Адипозо-генитальная дистрофия. Синдром Пехкранца — Фрелиха — Бабинского
Добавил пользователь Дмитрий К. Обновлено: 29.01.2026
Адипозо-генитальная дистрофия. Синдром Пехкранца — Фрелиха — Бабинского
Одним из первых это заболевание описал русский врач Пехкранц в 1899 г., и 1900 г.—М. Бабинский и в 1901 г. — А. Фрелих.
Этиология. Заболевание может возникать вследствие воспалительных процессов в диэнцефальной области, кровоизлияний, опухоли (хромофобная аденома или краниофарингиома), водянки желудочков головного мозга, сифилиса, внутриутробной инфекции (токсоплазмоз), инфекции, перенесенной в раннем детстве, паротита, гриппа, брюшного тифа. Во многих случаях причину заболевания установить не удается.
Патогенез. В патогенезе ожирения и недоразвития половой системы лежит поражение гипоталамуса. В дальнейшем по мере развития болезни присоединяются признаки гипофизарной недостаточности.
Снижение гонадотропной функции гипофиза ведет к снижению или прекращению андрогенной или эстрогенной секреции, что приводит к изменению соотношения между интенсивностью различных видов обмена, к изменению высшей нервной деятельности, вследствие чего по-разному наступает распределение жира.
В настоящее время принято считать, что адипозо-гепитальная дистрофия может выступать как самостоятельное заболевание в том случае, если оно возникло в детском возрасте и при этом не удается выявить причину ее возникновения. Во всех остальных случаях, когда выявлена причина заболевания, ожирение и гипогонадизм следует рассматривать как симптомы основного заболевания. Возможны семейные формы адипозо-генитальной дистрофии.
Патологическая анатомия. В ряде случаев морфологические изменения отсутствуют.
Клиника. Клиническое течение болезни определяется патогенетическим фактором. При быстро развивающейся опухоли гипоталамо-гипофизарной области синдром Пехкранца—Фрелиха—Бабинского может быстро прогрессировать и сопровождаться неврологическими нарушениями.
При вяло текущей инфекции диэнцефалыной области или остаточных явлениях воспалительного процесса наблюдается медленно прогрессирующее ожирение, гипогенитализм и задержка роста. У детей резко повышается аппетит, отмечается быстрая прибавка веса тела, появляется сонливость. В пубертатном периоде при задержке полового формирования могут наблюдаться вялость, апатия, эмоциональная неустойчивость. В случае опухоли беспокоит головная боль. Возможно ухудшение зрения. Чаще болеют мальчики и юноши. Интеллект обычно не нарушен.
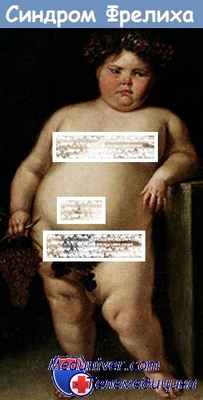
Для данного синдрома характерна локализация ожирения. Лицо округлое, отложение жира на шее, плечах, груди, животе, на бедрах и ягодицах. Конечности полные. У мужчин отложение жира по женскому типу. Иногда ожирение достигает значительного размера н вес ребенка 8—12 лет превышает 100 кг. Вес юноши или девушки при росте 160 см может быть иногда 160— 200 кг. Кожа мягкая, нормальной окраски. Через тонкую кожу могут просвечивать вены. Волосы у больных редкие, тонкие, шелковистые. Больные рано лысеют.
Описаны случаи, когда у больных наблюдался узловой липоматоз, нередко болезненный, как при болезни Деркума.
Гипогенитализм более выражен у мальчиков в препубертатном и пубертатном возрасте. Яички маленькие, атрофичпые, не опускаются в мошонку. Полосой член, мошонка и предстательная железа недоразвиты. У взрослых они остаются в размере детского возраста. Оволосение на лице и теле у взрослых мужчин отсутствует. Голос высокий, женоподобный. Наблюдается гинекомастия. Больные подобны евнухам. Потенция и половое влечение снижены.
Тучность и большие молочные железы придают девушкам облик женщин. Гипогенитализм проявляется недоразвитием матки, яичников. Клитор и большие половые тубы малы. Менструации отсутствуют. Оволосение на теле слабо выражено.
Со стороны внутренних органов изменения, как правило, отсутствуют. Артериальное давление нормальное или пониженное. Рост костей и длину обычно не нарушен. Иногда встречаются евнухоидные пропорции скелета (длинные кости конечностей и относительно короткое туловище).
При рентгенологическом исследовании можно обнаружить увеличение и деформацию турецкого седла, что говорит о наличии опухоли гипофиза. Обызвествление в супраселляриой области возникает при краниофарингиоме. Обнаруживается разъединение швов черепа и недоразвитие пазух. Известную трудность представляют стертые формы адипозо-генитальной дистрофии с преобладанием какого-либо одного симптома.
У больных нередко наблюдаются (неврологические нарушения вследствие перенесенных нейроинфекций, воспалительных процессов диэнцефальной области. Наряду с микросимптомами, указывающими на поражение нервной системы, могут быть нарушения зрения. Атрофия зрительного нерва, ограничение полей зрения и др.
Могут наблюдаться эпилептические припадки, шизоидные и депрессивные состояния. У некоторых больных иногда бывают псевдопараличи, корсаковский синдром. При адипозо-генитальной дистрофии нередко наблюдается несахарный диабет.
В крови чаще всего отмечается лимфоцитоз. Нередко отмечается низкий уровень сахара в крови и низкая гликемичоская кривая после нагрузки глюкозой. Холестерин и липиды крови могут быть повышены. Количество мочевой кислоты в крови иногда также повышено.
Исследование половых гормонов и гонадотропинов в детском возрасте не дает четких данных и поэтому не имеет особого значения. При исследовании в юношеском возрасте отмечается снижение ГТГ, АКТГ и эстрогенов. При исследовании 17-кетостероидов и 17-оксикортикостероидов в анализах мочи отмечаются или нормальные показатели, или некоторое их снижение.
Адипозогенитальная дистрофия (синдром пехкранца-бабинского-фрелиха)
Адипозогенитальная дистрофия - одна из форм гипоталамического ожирения. Характеризуется сочетанием ожирения с гипогенетализмом. Заболевание встречается у детей дошкольного возраста и подростков в препубертатном периоде. Если заболевание развивается у взрослых на почве травмы, восполнения или опухоли и сопровождается ожирение и вторичной генитальной атрофией, следует говорить о гипоталамическом синдроме (Юлес М., Холло И., 1963). Заболевание встречается с одинаковой частотой у мальчиков и девочек с той лишь разницей, что у мальчиков раньше обнаруживается генитальная гипоплазия.
Первое описание синдрома связывают с именем русского врача Пехкранца (1889). В последующем подобные синдромы, характеризующиеся сочетанием ожирения и гипогенитализма, описаны Бабинским (1900) и Фрелихом (1901). Именем этих исследователей и назван синдром.
ЭТИОЛОГИЯ. Причины заболевания - те же воспитательные или опухолевые процессы в гипоталамусе, которые лежат в основе других форм гипоталамического ожирения. Из острых воспалительных процессов чаще отмечается вирусная инфекция (грипп, скарлатина и др.), из хронических - туберкулез. Следует помнить также о возможных внутриутробных инфекциях (энцефалите), родовые травмах, токсоплазмозе.
ПАТОГЕНЕЗ. Кроме поражения вентромедиальных ядер, регулирующих аппетит, при адипозогенитальной дистрофии вовлекаются медиобазальные отделы гипоталамуса, ответственные за секрецию гонадотропинов. Поэтому наряду с ожирением у больных развивается гипоплазия и атрофия половых признаков, недоразвитие вторичных половых признаков. Возможна гипофункция щитовидной железы и недостаточность коры надпочечников. Пе-рекармливание и выраженное ожирение в детском возрасте может приводить к вторичному нарушению гипоталамическому регуляции эндокринных желез и развитию гипогонадизма.
Несахарный диабет
Несахарный диабет (НСД) - заболевание, характеризующееся мочеизнурением, повышением осмолярности плазмы, возбуждающим механизм жажды и компенсаторным потреблением большого количества жидкости.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ. Заболевание вызывается недостаточностью вазопрессина (ВП), контролирующего реабсорбцию воды в дистальных канальцах почечного нефрона, где в физиологических условиях обеспечивается отрицательный клиренс "свободной" воды в масштабах, необходимых для гемостаза, и завершается концентрация мочи.
Существует ряд этиологических классификаций несахарного диабета. Чаще других используют разделение на центральный (нейрогенный, гипоталамический) несахарный диабет с недостаточной продукцией вазопрессина (полной или частичной) и периферический. К центральным формам относятся истинный, симтоматический иидиопатический (семейный или приобретенный) несахарный диабет. При периферическом несахарном диабете сохраняется нормальная продукция ВП, но снижена или отсутствует чувствительность к гормону рецепторов почечных канальцев (нефрогенный вазопрессинрезистентный несахарный диабет) или вазопрессин усиленно инактивируется в печени, почках, плаценте.
Причиной центральных форм несахарного диабета могут быть воспалительные, дегенеративные, травматические, опухолевые и др. поражения гипоталамо-нейрогипофизарной системы
(передние ядра гипоталамуса, супраоптикогипофизарный тракт, задняя доля гипофиза). Конкретные причины заболевания весьма разнообразны. Истинному несахарному диабету предшествуют ряд острых и хронических инфекций и заболеваний: грипп, менингоэнцефалит (диэнцефалит), ангина, скарлатина, коклюш, все виды тифов, септические состояния, туберкулез, сифилис, малярия, бруцеллез, ревматизм. Грипп с его нейротропным влиянием встречается чаще других инфекций. По мере снижения общей заболеваемости туберкулезом, сифилисом и другими хроническими инфекциями, их причинная роль в возникновении несахарного диабета значительно уменьшилась. Заболевание может возникать после черепно - мозговой (случайной или хирургической), психической травм, поражения электрическим током, переохлаждения, во время беременности, вскоре после родов, абортов. Причиной сахарного диабета у детей может послужить родовая травма. Симптоматический несахарный диабет вызывается первичной и метастатической опухоли гипоталамуса и гипофиза, аденомой, тератомой, глиомой и особенно часто краниофарингиомой, саркоидозом. Метастазирует в гипофиз чаще рак молочной и щитовидной желез, бронхов. Известен также ряд гемобластозов - лейкоз, эритромиелоз, лимфогранулематоз, при которых инфильтрация патологическими элементами крови гипоталамуса или гипофиза вызывала несахарный диабет. Несахарный диабет сопровождает генерализованный ксантоматоз (болезнь Хенда-Шюллера-Крисчена) и может быть одним из симптомов эндокринных заболеваний или поврежденных синдромов с нарушением гипоталамо-гипофизарных функций: синдром Симмондса, Шихана и Лоуренса-Муна-Бидля, гипофизарного нанизма, акромегалии, гигантизма, адипозогенитальной дистрофии.
Вместе с тем, у значительного числа больных (60-70%) этиология заболеваний остается неизвестной - идиопатический несахарный диабет. Среди идиопатических форм следует выделить генетический, наследственный, наблюдаемые иногда в трех, пяти даже семи последующих поколениях. Тип наследования как аутосомно-доминантный, так и рецессивный.
Сочетание сахарного и несахарного диабета также встречается чаще среди семейных форм. В настоящее время предполагают, что участие больных с идиопатическим несахарным диабетом возможно аутоиммунная природа заболевания с поражением ядер гипоталамуса подобно деструкции других эндокринных органов при аутоиммунных синдромах. Нефрогенный несахарный диабет чаще наблюдается у детей и обусловлен либо анатомической неполноценностью почечного нефрона (врожденные уродства, кистозно-дегенеративные и инфекционно-дистрофические процессы): амилоидоз, саркоидоз, отравления метоксифлюраном, литием, либо функциональным ферментативным дефектом: нарушения продукции цАМФ в клетках почечных канальцев или снижением чувствительности его эффекта.
Гипоталамо-гипофизарные формы несахарного диабета с недостаточностью секреции вазопрессина могут быть связано с поражением любого отдела гипоталамо-нейрогипофизарной системой. Парность нейросекреторных ядер гипоталамуса и тот факт, что для клинической манифестации должно быть поражено не менее 80% клеток, секретирующих ВП, обеспечивают большие возможности внутренней компенсации. Небольшая вероятность возникновения несахарного диабета - при поражениях в области воронки гипофиза, где соединяются нейросекреторные пути, идущие от ядер гипоталамуса.
Недостаточность ВП снижает резорбцию жидкости в дистальном отделе почечного нефрона и способствует выделению большого количества гипоосмолярной неконцентрированной мочи. Первично возникающая полиурия влечет за собой общую дегидратацию с потерей внутриклеточной и внутрисосудистой жидкости с гиперосмолярностью (выше 290 мосм/кг) плазмы и жаждой, свидетельствующей о нарушении гомеостаза. В настоящее время установлено, что ВП вызывает не только антидиурез, но и натрийурез. При недостаточности гормона, особенно в период обезвоженности, когда стимулируется также натрийзадерживающий эффект альдостерона, натрий задерживается в организме, вызывая гипернатриемию и гипертоническую (гиперосмолярную) дегидротацию.
Усиленная ферментативная инактивация ВП в печени, почках, плаценте (во время беременности) вызывает относительную недостаточность гормона. Несахарный диабет при беременности (трансзиторный или в дальнейшем стабильный) может быть также связан со снижением осмолярного порога жажды, что усиливает потребление воды, "разводит" плазму и снижает уровень ВП. Беременность нередко ухудшает течение ранее существовавшего несахарного диабета и увеличивает потребность в лечебных препаратах. Врожденная или приобретенная рефрактерность почек к эндогенному и экзогенному ВП также создает относительную недостаточность гормона в организме.
Адипозогенитальная дистрофия. Болезнь Пехкранца-Бабинского-Фрелиха
Адипозогенитальная дистрофия – патология физического развития, при которой особый тип ожирения сочетается с гипогонадизмом, т.е. недостаточным формированием и функционированием половых органов. Специфика ожирения состоит в том, что липидные отложения концентрируются преимущественно в области живота по типу «фартука», на бедрах, ягодицах, молочных железах и в плечевом поясе, вследствие чего фигура больного приобретает характерную грушевидную форму.
Болезнь Пехкранца-Бабинского-Фрелиха чаще носит характер синдрома, а не самостоятельного заболевания, т.е. встречается в рамках более общей эндокринной патологии – как правило, в гипофизарно-гипоталамической системе. При начале в раннем детском возрасте и отсутствии установленной причины рассматривается как собственно болезнь. Интересно отметить, что в англоязычной литературе применительно к данному состоянию термин «болезнь» не употребляется вовсе: говорят о половом инфантилизме, гипоталамическом синдроме инфантилизма-ожирения, церебральном ожирении и т.п.
Точных эпидемиологических данных о распространенности адипозогенитальной дистрофии нет. Среди лиц, страдающих этим синдром/заболеванием, представлены оба пола, однако в ранней возрастной группе преобладают мальчики. Возрастные пики наиболее частого установления диагноза приходятся на периоды 6-7 лет и 10-13 лет.
2. Причины
Многочисленными исследованиями выявлено и доказано, что в основе синдрома гипогонадизма лежит гормональный дисбаланс, обусловленный поражением гипоталамуса и ассоциированной гонадотропной несостоятельностью гипофиза, – чем и формируется клиника. Зачастую в анамнезе обнаруживается внутриутробный токсоплазмоз или родовая травма, тяжелые пережитые или хронические инфекции, черепно-мозговые травмы, а при обследовании – всевозможные опухоли мозговых структур, признаки внутричерепной геморрагии, облитерации цереброваскулярных сосудов и пр.
Однако в некоторых случаях никаких органических изменений в гипоталамо-гипофизарной системе выявить не удается.
Изучается влияние наследственности и факторов, связанных с образом жизни (переедание в сочетании с гиподинамией).
3. Симптомы и диагностика
Ожирение является наиболее очевидным, выраженным и психотравмирующим пациента проявлением адипозогенитальной дистрофии. Для синдрома / болезни Пехкранца-Бабинского-Фрелиха характерны круглая форма лица, ложная гинекомастия (увеличенные и отвисшие молочные железы), инфантильные размеры гениталий, бесплодие, анлибидемия, импотенция. У мужчин нередко наблюдается также неопущение яичек (крипторхизм), у женщин – аменорея. Слабо развиты все вторичные половые признаки, включая степень и характер обволошения. Недоразвиты костные структуры и опорно-двигательный аппарат в целом, типична вальгусная (Х-образная) деформация нижних конечностей, плоскостопие.
Наличие задержек и дефицитов интеллектуального развития как характерного признака одними источниками отрицается (акцентируются лишь депрессивные, социо- и дисморфофобические развития в связи с сохранным чувством физической неполноценности), однако другие авторы описывают развернутый синдром психофизического инфантилизма (т.е. включают личностное, эмоционально-волевое и интеллектуальное отставание наряду с физическим) в рамках болезни Пехкранца-Бабинского-Фрелиха.
Нередко в сочетании с гонадотропной недостаточностью обнаруживается также гипотиреоз, проявляющийся специфической для него симптоматикой.
Клиническая картина болезни Пехкранца-Бабинского-Фрелиха, безусловно, зависит от возраста манифестации: при начале в постпубертатном периоде и нормальном преморбидном физическом развитии наблюдается тенденция к приобретению внешних черт, присущих противоположному полу (напр., фигура мужчины обретает евнухоидные формы), облысению, изменениям свойств кожи и пр.
Диагноз устанавливается клинически, анамнестически и лабораторно (в первую очередь исследуется гормональная формула крови и уровень гонадотропных гормонов в моче, различные параметры метаболизма и т.п.). Разработаны подробные критерии дифференциальной диагностики от заболеваний со сходной симптоматикой (болезнь Иценко-Кушинга, гиперкортицизм, гипотиреоз, первичный гипогонадизм и т.д.). Инструментальные исследования назначаются с целью обнаружения органической патологии в эндокринных мозговых структурах (КТ, МРТ, рентгенография).
4. Лечение
Терапевтическая стратегия определяется результатами диагностики и нацеливается на устранение первопричины заболевания, – если она достоверно установлена и устранима в принципе.
Применяют диетологическое лечение, заместительную гормональную терапию, препараты для подавления аппетита, диуретики. Лечение должно осуществляться строго по назначению и под контролем врача-эндокринолога. Как правило, курс занимает достаточно продолжительное время (1-2 года и более). В случаях, когда болезнь Пехкранца-Бабинского-Фрелиха выступает как самостоятельное заболевание неизвестной этиологии, лечение, как правило, эффективней и прогноз благоприятней, чем при наличии тяжелой системной эндокринной, наследственной, онкологической или иной патологии.
Заболевания- Заболевания щитовидной железы (гипотериоз, йододефицитные заболевания, опухоли щитовидной железы, хронический аутоиммунный тиреоидит, тиреотоксикоз) и сахарный диабет 2 типа
- Заболевания надпочечников (недостаточность коры надпочечников, гормонально-активные образования, феохромоцитома, альдостерома, артериальная гипертензия эндокринного генеза)
- Гиперпролактинемия
- Заболевания паращитовидных желез (гипопаратиреоз, гиперпаратиреоз)
- Эндокринные заболевания репродуктивной системы (бесплодие, аменорея, поликистоз у женщин, возрастной андрогенный дефицит у мужчин, гипогонадизм, гинекомастия)
- Нейроэндокринные нарушения (остеопороз, аденома гипофиза, гипопитуитаризм)
- Беспричинное снижение или увеличение веса
- Общее недомогание, слабость
- Нарушения сна
- Постоянное чувство жажды, сухость во рту
- Частые простудные заболевания
- Головные боли
- Склонность к депрессии
- Дискомфорт в области шеи, чувство «кома в горле» при глотании
- Сухость кожи, ломкость ногтей, выпадение волос, угревая сыпь
- Отеки
- Бесплодие, нарушение менструального цикла, выделения из груди при надавливании у женщин
- Бесплодие и снижение потенции у мужчин
- Повышение артериального давления, не поддающееся лечению традиционными методами
Если вы обнаружили у себя подобные симптомы, возможно, это сигнал заболевания, поэтому рекомендуем проконсультироваться с нашим специалистом.
Диагностика- Диагностика остеопороза
- УЗИ щитовидной железы
Мы стараемся оперативно обновлять данные по ценам, но, во избежание недоразумений, просьба уточнять цены в клинике.
Данный прайс-лист не является офертой. Медицинские услуги предоставляются на основании договора.
Адипозогенитальная дистрофия

Адипозогенитальная дистрофия (или синдром Пехкранца-Бабинского-Фрелиха) - это нейроэндокринное заболевание, характеризующееся ожирением гипоталамо-гипофизарного происхождения и нарушением функции половых желез (гипогонадизм).
Адипозогенитальная дистрофия вызвана поражением ядер гипоталамуса, которые отвечают за регуляцию аппетита и ядер гипоталамуса, в которых происходит синтез гормонов гонадолиберинов, стимулирующих выработку в гипофизе гормонов лютропина и фоллитропина. В условиях отсутствия или сниженного количества лютропина и фоллитропина стимуляции выработки гормонов половых желез не происходит и в конечном итоге возникает недостаточность функции половых желез.
Причинами адипозогенитальной дистрофии могут послужить:
- перенесенные инфекционные поражения головного мозга – вирусные и бактериальные менингиты, энцефалиты
- менингоэнцефалиты
- арахноидиты
- токсоплазмоз
К разрушению ядер гипоталамуса могут привести внутриутробные инфекции и интоксикации, родовые травмы головы, перенесенный матерью во время беременности токсоплазмоз. Иногда к поражению гипоталамуса приводят опухоли головного мозга в этой области.
Адипозогенитальная дистрофия возникает в детстве, но чаще всего проявляется в период пубертата, в подростковом возрасте, когда должно происходить половое созревание. Одинаково часто болеют мальчики и девочки. Самым ранним признаком заболевания бывает повышенный вес.
У мальчиков возникает увеличение молочных желез (гинекомастия) может быть уменьшен размер яичек и полового члена. Часто у мальчиков встречается крипторхизм (неопущение яичка в мошонку). В подростковом периоде половое созревание задерживается. Вторичные женские и мужские половые признаки формируются слабо. У мальчиков скудное оволосение на лобке и в подмышечных впадинах, часто отсутствие роста бороды и усов. Скудное оволосение сохраняется и во взрослом возрасте. Половые органы недоразвиты.
У девочек наружные половые органы отстают в развитии, наступление менструации задерживается (позднее менархе) или совсем не возникает.
У обоих полов нарушается развитие скелета, который формируется по евнухоидному типу: высокий рост, длинные конечности, размах которых превышает рост на несколько сантиметров.
Кроме вышеназванного также:
- Возникает неполноценность связочно-суставного аппарата: суставы патологически подвижны, переразогнуты, формируется плоскостопие.
- У пациентов выраженное ожирение. Подкожно-жировая клетчатка накапливается в основном в области живота, бедер, груди и лица.
- Кожа у пациентов тонкая и сухая.
- Возникают нарушения со стороны желудочно-кишечного тракта могут быть нарушения оттока желчи, запоры
- Со стороны сердечно-сосудистой системы происходят дистрофические изменения в сердечной мышце – появляется миокардиодистрофия.
Интеллектуальное развитие пациентов в большинстве случаев нормальное, соответствует возрасту и образованию.
Диагноз адипозогенитальной дисторфии устанавливают на основании характерных жалоб, внешнего вида пациентов, в крови обнаруживают снижение концентрации: фоллитропина, лютропина, тестостерона, эстрогенов.
Лечение адипозогенитальной дистрофии
Прежде всего назначается, если это возможно, лечение основного заболевания, которое привело к поражению гипоталамуса.
При опухолевых процессах назначается лучевая терапия или оперативное лечение.
Проводится лечение воспалительных заболеваний:
- назначаются антибиотики
- противовирусные препарататы
Кроме этого назначаются препараты, улучшающие состояние тканей головного мозга: актовегин, ноотропил, диавитол, церебролизин, кавинтон.
Для ограничения веса назначается диета с пониженной калорийностью пищи, ограничением жиров и легкоусвояемых углеводов.
Обязательно назначается лечебная физкультура с достаточной физической нагрузкой.
Мальчикам назначается заместительная гормональная терапия хорионическим гонадотропином с возраста 12 лет. Это позволяет достигнуть полового созревания. А в возрасте 15-16 лет назначаются мужские половые гормоны: тестостерон, метилтестостерон, сустанон.
Девочкам в подростковом возрасте в течение трех месяцев назначаются женские половые гормоны – эстрогены. Потом проводится лечение сложными эстроген-прогестиновыми препаратами.
При своевременно назначенном лечении адипозогенитальной дистрофии и соблюдении диеты состояние пациентов остается хорошим на протяжении всей жизни.
Адипозогенитальная дистрофия ( синдром Пехкранца-Бабинского-Фрелиха )
Адипозогенитальная дистрофия – нейроэндокринная патология, проявляющаяся избыточным весом и недоразвитием половых желез. Сопровождается ожирением по гиноидному типу и отставанием в половом развитии. Диагностика основывается на физикальном осмотре педиатра или эндокринолога, исследовании гормонального фона. Дополнительно назначается рентгенография и томография гипоталамо-гипофизарной области. Показана терапия основного заболевания, вызвавшего дистрофию. Симптоматическое лечение включает введение хорионического гонадотропина, заместительную гормонотерапию, диетическое питание.
МКБ-10
Общие сведения
Адипозогенитальная дистрофия (синдром Пехкранца-Бабинского-Фрелиха) – патологическое состояние, которое возникает в результате нарушений работы гипоталамо-гипофизарной системы. Заболевание было впервые описано в конце ХIХ века российским врачом Пехкранцем. Через несколько лет симптомы болезни были отражены в работах французского невропатолога Джозефа Бабинского и австрийского фармаколога Альфреда Фрелиха. Патология возникает в детстве или пубертатном периоде и чаще поражает мальчиков в возрасте от 6 до 13 лет. Синдром может быть самостоятельным заболеванием или проявлением другого патологического процесса. В последнем случае симптомы могут появиться в любом возрасте.
Причины
Несмотря на то, что симптомы болезни обычно проявляются в подростковом возрасте, формирование патологического процесса может начаться еще на этапе внутриутробного развития. К основным причинам возникновения патологии относят:
- Опухоли головного мозга. Синдром может возникать при краниофарингиоме, хромофобной аденоме гипофиза, других злокачественных и доброкачественных новообразованиях, расположенных в зоне гипоталамуса и гипофиза.
- Внутриутробные инфекции. Патологию провоцируют перенесенные женщиной во время беременности вирусные и бактериальные инфекции: грипп, токсоплазмоз, хламидиоз, скарлатина, корь, тиф.
- Некоторые инфекционные заболевания. К числу инфекций, способных вызвать развитие дистрофии, относят менингит, энцефалит, туберкулез, сифилис, перенесенные в детском возрасте.
- Травматические повреждения. К развитию синдрома приводят родовые травмы новорожденных, закрытые и открытые ЧМТ с поражением гипоталамуса и гипофиза.
Патогенез
Основной причиной возникновения патологии являются нарушения работы гипофиза и гипоталамуса. В результате расстройства эндокринной функции уменьшается секреция гонадотропных гормонов аденогипофизом, формируется гипогонадизм. В некоторых случаях отставание полового развития сочетается с нарушением продукции тиреотропного, соматотропного и антидиуретического гормонов. Поражение гипоталамуса приводит к патологическому раздражению его ядер и повышению аппетита. Возникают эпизоды булимии, пациенты потребляют избыточное количество пищи, развивается ожирение.
Симптомы адипозогенитальной дистрофии
Заболевание начинается в детском и подростковом возрасте, проявляется прибавкой веса, повышенной утомляемостью, сонливостью, снижением способности к обучению. У пациентов наблюдается нарушение аппетита по типу булимии. В пубертатный период у мальчиков возникает гинекомастия, отмечается крипторхизм, недоразвитие полового члена. У девочек выявляется задержка начала менструаций вплоть до аменореи, недоразвитие молочных желез, матки и придатков. Отсутствуют вторичные половые признаки (наличие волос в подмышечных впадинах и на лобке, на лице у мальчиков).
Нарушается формирование костно-суставной системы, обнаруживается плоскостопие, вальгусное искривление голеней. Характерно формирование скелета по евнухоидному типу. Больные имеют высокий рост, непропорционально длинные конечности, большой размер ноги. Либидо снижено или отсутствует. В связи с ожирением и недостаточным питанием сердечной мышцы развивается миокардиодистрофия, которая приводит к уменьшению сердечного выброса и нарушениям ритма. Синдром не влияет на умственные способности, интеллектуальное развитие соответствует возрасту.
Кожные покровы бледные, гладкие, сухие. Часто наблюдается нарушение пигментации кожи, воспаление роговицы и сухость глаз. Жировые отложения локализуются в области живота, бедер, груди, лица. У мальчиков увеличение веса протекает по гиноидному (женскому) типу. Адипозогенитальная дистрофия часто сочетается с другими эндокринными патологиями: аутоиммунным тиреоидитом, гипотиреозом, акромегалией, гиперсомнией. В большинстве случаев пациенты ведут малоподвижный образ жизни, что усугубляет ожирение.
Осложнения
При поздней диагностике или невосприимчивости к терапии возникают осложнения со стороны сердечно-сосудистой системы: сердечная недостаточность, склеротические изменения сосудов, нарушения ритма. При увеличении размера опухоли отмечается сдавление окружающих тканей, сопровождающееся нарушениями зрения (высокая степень миопии и гиперметропии, язвы роговицы) и повышением внутричерепного давления.
Отставание в формировании половых желез приводит к эректильной дисфункции и импотенции у мужчин, аменорее и бесплодию у женщин. Характерны осложнения, связанные с развитием ожирения: сахарный диабет 2 типа, ишемическая болезнь сердца. Дискинезия желчевыводящих путей способствует застою желчи и возникновению желчекаменной болезни. При росте опухоли возможно изменение поведения, повышение нервной возбудимости, нарушение сознания и развитие комы.
Диагностика
Диагностика адипозогенитальной дистрофии основывается на данных осмотра педиатра и эндокринолога, изучении анамнеза жизни и течения беременности матери, лабораторных и инструментальных исследованиях. При подозрении на задержку полового развития пациенты направляются на консультацию к гинекологу или андрологу. Диагноз подтверждается при наличии предожирения или ожирения с преимущественным отложением жира в верхней половине тела, гипоплазии половых желез и отсутствии вторичных половых признаков. В ходе обследования проводят:
- Определение гормонального статуса. Выполняют анализ крови на гормоны гипофиза, половые гормоны (ФСГ, ЛГ, тестостерон и эстроген).
- Рентгенологические и томографические исследования. В ходе рентгенографии черепа, КТ турецкого седла, МРТ головного мозга и гипофиза могут визуализироваться опухоли, кровоизлияния, водянка, увеличение размеров и деформация турецкого седла.
Дифференциальную диагностику дистрофии осуществляют с синдромом и болезнью Иценко-Кушинга, для которых характерно нормальное половое развитие. Патологию следует отличать от наследственного алиментарно-конституционального ожирения, имеющего семейный характер и нормальные этапы полового созревания. Болезнь также дифференцируют с синдромами Лоренса-Муна-Бидля, Морганьи-Стюарта-Мореля, Шерешевского-Тернера, Клайнфельтера. Для диагностики перечисленных патологий используются генетические исследования с выявлением хромосомных аномалий.
Лечение адипозогенитальной дистрофии
Этиотропное лечение направлено на устранение заболевания, повлекшего развитие патологического синдрома. При опухолевом процессе проводится хирургическое вмешательство, рентгенотерапия, химиотерапия. Медикаментозная терапия инфекционных и воспалительных заболеваний включает назначение антибактериальных или противовирусных препаратов, витаминных и минеральных комплексов. Симптоматическое лечение предполагает изменение режима питания и образа жизни, введение гормональных препаратов. При отсутствии противопоказаний со стороны опорно-двигательного аппарата показана лечебная физкультура, плавание, скандинавская ходьба.
Всем пациентам назначается низкокалорийная диета с пониженным содержанием легкоусвояемых углеводов и растительных жиров. В рацион вводят свежие овощи и сложные углеводы, которые увеличивают чувство насыщения. Пищу рекомендовано принимать 6-7 раз в день, не допуская длительного голодания. При развитии булимии показаны анорексигенные препараты, подавляющие аппетит. Терапия гипогонадизма предполагает введение ХГЧ. При достижении пубертатного возраста мальчикам назначают гонадотропные препараты в сочетании с тестостероном, девочкам – эстрогены с прогестероном.
Прогноз и профилактика
Течение синдрома зависит от этиологии основного заболевания. При своевременной диагностике и лечении прогноз благоприятный. Позднее обнаружение синдрома, развитие осложнений и прогрессирующее ожирение вызывают утрату трудоспособности, затрудняют нормальную жизнедеятельность.
Профилактика заболевания заключается в санации хронических очагов воспаления у детей и планировании беременности будущей матери. Правильное питание, отказ от вредных привычек и заместительная гормонотерапия могут замедлить развитие болезни. При грамотном лечении в пубертатном периоде пациенты развиваются по возрасту. При соблюдении диеты удается контролировать массу тела и избежать осложнений, связанных с ожирением.
Читайте также: