в результате чего в крови снижается концентрация гемоглобина, уменьшается содержание эритроцитов, т. е. развивается анемия. Другими общими признаками, характерными для всех видов гемолитических анемий, являются:
- лихорадка с ознобом;
- боли в животе и пояснице;
- нарушения микроциркуляции;
- спленомегалия (увеличение селезенки);
- гемоглобинурия (присутствие в моче гемоглобина);
- желтуха.
Гемолитической анемией страдает примерно 1% населения. В общей структуре анемий на долю гемолитических приходится 11%.
Причины гемолитической анемии и факторы риска
Гемолитические анемии развиваются либо под воздействием внеклеточных (внешних) факторов, либо в результате дефектов эритроцитов (внутриклеточные факторы). В большинстве случаев внеклеточные факторы являются приобретенными, а внутриклеточные – врожденными.
К внутриклеточным факторам относятся аномалии мембран эритроцитов, ферментов или гемоглобина. Все эти дефекты являются наследуемыми, за исключением пароксизмальной ночной гемоглобинурии. В настоящее время описано свыше 300 заболеваний, связанных с точечными мутациями генов, кодирующих синтез глобинов. В результате мутаций изменяется форма и мембрана эритроцитов, увеличивается их подверженность гемолизу.
Более обширную группу представляют внеклеточные факторы. Эритроциты находятся в окружении эндотелия (внутренняя оболочка) кровеносных сосудов и плазмы. Присутствие в плазме инфекционных агентов, токсических веществ, антител способно вызывать изменения в стенках эритроцитов, приводящие к их разрушениям. По такому механизму развивается, например, аутоиммунная гемолитическая анемия, гемолитические трансфузионные реакции.
Дефекты эндотелия кровеносных сосудов (микроангиопатии) также способны повреждать эритроциты, приводя к развитию микроангиопатической гемолитической анемии, у детей протекающей остро, в виде гемолитико-уремического синдрома.
Стать причиной гемолитической анемии может и прием некоторых лекарственных препаратов, в частности, противомалярийных средств, анальгетиков, нитрофуранов и сульфаниламидов.
Формы заболевания
Все гемолитические анемии подразделяются на приобретенные и врожденные. К врожденным, или наследственным формам относятся:
- эритроцитарные мембранопатии – результат аномалий строения эритроцитарных мембран (акантоцитоз, овалоцитоз, микросфероцитоз);
- энзимопении (ферментопении) – связаны с недостатком в организме определенных ферментов (пируваткиназы, глюкозо-6-фосфатдегидрогеназы);
- гемоглобинопатии – обусловлены нарушением структуры молекулы гемоглобина (серповидноклеточная анемия, талассемия).
Наиболее часто встречающейся в клинической практике наследственной гемолитической анемией является болезнь Минковского – Шоффара (микросфероцитоз).
Приобретенные гемолитические анемии, в зависимости от вызвавших их причин, делятся на следующие виды:
- приобретенные мембранопатии (шпороклеточная анемия, пароксизмальная ночная гемоглобинурия);
- изоиммунные и аутоиммунные гемолитические анемии – развиваются в результате повреждения эритроцитов собственными или полученными извне антителами;
- токсические – ускоренное разрушение эритроцитов происходит из-за воздействия на них бактериальных токсинов, биологических ядов или химических веществ;
- гемолитические анемии, связанные с механическими повреждениями эритроцитов (маршевая гемоглобинурия, тромбоцитопеническая пурпура).
Симптомы гемолитической анемии
Для всех видов гемолитических анемий характерны:
- анемический синдром;
- увеличение селезенки;
- развитие желтухи.
При этом каждый отдельный вид заболевания обладает особенностями.
Наиболее часто встречающейся в клинической практике наследственной гемолитической анемией является болезнь Минковского – Шоффара (микросфероцитоз). Она отслеживается в нескольких поколениях семьи и наследуется по аутосомно-доминантному типу. Генетическая мутация приводит к недостаточному содержанию в мембране эритроцитов определенного типа белков и липидов. В свою очередь это вызывает изменения в размерах и форме эритроцитов, их преждевременное массивное разрушение в селезенке. Микросфероцитарная гемолитическая анемия может манифестировать у пациентов в любом возрасте, но чаще всего первые симптомы гемолитической анемии возникают в 10–16 лет.
Заболевание может протекать с разной выраженностью. У некоторых больных отмечается субклиническое течение, а у других развиваются тяжелые формы, сопровождающиеся частыми гемолитическими кризами, которые имеют следующие проявления:
- повышение температуры тела;
- озноб;
- общая слабость;
- головокружение;
- боли в пояснице и животе;
- тошнота, рвота.
Основным симптомом микросфероцитоза является разной степени выраженности желтуха. Из-за высокого содержания стеркобилина (конечного продукта обмена гема) кал интенсивно окрашивается в темно-коричневый цвет. У всех пациентов, страдающих микросфероцитарной гемолитической анемией, увеличивается селезенка, а у каждого второго увеличивается и печень.
Микросфероцитоз повышает риск образования в желчном пузыре конкрементов, т. е. развития желчнокаменной болезни. В связи с этим нередко возникают желчные колики, а при закупорке желчного протока камнем – обтурационная (механическая) желтуха.
В клинической картине микросфероцитарной гемолитической анемии у детей присутствуют и другие признаки дисплазии:
- брадидактилия или полидактилия;
- готическое небо;
- аномалии прикуса;
- седловидная деформация носа;
- косоглазие;
- башенный череп.
У пациентов пожилого возраста из-за разрушения эритроцитов в капиллярах нижних конечностей возникают устойчивые к традиционной терапии трофические язвы стоп и голеней.
Гемолитические анемии, связанные с дефицитом определенных ферментов, обычно манифестируют после приема некоторых медикаментов или перенесенного интеркуррентного заболевания. Их характерными признаками являются:
- бледная желтуха (бледный цвет кожи с лимонным оттенком);
- сердечные шумы;
- умеренно выраженная гепатоспленомегалия;
- темный цвет мочи (обусловлен внутрисосудистым распадом эритроцитов и выделением с мочой гемосидерина).
При тяжелом течении заболевания возникают ярко выраженные гемолитические кризы.
К врожденным гемоглобинопатиям относятся талассемия и серповидноклеточная анемия. Клиническая картина талассемии выражена следующими симптомами:
- гипохромная анемия;
- вторичный гемохроматоз (связан с частыми гемотрансфузиями и необоснованным назначением железосодержащих препаратов);
- гемолитическая желтуха;
- спленомегалия;
- холелитиаз;
- поражение суставов (артрит, синовит).
Серповидноклеточная анемия протекает с рецидивирующими болевыми кризами, умеренно выраженной гемолитической анемией, повышенной восприимчивостью больного к инфекционным заболеваниям. Основными симптомами являются:
- отставание детей в физическом развитии (особенно мальчиков);
- трофические язвы нижних конечностей;
- умеренная желтуха;
- болевые кризы;
- апластические и гемолитические кризы;
- приапизм (не связанная с половым возбуждением самопроизвольная эрекция полового члена, сохраняющаяся несколько часов);
- холелитиаз;
- спленомегалия;
- аваскулярные некрозы;
- остеонекроз с развитием остеомиелита.
Из приобретенных гемолитических анемий чаще всего встречаются аутоиммунные. К их развитию приводит выработка иммунной системой пациентов антител, направленных против собственных эритроцитов. Т. е. под влиянием некоторых факторов происходит нарушение деятельности иммунной системы, в результате которой она начинает воспринимать собственные ткани как чужеродные и разрушать их.
При аутоиммунной анемии гемолитические кризы развиваются внезапно и остро. Их возникновению могут предшествовать предвестники в виде артралгий и/или субфебрильной температуры тела. Симптомами гемолитического криза являются:
- повышение температуры тела;
- головокружение;
- резкая слабость;
- одышка;
- сердцебиение;
- боли в пояснице и эпигастрии;
- быстрое нарастание желтухи, не сопровождающейся зудом кожи;
- увеличение селезенки и печени.
Существуют формы аутоимунных гемолитических анемий, при которых пациенты плохо переносят холод. При переохлаждении у них развивается гемоглобинурия, холодовая крапивница, синдром Рейно (сильный спазм артериол пальцев).
Особенностями клинической картины токсических форм гемолитических анемий являются:
- быстропрогрессирующая общая слабость;
- высокая температура тела;
- рвота;
- сильная боль в пояснице и животе;
- гемоглобинурия.
На 2-3 сутки от начала заболевания у больного начинает нарастать уровень билирубина в крови и развивается желтуха, а еще через 1-2 дня возникает гепаторенальная недостаточность, проявляющаяся анурией, азотемией, ферментемией, гепатомегалией.
Еще одной формой приобретенной гемолитической анемии является гемоглобинурия. При этой патологии происходит массовое разрушение эритроцитов внутри кровеносных сосудов и гемоглобин попадает в плазму, а затем начинает выделяться с мочой. Основным симптомом гемоглобинурии является темно-красный (иногда черный) цвет мочи. Другими проявлениями патологии могут стать:
Гемолиз эритроцитов при гемолитической болезни плода и новорожденных связан с проникновением в кровоток плода через плаценту антител из крови матери, т. е. по патологическому механизму данная форма гемолитической анемии относится к изоиммунным заболеваниям.
В норме средняя продолжительность жизни эритроцитов составляет 110–120 суток. При гемолитической анемии жизненный цикл красных клеток крови укорачивается в несколько раз и составляет 15–20 суток.
Гемолитическая болезнь плода и новорожденного может протекать по одному из следующих вариантов:
- внутриутробная гибель плода;
- отечная форма (иммунная форма водянки плода);
- желтушная форма;
- анемическая форма.
Общими признаками, характерными для всех форм этого заболевания, являются:
- гепатомегалия;
- спленомегалия;
- увеличение в крови эритробластов;
- нормохромная анемия.
Диагностика
Обследование пациентов с гемолитическими анемиями проводится гематологом. При опросе пациента выясняют частоту образования гемолитических кризов, их тяжесть, а также уточняет наличие подобных заболеваний в семейном анамнезе. В ходе осмотра пациента обращают внимание на окраску склер, видимых слизистых оболочек и кожных покровов, пальпируют живот с целью выявления возможного увеличения печени и селезенки. Подтвердить гепатоспленомегалию позволяет УЗИ органов брюшной полости.
Изменения в общем анализе крови при гемолитической анемии характеризуются гипо- или нормохромной анемией, ретикулоцитозом, тромбоцитопенией, лейкопенией, увеличением СОЭ.
Большое диагностическое значение при аутоиммунных гемолитических анемиях имеет положительная проба Кумбса (наличие в плазме крови или прикрепленных к поверхности эритроцитов антител).
В ходе биохимического исследования крови определяют увеличение активности лактатдегидрогеназы, наличие гипербилирубинемии (преимущественно за счет увеличения непрямого билирубина).
В общем анализе мочи выявляют гемоглобинурию, гемосидеринурию, уробилинурию, протеинурию. В кале отмечается повышенное содержание стеркобилина.
При необходимости выполняют пункционную биопсию костного мозга с последующим гистологическим анализом (обнаруживают гиперплазию эритроидного ростка).
Гемолитической анемией страдает примерно 1% населения. В общей структуре анемий на долю гемолитических приходится 11%.
Дифференциальная диагностика гемолитических анемий проводится со следующими заболеваниями:
- гемобластозы;
- гепатолиенальный синдром;
- портальная гипертензия;
- цирроз печени;
- гепатиты.
Лечение гемолитических анемий
Подходы к лечению гемолитических анемий определяются формой заболевания. Но в любом случае первостепенной задачей является устранение гемолизирующего фактора.
Схема терапии гемолитического криза:
- внутривенное вливание растворов электролитов и глюкозы;
- трансфузия свежезамороженной плазмы крови;
- витаминотерапия;
- назначение антибиотиков и/или кортикостероидов (по показаниям).
При микросфероцитозе показано хирургическое лечение – удаление селезенки (спленэктомия). После оперативного вмешательства у 100% пациентов наступает стойкая ремиссия, так как повышенный гемолиз эритроцитов прекращается.
Терапия аутоиммунных гемолитических анемий проводится глюкокортикоидными гормонами. При ее недостаточной эффективности может потребоваться назначение иммунодепрессантов, противомалярийных препаратов. Резистентность медикаментозной терапии является показанием к проведению спленэктомии.
При гемоглобинурии проводят трансфузию отмытых эритроцитов, инфузию растворов плазмозаменителей, назначают антиагреганты и антикоагулянты.
Лечение токсических форм гемолитических анемий требует введения антидотов (при их наличии), а также использования методов экстракорпоральной детоксикации (форсированный диурез, перитонеальный диализ, гемодиализ, гемосорбция).
Возможные последствия и осложнения
Гемолитические анемии могут приводить к развитию следующих осложнений:
Прогноз
При своевременно начатом и адекватном лечении гемолитических анемий прогноз в целом благоприятный. При присоединении осложнений он значительно ухудшается.
Профилактика
Профилактика развития гемолитических анемий включает следующие мероприятия:
- медико-генетическое консультирование пар при наличии в семейном анамнезе указаний на случаи гемолитической анемии;
- определение на этапе планирования беременности группы крови и резус-фактора будущей матери;
- укрепление иммунной системы.
Видео с YouTube по теме статьи:

Образование: окончила Ташкентский государственный медицинский институт по специальности лечебное дело в 1991 году. Неоднократно проходила курсы повышения квалификации.
Опыт работы: врач анестезиолог-реаниматолог городского родильного комплекса, врач реаниматолог отделения гемодиализа.
Информация является обобщенной и предоставляется в ознакомительных целях. При первых признаках болезни обратитесь к врачу. Самолечение опасно для здоровья!
Гемолитическая анемия — анемия, возникающая в результате повышенного эритродиереза, когда разрушение эритроцитов преобладает над их образованием.
Классификация. По этиологии гемолитические анемии подразделяются на приобретенные и наследственные. В свою очередь в зависимости от этиологических факторов, вызвавших гемолиз эритроцитов, приобретенные гемолитические анемии делятся на токсические, обусловленные действием экзогенных и эндогенных гемолитических ядов; иммунные (гетеро-, изо-, аутоиммунные), когда гемолиз происходит под влиянием комплекса антиген — антиэритроцитарное антитело; механические — при механическом повреждении эритроцитов; мембранопатии, связанные с соматической мутацией пролиферирующих клеток эритроцитарного ряда и образованием популяции эритроцитов с дефектом структуры мембраны.
На основании того, какие генетические нарушения привели к усилению гемолиза эритроцитов, наследственные гемолитические анемии подразделяют на наследственные мембранопатии, ферментопатии и гемоглобинопатии, вызванные генетическими дефектами структуры мембраны, активности ферментов эритроцитов и синтеза гемоглобина. Имеется две разновидности наследственных гемоглобинопатии: анемии, связанные с нарушением синтеза цепей глобина, и анемии, обусловленные наследственным дефектом первичной структуры цепей глобина.
Этиология приобретенных гемолитических анемий. Токсическая гемолитическая анемияможет развиться под влиянием гемолитических ядов (соединения мышьяка, свинца, нитробензол, фенилгидразин; алкоголь, желчные кислоты, токсические продукты азотистого обмена; змеиный, грибной, пчелиный яды и др.), а также при действии возбудителей инфекционных и паразитарных заболеваний (гемолитический стрептококк, анаэробная инфекция, малярийный плазмодий, лейшмания).
Иммунная (гетеро-, изо-, аутоиммунная) гемолитическая анемияразвивается при переливании видо-, группо- и резус-несовместимой крови; резус-несовместимости матери и плода; образовании аутоантител против собственных эритроцитов при изменении их антигенных свойств под влиянием лекарственных препаратов, вирусов, микроорганизмов или в результате соматической мутации иммуноцитов, когда возникает "запретный" клон лимфоцитов, продуцирующих антитела к нормальным антигенам эритроцитов (при лейкозе, системной красной волчанке и др.).
Механическое повреждение эритроцитовможет возникнуть при протезировании кровеносных сосудов и клапанов сердца, длительном марше или беге по твердому грунту (маршевая гемоглобинурия), спленомегалии.
Причиной приобретенной мембранопатииможет стать соматическая мутация эритробластов под действием вирусов, микроорганизмов, лекарственных препаратов с образованием патологической популяции эритроцитов, у которых нарушается структура мембраны и повышается чувствительность к комплементу (пароксизмальная ночная гемоглобинурия).
Патогенез.Механизм гемолиза при приобретенной гемолитической анемиизаключается в повреждении структуры мембран эритроцитов. Одни гемолитические факторы (например, механические) оказывают прямое повреждающее действие, другие (мышьяковистый водород, нитриты), являясь сильными окислителями, вызывают сначала метаболические, а затем функциональные и структурные изменения в мембране и строме эритроцитов, приводящие к их гемолизу. Многие гемолитические яды биологического происхождения обладают ферментной активностью (лецитиназная активность стрепто-, стафилолизинов, яда насекомых и змей), разрушая лецитин мембран. При иммунных гемолитических анемиях IgG и IgM присоединяют к эритроцитарной мембране комплемент, который при этом активируется и вызывает ее ферментативный лизис.
Под влиянием гемолитических агентов в мембранах эритроцитов образуются поры, через которые из клетки выходят ионы калия, фосфаты, а ионы натрия поступают в клетку. Вследствие сдвигов ионного баланса вода проникает в эритроцит, который при этом набухает, приобретает сферическую форму, его клеточная поверхность уменьшается, снижается способность к деформации. Такие сфероциты не могут пройти через межэндотелиальные поры синусов селезенки и фагоцитируются селезеночными макрофагоцитами. Когда объем эритроцита достигает критического (146 % первоначального), а размер пор мембраны превышает 6 нм, наступает гемолиз с выходом гемоглобина в плазму.
Гемолиз эритроцитов при приобретенных гемолитических анемиях происходит преимущественно в кровеносном русле. Однако при резус-конфликте (гемолитическая болезнь новорожденных) антирезусные агглютинины, образовавшиеся в организме резус-отрицательной матери, вызывают гемолиз резус-положительных эритроцитов плода или новорожденного не только внутри сосудов, но и в печени и селезенке (внутриклеточный гемолиз).
При наследственной гемолитической анемиигемолиз обусловлен снижением осмотической и механической резистентности эритроцитов с генетически детерминированными нарушениями структуры мембраны, метаболизма, синтеза гемоглобина.
Так, при наследственной мембранопатии(микросфероцитарная гемолитическая анемия или болезнь Минковского—Шоффара с аутосомно-доминантным наследованием) генетический дефицит в мембране - эритроцитов Са 2+ -зависимой АТФазы и фосфолипидов приводит к повышению проницаемости мембраны. В клетки поступают ионы натрия и вода, эритроциты превращаются в сфероциты с резко пониженной способностью деформироваться при прохождении через синусы селезенки. Отрыв части оболочки у таких эритроцитов ведет к образованию микросфероцитов с укороченной продолжительностью жизни (8—14 дней вместо 120 дней в норме) в связи с захватом их макрофагоцитами селезенки и печени (внутриклеточный гемолиз).
При наследственной ферментопатии, например глюкозо-6-фосфатдегидрогеназодефицитной анемии (доминантное, сцепленное с X-хромосомой наследование), острый внутрисосудистый гемолиз эритроцитов, возникающий при приеме лекарств с высокой окислительной способностью (противомалярийные препараты, фтивазиди др.), обусловлен повреждением клеточных мембран перекисями, так как в эритроцитах с дефицитом Г-6-ФДГ понижено содержание восстановленного глутатиона (антиоксиданта).
Внутриклеточный гемолиз эритроцитов при наследственной гемоглобинопатии связан с синтезом аномального или не свойственного данному возрасту гемоглобина 1 . Так, при серповидноклеточной анемии 2 образуется HbS (в β-цепи глобина глутаминовая кислота заменена валином), который в восстановленном состоянии выпадает в кристаллы и вызывает деформацию эритроцитов (серповидная форма); гипоксия способствует усилению гемолиза таких эритроцитов. При α-талассемии (генетический дефект синтеза α-цепей) происходит гемолиз эритроцитов с аномальным гемоглобином — Bart—НЬ(γ4) у новорожденных и НЬН(β4) у взрослых людей; при β-талассемии, когда нарушен синтез β-цепей и не образуется НЬА,(α2β2), гемолизируются эритроциты, содержащие фетальный гемоглобин (α2γ2) или же НbА2(α2δ2).
Следствием массивного гемолиза эритроцитов является анемия с нарушением дыхательной функции крови и развитием гипоксии. Образовавшийся при распаде эритроцитов гемоглобин циркулирует в крови (гемоглобинемия) и соединяется с гаптоглобином в крупномолекулярный комплекс, не проходящий через почечный фильтр. Если же содержание свободного гемоглобина в плазме превышает 20,9 ммоль/л (337 г/л) или исходный уровень гаптоглобина низкий, тогда не связанный с последним гемоглобин начинает выделяться с мочой (гемоглобинурия). Частично гемоглобин поглощается клетками макрофагоцитарной системы и расщепляется в них до гемосидерина. Гемосидероз селезенки, почек, печени, костного мозга сопровождается реактивным разрастанием соединительной ткани и нарушением функций этих органов. Повышенное образование из гемоглобина желчных пигментов обусловливает развитие гемолитической желтухи (см. раздел XXII — "Патологическая физиология печени"). Кроме того, внутрисосудистый распад эритроцитов может привести к появлению тромбов и нарушению кровоснабжения тканей, отсюда — трофические язвы конечностей, дистрофические изменения в селезенке, печени, почках. В результате поступления в сосудистое русло большого количества эритроцитарного тромбопластина возможно развитие ДВС-синдрома.
Картина крови. Приобретенная гемолитическая анемия по типу кроветворения является эритробластической, по степени регенерации костного мозга — регенераторной, по цветовому показателю — нормо-или гипохромной, реже — ложногиперхромной (вследствие абсорбции гемоглобина на эритроцитах). Степень уменьшения количества эритроцитов и гемоглобина зависит от интенсивности гемолиза. В мазке крови обнаруживаются клетки физиологической регенерации и дегенеративно измененные эритроциты (пойкилоцитоз; разорванные, фрагментированные эритроциты, анизоцитоз). Появление большого количества эритробластов и нормобластов характерно для гемолитической болезни новорожденных.
При наследственной гемолитической анемии отмечается усиленная регенерация эритроцитарного ростка часто с неэффективным эритропоэзом, когда в костном мозге разрушаются ядерные формы эритроцитов. В мазке крови наряду с регенеративными формами (высокий ретикулоцитоз, полихроматофилия, единичные ядерные формы эритроцитов) находятся дегенеративно измененные клетки (микросфероциты при болезни Минковского — Шоффара, серповидные при S-гемоглобинопатии, мишеневидные, базофильно пунктированные — при талассемии). При частых гемолитических кризах может возникнуть гипорегенераторная анемия.
Наследственная гемолитическая анемия
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
Приобретенная гемолитическая анемия
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Отдельные формы наследственной гемолитической анемии
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
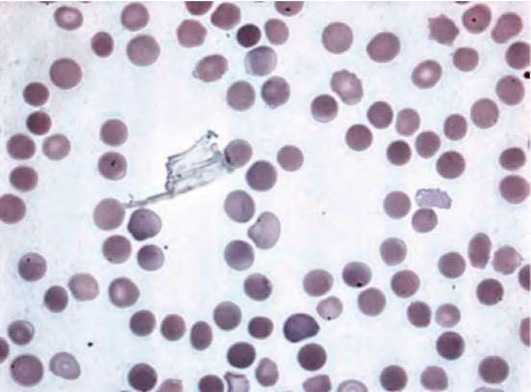
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
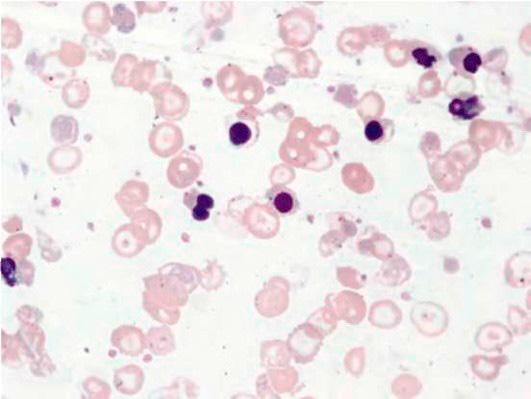
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
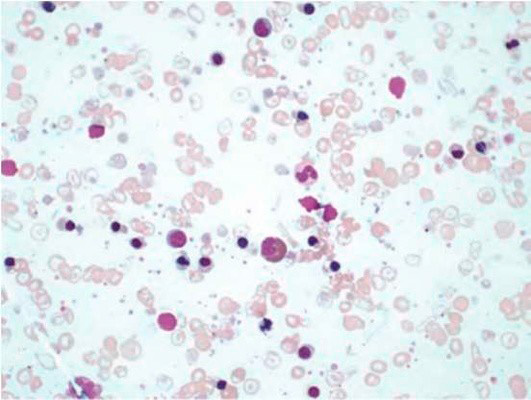
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
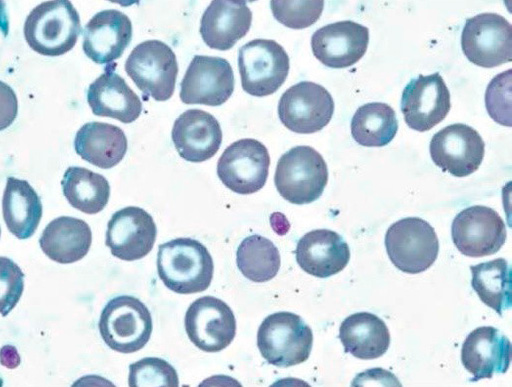
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
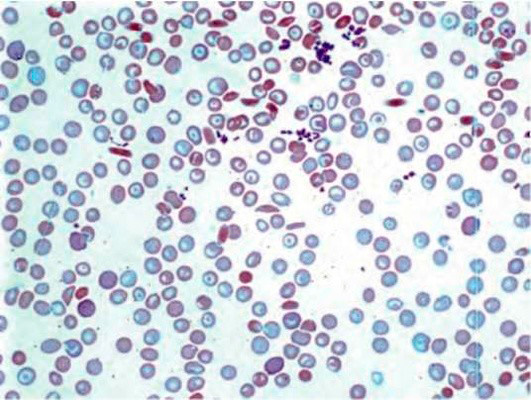
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
Читайте также: