Болезнь Фабри
Добавил пользователь Владимир З. Обновлено: 27.01.2026
Клинический случай демонстрирует кардиологический фенотип болезни Фабри - редкого наследственного заболевания, сцепленного с женским полом. Описана клиническая манифестация болезни Фабри у пациентки женского пола нарушениями ритма сердца в виде пароксизмальной формы фибрилляции предсердий. Изложены особенности дифференциальной диагностики, тактики ведения и показания к генетической диагностике и назначению генотип-специфической фермент-заместительной терапии.
Ключевые слова
Об авторах
Харлап Мария Сергеевна, кандидат медицинских наук, старший научный сотрудник Отдела нарушений сердечного ритма и проводимости
Мясников Роман Петрович, кандидат медицинских наук, старший научный сотрудник Отдела клинической кардиологии и молекулярной генетики
Павлунина Татьяна Олеговна, кандидат медицинских наук, врач-кардиолог 1 кардиологического отделения
Береговская Светлана Александровна, заведующая 1 кардиологическим отделением
Соничева Наталья Александровна, врач-кардиолог
Мершина Елена Александровна, кандидат медицинских наук, заведующий отделом рентгенодиагностики с кабинетами КТ и МРТ МНОЦ
Давтян Карапет Воваевич, доктор медицинских наук, руководитель Отдела нарушений сердечного ритма и проводимости
Список литературы
1. Lyon A, Ariga R, Minchole A. et al. Distinct ECG phenotypes identified in hypertrophic cardiomyopathy using machine learning associate with arrhythmic risk markers. Front Physiol 2018;13;9:213.
2. Pelto H, Owens D, Drezner J. et al. Electrocardiograhic findings suggestive of cardiomyopathy: what to look for and what to do next. Curr Sports Med Rep 2013;12(2):77-85.
3. Germain D.P. Fabry disease. Orphanet J Rare Dis 2010;22;5:30.
4. Arends M, Wanner C, Hughes D. et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J Am Soc Nephrol 2017; 28 (5):1631-164.
5. Ortiz A, Germain GP, Desnick RJ et al. Fabry disease revisited: managemet and treatment recommendations for adult patients. Mol Genet Metab 2018;123(4):416-427.
Рецензия
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
М. С. Харлап
ФГБУ "Национальный медицинский исследовательский центр профилактической медицины" МЗ РФ
Россия
Харлап Мария Сергеевна, кандидат медицинских наук, старший научный сотрудник Отдела нарушений сердечного ритма и проводимости
Р. П. Мясников
ФГБУ "Национальный медицинский исследовательский центр профилактической медицины" МЗ РФ
Россия
Мясников Роман Петрович, кандидат медицинских наук, старший научный сотрудник Отдела клинической кардиологии и молекулярной генетики
Т. O. Павлунина
ФГБУ "Национальный медицинский исследовательский центр профилактической медицины" МЗ РФ
Россия
Павлунина Татьяна Олеговна, кандидат медицинских наук, врач-кардиолог 1 кардиологического отделения
С. А. Береговская
ФГБУ "Национальный медицинский исследовательский центр профилактической медицины" МЗ РФ
Россия
Береговская Светлана Александровна, заведующая 1 кардиологическим отделением
Н. А. Соничева
Научный комитет Генетической Лаборатории «Health in Code»
Испания
Соничева Наталья Александровна, врач-кардиолог
Е. А. Мершина
Московский Государственный Университет им. М.В.Ломоносова
Мершина Елена Александровна, кандидат медицинских наук, заведующий отделом рентгенодиагностики с кабинетами КТ и МРТ МНОЦ
К. В. Давтян
ФГБУ "Национальный медицинский исследовательский центр профилактической медицины" МЗ РФ
Россия
Давтян Карапет Воваевич, доктор медицинских наук, руководитель Отдела нарушений сердечного ритма и проводимости
Орфанные заболевания: болезнь Фабри
Болезнь Фабри или болезнь Андерсона-Фабри – орфанное наследственное заболевание, относящееся к группе лизосомных болезней накопления, причиной которого является мутация гена мутации гена GLA, контролирующего структуру α-галактозидазы А, расположенный на Х хромосоме (Хq22.1). К настоящему времени идентифицировано почки 600 мутаций и полиморфизмов в гене GLA и большинство мутаций являются уникальными для каждой семьи. Значительное снижение активности или отсутствие фермента α-галактозидазы А из-за мутации гена GLA приводит к накоплению глоботриаозилцерамида и других гликофосфолипидов в лизосомах клеток различных органов, включая сердце, почки, нервную систему и эндотелий сосудов.
Болезнь была впервые описана в 1898г английским дерматологом Андерсоном и немецким дерматологом Фабри.
Болезнь Фабри относится к редким заболеваниям. Частота заболевания составляет 1:80 000 новорожденных.
Так как болезнь Фабри наследуется по рецессивному X-сцепленному типу, мужчины имеют единственную Х-хромосому с мутацией, и поэтому у них развивается тяжелая классическая форма болезни. Они передают хромосому с мутацией только своим дочерям, но не сыновьям. Таким образом дочери больных отцов имеют одну нормальную и одну мутантную хромосому, т.е. являются гетерозиготными. Женщины носительницы передают хромосому с мутацией половине своих потомков обеих полов. У женщин носительниц течение болезни, как правило, более легкое, с более поздним началом и медленным прогрессированием.
Выделяют две формы болезни – классическую и атипичную. Атипичные формы могут быть с поздним началом, изолированным поражением сердца, почек или головного мозга.
Проявления болезни может быть чрезвычайно разнообразными как у мужчин, так и у женщин, и даже у членов одной семьи.
В детском возрасте заболевание проявляется болями в кистях и стопах, ангиокератомами, снижением потоотделения, астенией, позднее присоединяются боли в животе, поражение почек, сердца, возможны транзиторные ишемические атаки и инсульт.
Ангиокератомы – это мелкие, плоские, или слегка приподнятые кожные образования, имеющие оттенок от темно-красного до синего цвета, обычно расположенные в паховой области, на ягодицах, бедрах и в области пупка. Ангиокератомы проявляются в детстве и постепенно с годами увеличиваются в количестве и в размерах. Известны случаи болезни без ангиокератом. Боли в кистях и стопах носят характер изнуряющей невропатической боли, т.е. это жгучая боль, с чувством покалывания или ползания мурашек в конечностях, может быть сильная боль, возникающая при небольшом болевом раздражении, или боль, возникающая при повышении температуры окружающей среды. Первые симптомы поражения почек (микроальбуминурия, протеинурия) появляются в возрасте 20-30 лет, постепенно прогрессируют, снижается функции почек, что приводит к развитию терминальной почечной недостаточности. Это одна из основных причин смерти пациентов с болезнью Фабри. Признаком поражения сердца при болезни Фабри являются гипертрофия левого желудочка, фиброз миокарда часто с диастолической дисфункцией левого желудочка. Нарушения диастолической функции могут привести к развитию сердечной недостаточности, часто развиваются предсердные и желудочковые аритмии и нарушения проводимости. У больных высок риск развития транзиторных ишемических атак, ишемических и геморрагических инсультов уже в молодом возрасте, так как болезнь Фабри – это болезнь малых сосудов. Результатом хронической ишемии головного мозга может быть сосудистая деменция (снижение памяти, изменение поведения). Типичным симптомом болезни является помутнение роговицы в виде завитков (у 70-90% больных), задняя субкапсулярная катаракта и поражение сосудов сетчатки, что иногда вызывает тяжелую потерю зрения. При болезни Фабри часто бывает снижение слуха, шум в ушах, головокружение.
Заболевание носит прогрессирующий характер. Без лечения смерть пациентов, как правило, наступает на 4-м десятилетии жизни от почечной недостаточности, сердечно-сосудистых или цереброваскулярных осложнений.
С целью диагностики болезни Фабри определяют активность альфагалактозидазы А в высушенных пятнах крови, в плазме или лейкоцитах крови. Новым эффективным биомаркером заболевания является показатель уровня глоботриаозилсфингозина (лизо-Gb3) крови и экскреция лизо-Gb3 с мочой. Диагноз подтверждают путем молекулярной диагностики, исследования гена альфа-галактозидазы А. В семьях, где есть больной с установленной мутацией, вызвавшей болезнь, существует возможность проведения преимплантационной и пренатальной диагностики.
С внедрением ферментозаместительной терапии (ФЗТ) рекомбинантными препаратами альфагалактозидазы А в 2001 года, качество жизни и прогноз для больных значительно улучшились. Применяют препараты агалсидазу альфа (Реплагал, Shire Human Genetic Therapies) и агалсидазу бета (Фабразим, Genzyme Corporation). ФЗТ уменьшает выраженность невропатической боли, вызывает регресс гипертрофии левого желудочка и стабилизацию функции почек, задерживает развитие почечной и сердечно-сосудистой недостаточности заболеваний. Препараты имеют хорошую переносимость. Так же используется симптоматическая терапия (антиконвульсанты, ингибиторы АПФ, блокаторы рецепторов ангиотензина II, антиагреганты, антикоагулянты, гемодиализ, трансплантация почки).
Болезнь Фабри
Болезнь Фабри, или болезнь Андерсона - Фабри – наследственное заболевание, относящееся к группе лизосомных болезней накопления, обусловленное значительным снижением активности или отсутствием фермента α-галактозидазы А. Отсутствие или дефицит фермента приводит к накоплению глоботриаозилцерамида и родственных гликофосфолипидов в лизосомах клеток различных органов, включая сердце, почки, нервную систему и эндотелий сосудов. Заболевание обладает широким спектром клинических симптомов. В детском возрасте часто сопровождается болями в кистях и стопах, ангиокератомами, гипогидрозом, астенией, а в более старшем возрасте дополняется болью в животе, поражением почек, сердца, возможны транзиторные ишемические атаки, инсульт. Секвенирование ДНК гена GLA - наиболее точный метод диагностики болезни Фабри.
Синонимы русские
Диффузная ангиокератома, болезнь Андерсона – Фабри.
Метод исследования
Полимеразная цепная реакция (ПЦР).
Какой биоматериал можно использовать для исследования?
Общая информация об исследовании
Болезнь Фабри, или болезнь Андерсона - Фабри – наследственное заболевание, относящееся к группе лизосомных болезней накопления, обусловленное значительным снижением активности или отсутствием фермента α-галактозидазы А. Отсутствие или дефицит фермента приводит к накоплению глоботриаозилцерамида и родственных гликофосфолипидов в лизосомах клеток различных органов, включая сердце, почки, нервную систему и эндотелий сосудов. Заболевание обладает широким спектром клинических симптомов. В детском возрасте часто сопровождается болями в кистях и стопах, ангиокератомами, гипогидрозом, астенией, а в более старшем возрасте дополняется болью в животе, поражением почек, сердца, возможны транзиторные ишемические атаки, инсульт. Заболевание носит прогрессирующий характер, сопровождается снижением качества и продолжительности жизни.
Относится к редким заболеваниям. Распространенность болезни в различных странах мира варьируетсся в широких пределах (от 1 на 117000 до 1 на 476000 населения). У мужчин и женщин с ранним инсультом частота болезни Фабри составила 4,2% и 2,15 соответственно, с гипертрофией левого желудочка неясного происхождения – 0,9-3,9% и 1,1-11,8%, с терминальной почечной недостаточностью – 0,33% и 0,10%. Вероятно, что более распространенным является легкое, атипичное течение болезни с признаками поражения одного органа. Таким образом, является одной из наиболее распространённых (после болезни Гоше) лизосомных болезней накопления. Обычно (примерно в 95% случаев) больные наследуют дефектный ген от одного из родителей (мужчина - от матери, женщина - от матери или отца), но около 5% случаев связаны с так называемыми мутациями de novo. Таким образом, отсутствие семейной истории заболевания не исключает наличия болезни Фабри.
Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру α-галактозидазы А (GLA; EC 3.2.1.22). Ген α-галактозидазы А картирован на длинном плече хромосомы Хq 22.1, имеет размер 12 kb и состоит из 7 экзонов, протяженностью от 92 до 291 пар оснований. Кодирующий регион гена содержит 1290 пар оснований, определяющий полипептидную структуру фермента (429 аминокислотных остатков). К настоящему времени идентифицировано 599 мутаций и полиморфизмов в гене GLA, в том числе 435 патогенетических "точковых" мутаций, изменяющих кинетические свойства и стабильность галактозидазы А. Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную мутантную Х-хромосому, что определяет классический фенотип болезни. Они передают мутантную хромосому только своим дочерям, но не сыновьям. Таким образом, дочери больных болезнью Фабри отцов имеют одну нормальную и одну мутантную хромосому, т.е. являются гетерозиготами. Оставаясь, как правило, клинически здоровыми, они могут передать мутантную хромосому и, следовательно, патологический аллель половине своим потомкам. Течение болезни у них, как правило, умеренно выраженное с более поздним началом, медленным прогрессированием и легкими клинико-патологическими изменениями. Вместе с тем было показано, что у части гетерозиготных женщин с мутацией гена α-галактозидазы А развиваются тяжелые проявления болезни Фабри, требующие медицинской помощи и вмешательства. Механизм, посредством которого у гетерозиготных женщин развиваются жизнеугрожающие симптомы, неизвестен. У большинства из них имеется почти нормальный уровень циркулирующего фермента за счет того, что случайный процесс инактивации Х-хромосомы (лайонизация) приводит к образованию как дефицитных, так и нормальных клеток. Таким образом, гетерозиготных женщин не следует называть носителями, поскольку носительство подразумевает отсутствие клинических проявлений болезни Фабри. Гликосфинголипиды, такие как глоботриаозилцерамид и галабиозилцерамид, имеют терминальный остаток α-галактозила, который отщепляется ферментом α-галактозидазой А. Таким образом, недостаточность или отсутствие фермента приводит к накоплению различных гликосфинголипидов с терминальным α-галактозил-остатком.
Симптомы болезни нередко проявляются в раннем детстве, однако ввиду своей неспецифичности задержка с постановкой диагноза составляет годы и даже десятилетия. Но с возрастом количество симптомов и степень их выраженности обычно увеличивается. Различают классическую и неклассическую форму болезни Фабри.
Мужчины часто обладают характерным фенотипом, напоминающим внешний вид пациентов с акромегалией: выступающие супраорбитальные дуги и лобные бугры, выступающая нижняя челюсть, увеличенные губы, запавшая переносица (с 12-14-летнего возраста). Боли необычного характера - один из самых частых и ранних симптомов болезни Фабри. Выделяют два основных типа болей: нейропатическая боль (акропарестезии), характеризующаяся постоянным жжением и покалыванием, ощущением дискомфорта в ступнях и ладонях, и кризы Фабри, возникающие время от времени сильные, жгучие боли, возникающие в ступнях или ладонях и иногда распространяющиеся на другие части тела. Значительная часть больных отмечает нарушение потоотделения, они потеют меньше нормы (гипогидроз) или не потеют вообще (ангидроз). Это может приводить к перегреву тела и непереносимости жары.
Одни из самых ранних и заметных признаков болезни Фабри - мелкие, красновато-фиолетовые безболезненные папулы на коже - ангиокератомы. С возрастом количество ангиокератом у больных увеличивается, также иногда увеличиваются размеры отдельных элементов сыпи (до 10 мм). Наиболее часто ангиокератомы располагаются в области губ, пальцев рук и ног, ано-генитальной области (при достижении половой зрелости).
Больные жалуются на затруднение дыхания и боли в груди, боли по типу стенокардии, тахикардию. Наиболее часто наблюдается гипертрофия левого желудочка, аритмии. Нарушение функции почек наблюдается у значительной части детей, многих женщин и большинства мужчин с болезнью Фабри и в конечном итоге приводит к терминальной стадии хронической почечной недостаточности у большинства больных мужчин и некоторых женщин.
Начальные стадии почечной недостаточности остаются практически незамеченными, так как нет симптомов. В отличие от "классических" нефрологических больных, у больных Фабри обычно нормальное артериальное давление, нормальный уровень сывороточного креатинина, "минимальная" протеинурия, что затрудняет оценку степени почечной недостаточности нефрологом.
Также болезнь Фабри имеет много неврологических проявлений. Больные жалуются на головокружение, нарушения кровообращения, головные боли. Нарушение мозгового кровообращения вследствие накопления сфинголипидов выливается в транзиторные ишемические атаки, инсульты. Пациенты могут жаловаться на звон в ушах, в отдельных случаях наблюдается ухудшение слуха, которое может медленно прогрессировать или появиться внезапно.
Диагностика болезни Фабри включает комплексную оценку клинический картины, лабораторные тесты и другие исследования (например, исследование биоптата почки на наличие подоцитов, лизосомы которых заполнены специфическим субстратом, характерным для болезни Фабри). На МРТ головного мозга могут присутствовать характерные признаки: на Т2-изображении гиперинтенсивный сигнал в белом веществе фронтальных и теменных долей, на Т1-взвешенном изображении наблюдается высокий сигнал от серого вещества глубинных структур, особенно заднего бугорка таламуса. Изолированное поражение заднего бугорка таламуса считается патогномоничным для болезни Фабри. Также на МР-картине часты сосудистые мальформации, преимущественно представленные долихоэктазиями вертебробазилярной артерии. На ЭКГ наблюдается гипертрофия левого желудочка, укорочение P-R интервала (на ранних стадиях болезни) и предсердножелудочковая блокада (на более поздних стадиях).
Наиболее точным исследованием для постановки диагноза является лабораторная диагностика, где применяются следующие исследования:
1. Определение активности альфа-галактозидазы (энзимодиагностика)
При болезни Фабри активность альфа-галактозидазы в крови у мужчин всегда снижена, а у женщин может быть около нижней границы нормы, чуть ниже её или нормальной, из-за чего ферментный анализ для женщин не показателен в отношении наличия/отсутствия болезни Фабри.
2. Количественное определение сфинголипидов
Относительно новый тест, рекомендованный для использования в комплексной лабораторной диагностике болезни Фабри. Количественное определение глоботриазилсфингозина (Lyso-GB3, Lyso-GL3) более показательно, нежели глоботриазилцерамида (GB3, GL3). Тест позволяет не только разъяснить трудные диагностические случаи болезни, но и определить наиболее вероятную форму болезни (классическая или неклассическая). Полезен для мониторинга состояния пациента, т.к. показано, что улучшение на фоне патогенетической терапии сопровождается снижением содержания Lyso-GL3 в плазме пациентов.
3. Генодиагностика болезни Фабри (ген GLA) (секвенирование экзонов и приэкзонных участков интронов гена GLA)
Идентифицировано более 400 мутаций, приводящих к развитию болезни Фабри, большинство из которых являются уникальным для каждой семьи. У больного мужчины мутация выявляется в гемизиготном состоянии, у женщины - в гетерозиготном состоянии. Секвенирование ДНК гена GLA - наиболее точный метод диагностики болезни Фабри. Для женщин предпочтительно проведение ДНК-диагностики, т.к. энзимодиагностика не всегда позволяет выявить болезнь. Большинство мутаций у пациентов с болезнью Фабри могут быть выявлены с помощью стандартного секвенирования (после выделения ДНК используют полимеразную цепную реакцию для того, чтобы амплифицировать кодирующую область гена; полученный ПЦР-продукт затем подвергается секвенированию (определению нуклеотидной последовательности полученного фрагмента ДНК); последовательность ДНК гена α-галактозидазы А пациента сравнивают с референсной). Молекулярно-генетические исследования гена α-галактозидазы А позволяют выявлять замены нуклеотидов, делеции, инсерции (вставки), частичные дупликации, сложные мутации, вовлекающие более чем одно молекулярное изменение. При подтверждении диагноза и выявлении мутации у пробанда целесообразно обследовать на наличие этой мутации всех родственников пробанда, которые могут нести одну с ним X-хромосому.
Для чего используется исследование?
- Для наиболее точного определение болезни Фабри;
- для ранней диагностики болезни Фабри до появления многих клинических симптомов;
- для определения рисков передачи генетических патологий при беременности.
Когда назначается исследование?
- При наличие симптомов заболевания Фабри (боли в кистях и стопах, ангиокератомы, гипогидроз, астения, боль в животе, поражение почек, сердца, транзиторные ишемические атаки, гипертрофия миокарда неясного происхождения и инсульт в молодом возрасте);
- планируемая беременность;
- при наличии наследственных рисков (подтвержденная или неподтвержденная болезнь Фабри у родственников).
Что означают результаты?
Референсные значения: мутаций в гене GLA не обнаружено.
Кто назначает исследование?
Акушер-гинеколог, неонатолог, педиатр, терапевт, кардиолог, уролог, невропатолог.
Болезнь Фабри
ГБУЗ МО «Московский областной научно-исследовательский институт им. М.Ф. Владимирского», Москва, Россия
Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского
Неврологические проявления при болезни Фабри
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2016;116(9): 98‑105
Болезнь Фабри (болезнь Андерсона—Фабри) — Х-сцепленная рецессивная лизосомальная болезнь накопления, возникающая вследствие недостаточной активности лизосомальной гидролазы (альфа-галактозидазы А), приводящей к прогрессирующей аккумуляции глоботриаозилцерамида (Gb3) в различных клетках, преимущественно эндотелиальных и клетках гладкой мускулатуры сосудов, с поражением органов, включая ЦНС. Болезнь Фабри проявляется прогрессирующей почечной и сердечной недостаточностью, нейропатической болью, инсультами, кожными и желудочно-кишечными симптомами. Клинические проявления начинаются в детстве, но диагностируются у многих пациентов только во взрослом возрасте. Женщины болеют также тяжело, как и мужчины. Возможен летальный исход вследствие раннего инсульта, болезней сердца и почек. Раннее распознавание симптомов, определение уровня активности ферментов, концентрации Gb3 в крови, моче, биопсия кожи, генетическое тестирование (GLA ген) позволяют осуществлять раннюю диагностику и фермент-заместительную терапию. Данная терапия способствует стабилизации или редуцированирю прогрессирования болезни. Раннее начало лечения позволяет предотвратить развитие осложнений.
ГБУЗ МО «Московский областной научно-исследовательский институт им. М.Ф. Владимирского», Москва, Россия
Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского
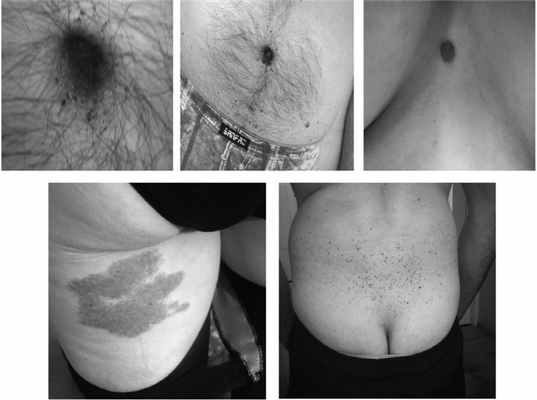
Примеры ангиокератомы у мужчин с «классической» болезнью Фабри.
В 1898 г. немецкий врач Дж. Фабри и английский У. Андерсон независимо друг от друга описали кожные изменения в сочетании с признаками поражения других органов у 2 пациентов мужского пола (мальчика и взрослого мужчины). В дальнейшем заболевание получило название «болезнь Фабри» или «болезнь Андерсона—Фабри» [1]. В 1967 г. Brady и соавт. открыли первичный биохимический дефект, лежащий в основе заболевания — недостаточность α-галактозидазы А, позднее в 1989 г. был секвенирован ген α-галактозидазы А, что дало возможность с помощью генно-инженерных методов синтезировать данный фермент in vitro и создать препараты для фермент-заместительной терапии заболевания. При болезни Фабри происходят мутации в гене GLA, расположенном на Х хромосоме и кодирующем фермент α-галактозидазу А. От больного мужчины мутантная Х-хромосома передается дочерям. Оставаясь, как правило, клинически здоровыми, дочери могут передать поврежденную хромосому и, следовательно, патологический аллель половине своих потомков. Однако женщины-носительницы также могут иметь проявления заболевания и в некоторых случаях столь же тяжелые, как и больные мужчины. Мужчины имеют одну Х-хромосому, следовательно, при наличии у мальчиков мутантной Х-хромосомы всегда развивается болезнь. Около 5% случаев связаны с так называемыми мутацииями de novo из-за чего болезнь Фабри может встречаться у людей, не имеющих отягощенного семейного анамнеза.
В результате мутации в гене α-галактозидазы, А при болезни Фабри имеется уменьшение активности данного фермента в лизосомах, результатом чего является неполное расщепление гликосфинголипидов. В результате этого в лизосомах откладываются нейтральные сфинголипиды и другие соединения. Основной накапливаемый субстрат — глоботриаозилцерамид (Gb3) [2]. Эти вещества накапливаются в гладкомышечных и эндотелиальных клетках сосудов; клетках ЦНС, роговицы, почек, сердечной мышцы. Накопление Gb3 начинается с раннего возраста, происходит поражение различных тканей и органов, повреждение имеет прогрессирующий характер и может быть необратимым. Дисфункция эндотелия и стенок сосудов с последующим воспалением вызывает обструктивные процессы, аналогичные атеросклеротическим поражениям [3]. Накопление Gb3 запускает процесс поражения иммунной системы с явлениями местного и общего воспаления, вызывающего повреждение органов [4].
Гомизиготные больные мужчины без резидуальной активности α-галактозидазы, А имеют выраженные неврологические (боль), кожные (ангиокератомы), почечные (протеинурия, почечная недостаточность), кардиоваскулярные (кардиомиопатия, аритмия), кохлеовестибулярные, цереброваскулярные (инсульты) проявления болезни. У гетерозиготных женщин симптоматика варьирует от слабой до ярко выраженной [1].
Симптомы заболевания можно разделить на ранние и поздние [5]. К ранним относятся акропарестезии; пониженноепотоотделение, приводящее к перегреванию и снижению переносимости жары; ангиокератомы, расстройства ЖКТ;дискомфорт, боль в кистях и стопах, появляющиеся при повышении окружающей температуры или при физических упражнениях [6]. Выделяются два типа болей: нейропатическая (акропарестезии) — постоянное жжение, покалывание, ощущение дискомфорта и кризы Фабри — периодически возникающие, сильные, жгучие, изнуряющие боли, продолжающиеся от нескольких минут до нескольких дней [7].
По мере прогрессирования болезни могут присоединяться следующие симптомы: повышенная утомляемость; желудочно-кишечные расстройства, образование дивертикулов в кишечнике [8—10]; головные боли; шум в ушах, снижение слуха [11, 12]; отеки голеней; боль в груди или сердцебиение.
Поздними осложнениями, определяющими смертность после 30 лет, являются: поражение почек [13, 14]; поражение сердца [15—17]; нарушения мозгового кровообращения [18—21].
В головном мозге отмечаются прогрессирующая многоочаговая окклюзия сосудов малого калибра, которая обусловливает клиническую симптоматику болезни вследствие ишемии и инфарктов в различных органах, изменение формы артерий, эндотелиальная дисфункция, явления тромбоза, церебральная гипоперфузия, кардиальные эмболы, повреждение белого вещества. Поражаются небольшие перфорантные артерии в перивентрикулярной области, области ствола мозга и базальных ганглиев. Для болезни Фабри специфичными являются пульвинарные кальцификаты. Вследствие отложения гликосфинголипидов в эндотелиальных клетках сосудов происходит замедление скорости кровотока [22, 23]. Также имеется неспецифическое поражение белого вещества головного мозга, нарастающее с возрастом, встречающееся одинаково часто у мужчин и женщин [24]. Субарахноидальные кровоизлияния при болезни Фабри встречаются редко [25], также как и асептический менингит [26].
У пациентов с болезнью Фабри в 10 раз чаще чем в популяции встречается обструктивная болезнь легких, характеризующаяся одышкой, хрипами в легких, сухим кашлем [27, 28]. Почечные, кардиоваскулярные, цереброваскулярные осложнения уменьшают на 20 и 10 лет продолжительность жизни у мужчин и женщин, не получающих заместительную терапию, по сравнению с популяцией соответственно [29].
При болезни Фабри преимущественно поражаются тонкие нервные волокна [30—33]. Клинически поражение тонких волокон проявляется в периодической боли и жжении в дистальных отделах конечностей [34], триггером может выступать повышение температуры окружающей среды. Помимо этого, дисфункция тонких миелиновых и безмиелиновых волокон может вызывать симпатические и парасимпатические расстройства. Симпатическая дисфункция объясняет гипогидроз, непереносимость физических упражнений и перегревания [35]. В отличии от нейропатии при других болезнях, при болезни Фабри страдает температурная чувствительность при сохранной вибрационной и тактильной [36]. Нейропатия при болезни Фабри также характеризуется поражением срединного нерва (в рамках туннельного синдрома), дисфункцией температурных афферентных волокон, при которой холодовая чувствительность нарушается больше, чем тепловая [37,38]. Описаны случаи нейропатии зрительного нерва [39—41]. Нейропатия тонких нервных волокон иногда может быть единственным симптомом болезни Фабри в зрелом возрасте [42]. Прямая зависимость между степенью выраженности боли и функционированием тонких безмиелиновых волокон отсутствует. По мере старения пациента и развития болезни боль может уменьшаться, в то время как функционирование волокон будет продолжать ухудшаться [43, 44].
Гетерозиготные пациентки с болезнью Фабри, даже без клинической манифестации заболевания, имеют периферическую нейропатию, подтверждаемую биопсией [45], однако при электронейромиографии патология может не выявляться [26]. Могут наблюдаться нарушения сна, обструктивное апное во время сна и связанные с нейропатической болью синдром «беспокойных ног» [46, 47]. Могут отмечаться кохлеовестибулярные расстройства, дисфункция вегетативной нервной системы [48, 49]. Пациенты имеют замедленные походку и движения рук. Однако у них отсутствуют двигательные экстрапирамидные симптомы, значительные когнитивные нарушения, гипоосмия, ортостатическая недостаточность, расстройства сна на стадии быстрого движения глаз, которые обычно имеются при нейродегенеративных заболеваниях.
Многие пациенты с болезнью Фабри находятся в состоянии постоянного стресса, связанного с болями, соматической патологией и психосоциальной стигматизированностью [50] они более подвержены депрессии, сонливы днем, имеют низкое качество жизни. Иногда отмечается наличие и более выраженной психической патологии, суицидальных попыток [50—52]. Однако выраженные когнитивные нарушения для болезни Фабри не характерны [53].
Сексуальная сфера у мужчин и автономная регуляция сердечно-сосудистой системы при болезни Фабри не страдают [54, 55].
Симптомы начинают проявлятся в детстве, а диагноз часто ставится только в развернутой стадии болезни у взрослых [56, 57]. В среднем проходит примерно 10 лет от начала появления симптомов до постановки диагноза [58]. Очень важно поставить правильный диагноз в детстве, чтобы начать раннюю заместительную терапию [59].
С болезнью Фабри связано одновременное поражение многих органов и систем [60]. Болезнь может мимикрировать под другую патологию, например, рассеянный склероз [61]. При наличии полиорганной патологии, необходимо шире подходить к проблеме и лечить не десяток одновременно протекающих болезней, а сосредоточиться на наследственном заболевании [62—65].
При наличии инсульта в раннем возрасте можно заподозрить болезнь Фабри, особенно при криптогенной этиологии, вертебробазилярной локализации, коморбидной дисфункции почек [66, 67].
Одним из ранних патогномоничных симптомов является мутовчатая кератопатия, легко обнаруживаемая при рутинном осмотре глаза с помощью щелевой лампы [68, 69]. Специфичными офтальмологическими проявлениями болезни Фабри являются патология сосудов конъюнктивы и сетчатки, помутнение роговицы и хрусталика. Как правило, они не вызывают значительного нарушения зрения, однако могут выступать в качестве маркеров болезни. Глаз является наружным органом легко доступным для изучения. Без применения инвазивных технологий. Он может использоваться для мониторирования протекания болезни и контроля за результатами лечения [70].
Дерматологическими проявлениями болезни Фабри являются ангиокератомы (см. рис.), телангиоэктазии, лимфодема, ангидроз или гипогидроз, псевдоакромегалические черты лица [71, 72]. Пациенты часто имеют характерный внешний вид, напоминающий больных с акромегалией — грубые черты лица, утопленный лоб, густые брови, выступающие супраорбитальные дуги и лобные бугры, выступающую нижнюю челюсть, увеличенные губы, широкую переносицу, грушевидный нос.
Снижение слуха отмечается у 90% пациентов. После 44 лет почти ни один больной не имеет нормального слуха [73].
Золотым стандартом диагностики болезни Фабри является определение активности лизосомного фермента α-галактозидазы, А в плазме, лейкоцитах, сыворотке крови, сухих пятнах крови, слезной жидкости, в любом биоптате или культуре клеток кожных фибробластов. У мужчин сниженная активность α-галактозидазы, А является достаточно информативным признаком болезни. Однако у 1/3 женщин с болезнью Фабри, а также у здоровых носителей активность этого фермента может быть нормальной, поэтому надежно исключить или подтвердить диагноз позволяет только генетический тест (секвенирование гена GLA). Также у всех гемизиготных и свыше 90% гетерозиготных носителей имеется в моче повышенный уровень Gb3. Для всех мужчин с пониженной активностью α-галактозидазы, А или атипичными симптомами болезни измерение уровня Gb3 в моче исеквенирование гена GLA являются обязательными [74]. При наличии мутации у пробанда необходимо проведение обследования всех его родственников, потенциально являющихся носителями мутантной X-хромосомы. Для мониторинга состояния пациента может использоваться количественное определение сфинголипидов, так как на фоне успешной патогенетической терапии происходит снижение их содержания в плазме.
Очень информативна диагностика заболевания у новорожденных [75, 76]. Возможна пренатальная диагностика с определением активности лизосомного фермента α-галактозидазы, А и секвенирование гена GLA в ворсинках хориона или культивированных амниотических клетках. Возможна предимплантационная диагностика [77].
Учитывая, что сильная нейропатическая боль, гипогидроз, нарушение холодовой перцепции характерны для болезни Фабри, их инструментальное тестирование может служить для оценки эффективности проводимого лечения [78, 79]. Для оценки степени выраженности и прогрессирования болезни у взрослых пациентов применяются также соответствующие шкалы: Mainz Severity Score Index (MSSI) и Fabry Outcome Survey Mainz Severity Score Index (FOS-MSSI) [80], а также стандартные опросники качества жизни пациентов [81].
Диффузионно-тензорная томография (ДТ МРТ, более чувствительна, по сравнению с рутинной МРТ, для обнаружения изменений в мозговой ткани при болезни Фабри. Она позволяет оценивать динамику болези и эффективность проводимой терапии [82]. С возрастом на МРТ обнаруживается ухудшение состояния нервной ткани [83].
При наличии органной патологии может применяться биопсия [84—86].
При болезни Фабри применяют симптоматическую терапию (обезболивающие препараты, гипотензивные средства, диализ, трансплантацию почек, искусственный водитель ритма, антидепрессанты) [87]. Для профилактики инсультов могут использоваться статины [88]. Для купирования нейропатической боли эффективны карбамазепин, фенитоин, габапентин [89].
Начиная с 2001 г. проводят фермент-заместительную терапию (ФЗТ) рекомбинантными препаратами α-галактозидазы, А [90]. Она позволяет уменьшить нейропатическую боль [91—97], улучшить функционирование различных типов нервных волокон иинтрадермальных рецепторов вибрации [98], также отмечается улучшение функции потооделения, переносимость жары, температурная чувствительность [99, 100]. Наблюдается положительная динамика при лечении синдрома беспокойных ног [101]. Кроме того при проведении заместительной терапии отмечается улучшение функционирования сердца [102—104]. Лечение эффективно при гипертрофии левого желудочка, [105, 106]. Заместительная терапия эффективна при легком и умеренном поражении почек, но неэффективна в тяжелых случаях (при протеинурии >1 г/сутки и грубом снижении уровня клубочковой фильтрации) [107—109]. При раннем начале лечения отмечается улучшение функционирования ЖКТ [110—112].
У пациентов с поздними стадиями болезни Фабри — при выраженном фиброзе и необратимом поражении органов, ФЗТ может быть неэффективна [113, 114], хотя есть данные [115], что ее раннее начало позволяет предотвратить поражение внутренних органов.
У некоторых больных, в ответ на введение данных препаратов, вырабатываются антитела класса IgG, что влияет на безопасность и эффективность лечения. При введении агалзидазы бета антител вырабатывается больше, чем при введении агалзидазы альфа. Антител класса IgE к агалсидазе альфа выявлено не было [122]. Лечение агалсидазой альфа в течении 10 лет предупреждает прогрессирование и уменьшает проявление кардиомиопатии при болезни Фабри, а также снижает накопление GB3 в миокарде [123]. При проведении ФЗТ происходят улучшения в течение заболевания, качестве жизни пациентов, а также снижается смертность. Согласно данным Программы оценки исходов болезни Фабри (FOS), медиана выживаемости у мужчин, получавших агалсидазу альфа в течении 5 лет составила 77,5 года, в сравнении с 60 годами у мужчин в контрольной группе, которые не получали заместительной ферментной терапии [124].
В настоящее время мужчинам с болезнью Фабри заместительная терапия показана сразу после установления диагноза. У мальчиков с бессимптомной болезнью Фабри лечение может быть начато в возрасте 10—13 лет, в то время как при наличии симптомов откладывать заместительную терапию не следует. У женщин показаниями к лечению считают выраженные симптомы или признаки прогрессирующего поражения органов-мишеней, в том числе хроническую нейропатическую боль в кистях и стопах, резистентную к стандартной терапии, персистирующую протеинурию, снижение скорости клубочковой фильтрации
В настоящее время развиваются новые терапевтические направления — лечение белками-шаперонами и генная терапия [126—134]. Также проводятся исследования комбинации сапосина В с модифицированной α-N-ацетил-галактозаминидазой [135]. Также рассматривается возможность использования энзимных активаторов и молекул, модифицирующих активность гена GLA [136].
Диагностика и комплексное лчение пациентов с болезнью Фабри требуют мультидисциплинарного подхода, включающего генетическое тестирование, интерпретацию тестов, генетическое консультирование, долговременное мониторирование симптомов болезни, выработку рекомендаций по лечению, координацию терапии проводимой врачами разных специальностей [137].
Болезнь Фабри: комплексная диагностика и лечение в Германии
Болезнь Фабри — это очень редкая патология, симптомы которой часто путают с другими схожими недугами. Большинство признаков специфичны для нарушений гормонального фона, дерматитов, аллергических реакций организма и прочего. Однако болезнь Фабри диагностируют по ряду симптомов и признаков, характерных как для взрослых, так и детей.
К симптомам болезни Фабри относятся:
- жгучая боль в области ладоней и стоп;
- острые болевые приступы (так называемые кризы Фабри);
- проблемы с потоотделением;
- непереносимость резких температурных проявлений (жары, холода);
- кожные проявления (ангиокератомы) ;
- ослабление зрения из-за помутнения роговицы;
- ухудшение слуха, шум в ушах;
- расстройство желудочно-кишечного тракта;
- кардиологическая симптоматика (нарушение сердечного ритма);
- почечная недостаточность;
- нарушение работы нервной системы и пр.
Болезнь Фабри: причины возникновения и характерные проявления
Болезнь Фабри является наследственным заболеванием, проявляющемся в результате нарушения обмена веществ, обусловленного недостатком фермента альфа-галактозидазы. Заболевание ведет к накоплению конечных продуктов обмена веществ в различных органах.
У пациента проявляется широкий спектр различных симптомов, за счет чего болезнь трудно диагностировать на протяжении многих лет. В начале появляются боли в пальцах рук и ног, непереносимость жары, значительно снижается потоотделение. Отложения в почках приводят к необходимости диализа, а в кровеносных сосудах — к риску возникновения инсульта или инфаркта.
Наиболее ранний и самый заметный признак болезни Фабри — красно-фиолетовые папулы на кожных покровах, абсолютно безболезненные. По мере взросления пациента их количество и размеры увеличиваются.
Неврологические проявления при болезни Фабри более свойственны пациентам старшего возраста. К таковым симптомам относятся головокружение, мигрени, нарушение кровообращения головного мозга.
Диагностика болезни Фабри
Диагностировать болезнь Фабри можно в ходе комплексного клинического обследования пациента в результате сложных лабораторных анализов и исследований (МРТ, ЭКГ).
В рамках лабораторной диагностики используют:
- энзимодиагностику — оценку уровня альфа-галактозидазы (при болезни Фабри у женщин активность данного фермента в крови практически равна норме);
- определение количества сфинголипидов;
- генодиагностику (исследование мутированного гена GLA).
Исследования данного типа успешно проводят специалисты в Германии, в Университетской клинике г. Фрайбурга. Необходимость комплексного анализа связана с:
- более точным определением болезни Фабри (в частности, на ее ранних стадиях);
- определением степени риска передачи генетической патологии при беременности;
- прогнозами развития болезни Фабри в конкретном случае;
- выбором индивидуальных методов лечения.
Важная роль в диагностике патологии отведена скринингу болезни Фабри в так называемых группах риска.
Особенности лечения болезни Фабри
Пациентам с диагностированной болезнью Фабри назначают ферментозаместительную терапию. В качестве лечения используются два препарата, замещающих фермент, которые вводят внутривенно или в виде инъекций каждые 14 дней. Это очень дорогостоящее лечение должно проводиться в течение всей жизни.
В лабораториях научной группы Университетской клиники г. Фрайбурга ведутся молекулярно-генетические исследования гена альфа-галактозидазы (GALA), мутацией которого обусловлена болезнь Фабри.
Результаты комплексных клинических исследований говорят об эффективности именно раннего лечения патологии, пока не сформировались тяжелые и необратимые поражения сердца, почек и других внутренних органов.
Своевременно назначенное лечение улучшает качество жизни пациентов с болезнью Фабри. Терапия с использованием инновационных препаратов позволяет снизить степень некоторых проявлений генного заболевания, приостановить, а то и полностью прекратить прогрессирование внутренних поражений.
Читайте также: