Болезнь Гиппеля-Линдау
Добавил пользователь Cypher Обновлено: 29.01.2026
Синдром Хиппеля-Линдау (ВVon Hippel-Lindau hereditary cancer syndrome (VHL) (церебро-ретино-висцеральный ангиоматоз, OMIM193300) - аутосомно-доминантный семейный опухолевый синдром, предрасполагающий к образованию различных злокачественных и доброкачественных опухолей. Наиболее часто при данном синдроме наблюдаются гемангиобластомы сетчатки, мозжечка и спинного мозга, carcinoma (RCC) почек, феохромоцитомы, опухоли поджелудочной железы. Кроме того, могут встречаться цисты в почках и поджелудочной железе, изменения в лёгких и печени. Эти опухоли характеризуются высокой васкуляризацией и избыточной продукцией VEGF и других ангиогенных белков, а также избыточной продукцией эритропоэтина, что может привести к увеличению продукции эритроцитов и, как следствие, к полицитемии. Синдром VHL - один из вариантов цилиопатий — генетически обусловленных заболевания, возникающие при нарушении структуры или функции цилий. Мутации в гене VHL приводят к разным типам заболевания: тип 1 характеризуется низким риском развития феохромоцитомы, тип 2 - высоким риском развития феохромоцитомы. При этом тип 2 делится ещё на три подтипа: 2А характеризуется низким риском развития RCC, 2В - высоким риском развития RCC, а при типе 2С развивается только феохромоцитома без гемангиобластом и RCC. Гемангиобластомы развиваются при типах 1, 2А и 2В. Мутации, приводящие к развитию синдрома VHL типа 1, представляют собой в основном микроделеции/вставки, нонсенс-мутации и крупные делеции (56% заболевших). К 96% больных типа 2 приводят миссенс-мутации, а мутации в кодоне 238 ответственны за развитие 43% опухолей при типе 2.
VHL - ген-супрессор опухолевого роста (VHL GENE), расположен на хромосоме 3 в регионе 3р25.3, содержит 3 экзона.
Мутации в данном гене приводят также к развитию семейного эритроцитоза тип 2, феохромоцитоме, а соматические мутации в данном гене приводят к развитию мозжечковой гемангиобластоме и почечноклеточной карциноме.
Наиболее часто при данном синдроме наблюдаются гемангиобластомы сетчатки, мозжечка и спинного мозга. Клинически проявляются головными болями в области затылка, головокружением, рвотой, атаксией, нистагмом, дизартрией, спастическими парапарезами. Следствием гемангиомы задней черепной ямки может быть гидроцефалия.
Типичен рак почек, феохромоцитомы, опухоли поджелудочной железы. Кроме того, могут встречаться цисты в почках и поджелудочной железе, изменения в легких и печени. В некоторых случаях выявляются гемангиомы лица, надпочечников. Эти опухоли характеризуются высокой васкуляризацией и избыточной продукцией VEGF и других ангиогенных белков, а также избыточной продукцией эритропоэтина, что может привести к увеличению продукции эритроцитов и, как следствие, к полицитемии.
Поражение глаз проявляется ангиомами сетчатки. Заболевание прогрессирует и осложняется отслойкой сетчатки, катарактой, глаукомой и слепотой.
Первые симптомы появляются на втором-четвертом десятилетии жизни, смерть наступает в среднем в 41 год.
Болезнь Гиппеля-Линдау: диагностика и современное лечение
Болезнь Гиппеля-Линдау — это редкая нейрокутанная патология, характеризующаяся полиморфным опухолевым синдромом. Первичная симптоматика болезни чаще всего проявляется наличием церебеллярных кист (кисты мозжечка). Болезнь Гиппеля-Линдау (VHL) является наследственным заболеванием, симптомы которого проявляются, как правило, в возрасте от 15 до 40 лет. Болезнь Гиппеля-Линдау характеризуется образованием опухолей в различных органах: ангиомы сетчатки глаза, гемангиобластомы мозжечка, мозгового ствола и спинного мозга, карциномы почки, феохромоцитомы, кисты в поджелудочной железе и опухоли из островковых клеток, опухоли внутреннего уха, придатков яичка и широкой связки матки.
При своевременном обнаружении опухоли хорошо поддаются лечению, поэтому очень важно проходить регулярные обследования. Зарекомендовавший себя молекулярно-генетический анализ гена VHL открывает новые возможности в выявлении, а также исключении наследственной предрасположенности к заболеванию.
Симптоматика VHL-синдрома
Заболевание Гиппеля-Линдау является наследственным в 80% случаев, а в 20% патология развивается на фоне новой генной мутации. Манифестация неврологических симптомов наблюдается в возрасте 30-40 лет, тогда как у детей недуг проявляется в виде неврологических симптомов на фоне имеющихся зрительных расстройств. Иногда болезнь сопровождается субарахноидальным кровоизлиянием.
Среди характерных общемозговых симптомов VHL-синдрома отмечаются:
- резкие головные боли;
- плохой аппетит, тошнота, рвота;
- повышенное внутричерепное давление;
- ухудшение слуха.
На ранних этапах часто наблюдаются и другие симптомы болезни Гиппеля-Линдау: эпилептические приступы, которые носят генерализованный либо фокальный характер. Постепенно проявляется симптомокомплекс, сигнализирующий о мозжечковой атаксии. Возникают проблемы с глотанием, диплопия, дизартрия. О спинальных опухолях свидетельствует отсутствие рефлексов сухожилий, корешковые синдромы, нарушения глубокой чувствительности.
Признаки болезни Гиппеля-Линдау выявляются также в ходе проведения офтальмоскопии. Глазные поражения на первичных стадиях развития патологии объясняются потерей резкости зрения, искажением изображения.
Увеличение ангиомы сетчатки, которое развивается постепенно, вызывает нарушение кровообращения, ишемический синдром, кистозную дегенерацию. На поздних стадиях часто наблюдаются катаракта, внутриглазное давление, отслоение сетчатки.
Важность диагностики болезни Гиппеля-Линдау
При наличии любого из признаков болезни Гиппеля-Линдау необходимо срочно пройти комплексную диагностику. Людям из группы риска в рамках профилактических мероприятий следует проходить осмотр у специалистов раз в год.
Постановка диагноза возможна только после обследований у офтальмолога, невропатолога и ряда других специалистов в силу коплексности заболевания. С целью выявления у пациента церебеллярных образований, врач назначает магнитно-резонансную (компьютерную) томографию участков головного мозга. Определить опухоли других локализаций позволяет ультразвуковое исследование, а для выявления генных мутаций назначается ДНК-диагностика.
Современное лечение болезни Гиппеля-Линдау
Лечение при установлении болезни Гиппеля-Линдау носит симптоматический характер. Терапия направлена на устранение опухолевых образований, недопущение возникновения их в будущем, а также улучшение качества жизни пациента с VHL-синдромом.
Современные инвазивные методы позволяют проводить эффективное удаление опухолей с максимальным сохранением нарушенных функций органа практически на любой стадии развития болезни.
Специалисты Университетской клиники г. Фрайбурга уже более 30 лет занимаются исследованиями синдрома Гиппеля-Линдау. Во Фрайбурге обслуживается большое количество пациентов с этим заболеванием. Для пациентов и их близких также создан портал группы взаимопомощи, содержащий подробную информацию о болезни и возможных терапиях.
Лечение состояний, связанных с VHL-синдромом, назначается индивидуально на основе комплексного обследования. Мы предлагаем нашим пациентам современную диагностическую базу, позволяющую точно классифицировать патологию и, в результате, подобрать наиболее эффективный протокол терапии.
Болезнь Гиппеля-Линдау
- Главная /
- Редкие болезни /
- Энциклопедия заболеваний /
- Болезнь Гиппеля Линдау
Болезнь Гиппеля-Линдау ( VHL ), Синдром Гиппеля-Линдау
Von Hippel–Lindau disease , Von Hippel–Lindau syndrome
OMIM – 193300 МКБ10 – Q 85.8
Болезнь Гиппеля-Линдау классифицируется как одно из более 7000 редких заболеваний, выявленных на сегодняшний день. Частота заболеваемости среди населения 1/25000-50000. VHL причисляется к наследственным (Генетическим) заболеваниям и наследуется аутосомно-доминантным путем (~80% случаев).
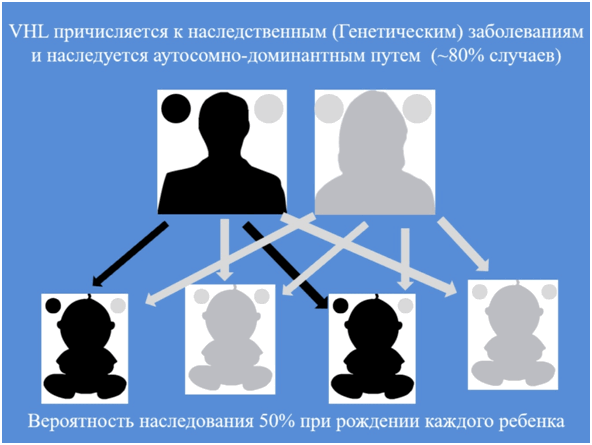
В 20% случаев заболевание носит спорадический характер (Мозаичный кариотип - как правило следствие мутаций на ранних стадиях эмбриогенеза). У некоторых пациентов данной группы генетическое тестирование не выявило мутаций в половых клетках, несмотря на то, что в соматических клетках мутации были подтверждены и как следствие у данных людей рождается здоровое потомство. Нельзя исключить также появление подобных случаев вследствие мозаицизма гонад у родителей (Как правило, в результате мутаций на поздних стадиях эмбриогенеза). У данных пациентов вероятность передачи мутантного гена потомству составляет 50%.
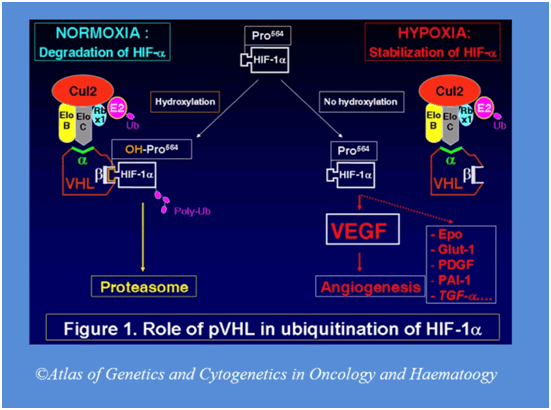
Генетика: Ген VHL расположен в 3-й хромосом (регион 3 p 25.3.), содержит 213 аминокислот и способствует выроботке белка pVHL , который является в целом ингибитором опухолевого роста (Ингибитором ангиогенеза).

Белок pHVL взаимодействует с 3-мя белками: Elongin B , Elongin C , Cullin 2 ( CUL 2) и играет ключевую роль в инактивации HIF -1 a . Также pVHL снижает продукцию таких факторов ангиогенеза как VEGF . Мутации в гене VHL приводят к нарушению синтеза данного белка, и отсутствие его ингибирующего влияния запускает каскад патологических процессов, что в итоге приводит к различным соматическим проявлениям.
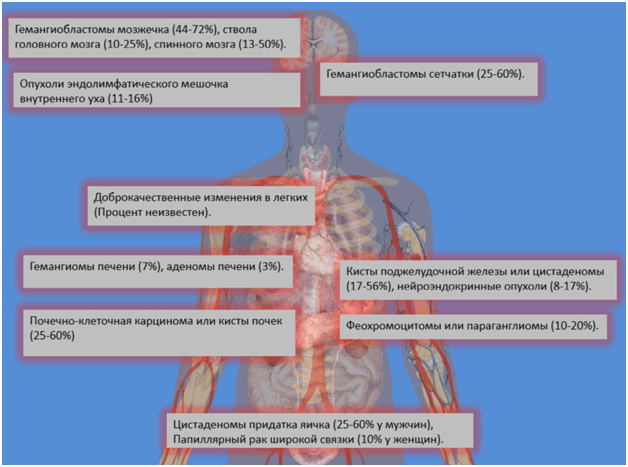
Клинические проявления: Основными проявлениями заболевания являются
- Гемангиобластомы мозжечка, ствола головного мозга, позвоночного канала,
- Гемангиобластомы сетчатки,
- Новообразования эндолимфатичесого мешочка внутреннего уха,
- Гемангиомы печени, Аденомы печени,
- Феохромоцитомы или параганглиомы,
- Кисты поджелудочной железы, цистаденомы или нейроэндокринные опухоли,
- Кисты почек, почечно-клеточная карцинома,
- Цистаденомы придатка яичка (У мужчин), Папиллярный рак широкой связки (У женщин),
- Доброкачественные изменения в легких.
Клиническая картина заболевания зависит от количества и степени вовлеченности «органов мишеней», и зачастую сильно варьирует даже у пациентов из одной семьи.
Манифестация: Манифестация заболевания может происходить практически в любом возрасте. Заболевание может начаться с поражения любого из «органов мишеней». Следует отметить, что VHL длительное время может протекать бессимптомно и время постановки диагноза не может считаться временем манифестации у большинства пациентов.
Типы заболевания: Различают несколько вариантов заболевания, которые протекают с вовлечением органов мишеней в различных комбинациях.
Диагностические мероприятия при подозрении на VHL
- Генетическое тестирование
- Нейровизуализация (МРТ головного мозга, в том числе с фокусировкой внимания на область внутреннего уха, МРТ шейного, грудного, пояснично-крестцового отделов позвоночника)
- Флюоресцентная ангиография, офтальмоскопия (Можно также провести дополнительные методы обследования: УЗИ, ЦДК, ОКТ)
- КТ брюшной полости с контрастным усилением
- Тест на фракционированные метанефрины, в частности выявление свободного норметанефрина в плазме крови или в анализе суточной мочи.
Лечение: Патогенетического лечения болезни Гиппеля-Линдау на данный момент нет. Наблюдением и лечением пациентов должна заниматься мультидисциплинарная команда специалистов, со знанием особенностей течения данного заболевания.
- Тактика лечения гемангиобластом головного мозга
- Хирургическое лечение
- Радиохирургия ( Cyber Knife )
- Наблюдение
- Тактика лечения гемангиобластом сетчатки
- Основным методом лечения является транспупиллярная лазерная коагуляция ангиоматозного узла/капиллярной гемангиомы (Эффективность высока при размерах опухоли до 3-х мм).
- Также применяются - Криотерапия, брахитерапия, наружная лучевая терапия, транпупиллярная термотерапия, фотодинамическая терапия
- В случае развития осложнений применяется (Отслойка сетчатки) - Витроретинальная хирургия.
- Тактика лечения поражений брюшной полости
- Кисты и цистаденомы поджелудочной железы обычно не требуют хирургического вмешательства
- PNET : Одним из возможных методов лечения является хирургический, В настоящий момент идет пересмотр рекомендация по лечению PNET .
- Почечно-клеточная карцинома: Основной метод лечения хирургический, возможно применение радиочастотной абляции и криохирургии.
- Феохромоцитомы: Хирургическое лечение ( Laparoscopic partial adrenalectomy ) с дальнейшим подбором консервативной терапии.
Образ жизни: Важное значение для качества жизни пациентов имеют - питание, физическая активность, эмоциональное состояние.
Наблюдение пациентов: осуществляется мультидисциплинарной командой, в зависимости от проявлений заболевания. Координируют деятельность команды
Болезнь Гиппеля-Линдау
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Болезнь фон Гиппеля-Линдау (VHL-синдром)
ФГУ Эндокринологический научный центр, Москва
Болезнь фон Гиппеля-Линдау является наследственным опухолевым синдромом, предполагающим развитие различных доброкачественных и злокачественных новообразований (гемангиобластома центральной нервной системы и сетчатки глаза, опухоль внутреннего уха, карцинома и кисты почек, феохромоцитома, нейроэндокринная опухоль и кисты поджелудочной железы, цистаденома придатка яичка у мужчин и широкой связки у женщин). Болезнь фон Гиппеля-Линдау - наиболее распространенная причина наследственного рака почки.
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
В 1895 г. Ю. фон Гиппель [1] описал пациента с ретинальной ангиомой, а в 1926 г. А. Линдау [2] — пациента с ретинальной ангиомой и гемангиоматозом центральной нервной системы. Год спустя тот же автор обнаружил ассоциацию этих проявлений с почечными и панкреатическими кистами [3]. Термин «синдром von Hippel—Lindau» (VHL) был введен Мелмоном и Роузеном [4]. Этот синдром выявляется приблизительно у 1 из 36 000 человек [5] и обусловлен мутацией в участке 3p25/26, где локализован ген подавления роста опухоли VHL [6—8]. 23% пациентов не имеют семейного анамнеза заболевания [9—13].
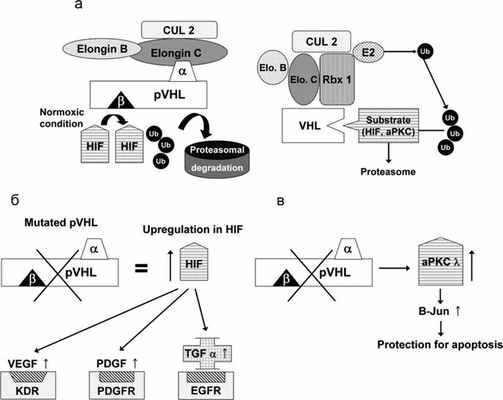
Ген VHL был идентифицирован в 1993 г. [8, 14]. Приблизительно у 20% пациентов выявляется делеция VHL-локуса в материнской или отцовской аллели [15, 16]. Герминальные мутации VHL наследуются по аутосомно-доминантному типу. Почти все мутации VHL у пациентов с феохромоцитомой являются миссенс-мутациями. Ген VHL состоит из 3 экзонов. Белок VHL (pVHL) включает 213 аминокислотных остатков, и его молекулярная масса равна приблизительно 28 кДa. Клетки с дефицитом pVHL накапливают фактор, индуцирующий гипоксию (HIF), что приводит к перепроизводству HIF-зависимых продуктов (которые вовлечены в адаптацию к гипоксии): сосудистого эндотелиального фактора роста (VEGF), эритропоэтина и трансформирующего ростового фактора альфа (TGF). Это объясняет сильную васкуляризацию VHL-ассоциированных опухолей [17—19]. Таким образом, продукт мутированного гена VHL приводит к сверхрегулированию различных генов, участвующих в патогенезе гипоксии, ускоряет ангиогенез, изменяет внеклеточный матрикс и регуляцию клеточного цикла [20—28]. Однако точные механизмы туморогенеза при синдроме VHL в настоящее время остаются неизвестными (рис. 1). Рисунок 1. Комплекс VBC (VHL protein and Elongin B, C) и механизм его действия. а — Normoxic condition — нормальное давление кислорода; Ub — убиквитин; HIF — фактор, индуцирующий гипоксию; pVHL — VHL-протеин; α — элонгин C-связывающий домен pVHL; β — β-домен (субстратсвязывающий домен) pVHL; CUL2 — куллин-2, формирующий комплекс с элонгином-B, C и pVHL; б — мутация гена VHL и, как результат, отсутствие регуляции HIF, VEGF, PDGF и TGF-α; в — мутация гена VHL и отсутствие регуляции aPKC λ (атипичная протеинкиназа C) [29].
VHL-синдром характеризуется развитием гемангиобластом сетчатки глаза (ангиомы сетчатки) и центральной нервной системы (ЦНС), билатеральной и мультифокальной дифференцированной карциномы почки, поликистоза почек, феохромоцитомы, кист и нейроэндокринных опухолей поджелудочной железы, папиллярной цистаденомы придатка яичка у мужчин и широкой связки у женщин, опухолей внутреннего уха. Поражение различных органов и степень этого поражения очень вариабельны (табл. 1, 2).

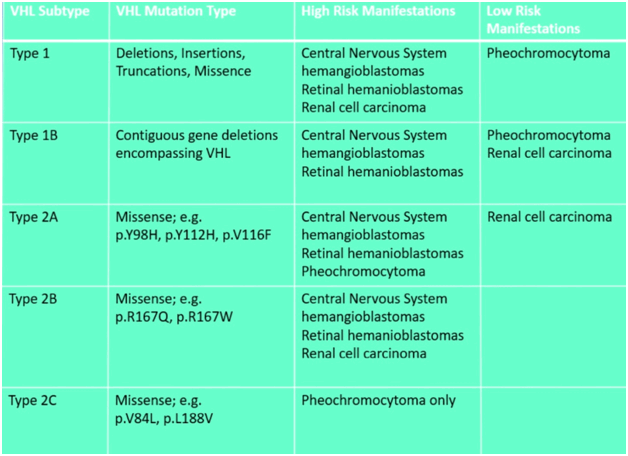
Клинически заболевание делится на две группы. Тип 1 включает главным образом большие делеции или мутации гена VHL и характеризуется полным фенотипом заболевания [поражение сетчатки, кисты или опухоли головного и спинного мозга, панкреатические, почечные, и селезеночные кисты, солидные панкреатические опухоли (реже аденокарциномы), карциномы почек, цистаденомы эпидидимуса и опухоль внутреннего уха], но без феохромоцитомы. Тип 2, при котором развивается феохромоцитома (миссенс-мутации гена VHL может иметь и неполный фенотип [13, 37, 40]. Тип 2 подразделяется на подтипы с низким (тип 2A) и высоким (тип 2B) риском развития рака почки, а также тип 2C, проявляющийся только феохромоцитомой [35, 38, 41—43] (табл. 3).
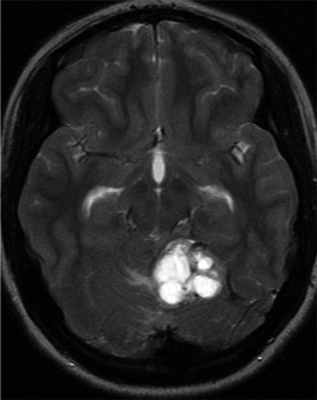
Гемангиобластомы ЦНС могут выявляться в детском возрасте, однако средний возраст диагностирования составляет 29 лет [32, 45, 46]. VHL-ассоциированные гемангиобластомы выявляются в среднем на 15 лет раньше, чем спорадические [47]. В зависимости от размера и местоположения опухоли клинические признаки гемангиобластомы ЦНС включают головную боль, тошноту, головокружение, атаксию, расстройство координации движений, нистагм, расстройства речи. Гемангиобластома спинного мозга может приводить к слабости конечностей и парестезиям. Диагноз устанавливается с помощью МРТ головного мозга и позвоночника. Гемангиобластомы обычно характеризуются медленным ростом и имеют высокий риск кровотечений, часто являются мультифокальными. Понимание патогенеза заболевания важно для выбора оптимального времени скрининга на опухоли и лечение [19]. Исследование тканей ЦНС умерших пациентов помогло пониманию гистогенеза гемангиобластом [49]. Активация фактора, индуцирующего гипоксию 2-альфа (HIF 2-α) происходит в маленьких мезенхимальных опухолях и в мезенхимальном компоненте больших опухолей. Активация HIF 1-α наблюдается в эпителиальном компоненте. Это позволило предполагать, что поражение ЦНС при VHL-синдроме — длительный процесс гемангиобластической пролиферации и дифференцировки [50] (рис. 2). Рисунок 2. Гемангиобластома ЦНС.
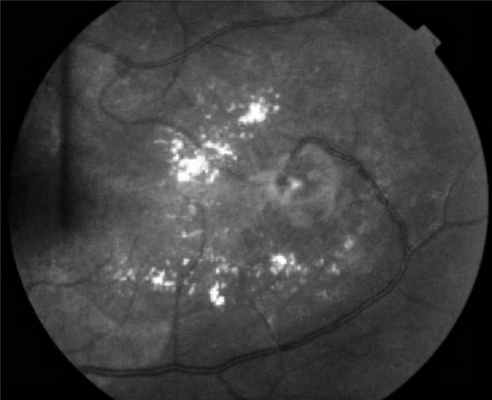
Поражения глаз выявляются примерно у 37% пациентов с VHL-синдромом, среди них только у 14% обнаруживается полная делеция VHL [51, 52]. Приблизительно у 8% пациентов снижена острота зрения [53]. Для лечения ангиомы сетчатки используют лазерную или криотерапию [32, 34, 54]. Недавние исследования [ 55, 56] показали, что при внутривенном введении антагониста сосудистого эндотелиального фактора роста (anti-VEGF) в течение 7 мес размер гемангиобластом не уменьшается (рис. 3). Рисунок 3. Ангиоматоз сетчатки.
У пациентов с синдромом VHL могут встречаться как кисты, так и рак почек [57—62]. Средний возраст манифестации — 37 лет. Для диагностики используют КТ и УЗИ [36, 63, 64]. Поскольку со`лидные раки могут содержать кистозные части (что затрудняет дифференцирование доброкачественных и злокачественных процессов с помощью визуализирующих методик), при отсутствии данных о метастазах лечение должно быть направлено на удаление этих образований по возможности с соблюдением принципа органосохраняющей операции. Это позволяет поддерживать почечную функцию максимально долго и избежать диализа [58, 65]. Опухоли почек отличаются медленным ростом (3 см (по стандартам США) или 5 см (по стандартам Европы) [58, 60, 67, 68]. Некоторые авторы [69] сообщают о высоком риске местного рецидива (приблизительно 50%) в среднем в течение 53 мес (диапазон 10—115 мес) и росте опухоли со скоростью 34 мм/год (диапазон 1—10,8 мм). «Золотым» стандартом лечения небольших опухолей является открытая и лапароскопическая частичная нефрэктомия. В настоящее время используются также альтернативные методы — криотерапия и радиочастотная аблация [70]. Последние методы могут повлиять на результат патоморфологического диагноза, хотя, по некоторым данным, патоморфологический диагноз после первого цикла криотерапии приблизительно в 91% случаев подтверждает результаты предварительной биопсии [71].
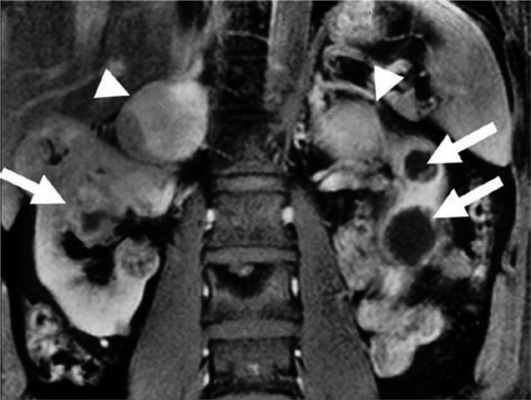
Феохромоцитома выявляется примерно у 26% пациентов с синдромом VHL [37]. У пациентов с очевидно спорадической феохромоцитомой в 3—11% случаев впоследствии выявляют мутацию VHL [10, 12, 13]. Феохромоцитома может быть первым проявлением синдрома [30, 72]. В большинстве случаев надпочечниковые феохромоцитомы при VHL-синдроме двусторонние (синхронные или метахронные) [37, 73]. Вненадпочечниковые феохромоцитомы встречаются примерно в 30% случаев [37, 74—76]. Феохромоцитомы как часть синдрома VHL имеют исключительно норадреналиновый фенотип. Биохимические маркеры опухоли могут помочь отличить VHL-ассоциированные феохромоцитомы от феохромоцитом при синдроме МЭН 2-го типа [75]. Различия в биохимическом фенотипе при VHL-синдроме и МЭН 2-го типа связаны с различной экспрессией тирозингидроксилазы (TH) — лимитирующего фермента синтеза катехоламинов, и фенилэтаноламин-N-метилтрансферазы (PNMT). У пациентов с синдромом VHL отмечена низкая экспрессия PNMT, преобразующей норадреналин в адреналин. Различия биохимического фенотипа также связаны с различиями хранения, транспорта и секреции катехоламинов [77]. МЭН 2-ассоциированные феохромоцитомы содержат более высокие концентрации катехоламинов из-за более выраженной экспрессии TH. VHL-ассоциированные феохромоцитомы, секретируют катехоламины непрерывно, тогда как при синдроме МЭН 2 отмечен эпизодический характер секреции. Это определяет и различия клинических проявлений двух синдромов. Например, пациенты с МЭН 2 чаще жалуются на кризовые подъемы АД [78]. Помимо генетических различий [26], регистрируется разная экспрессия эритропоэтина и его рецептора [79]. Кроме того, около 80% феохромоцитом бессимптомны и выявляются случайно при визуализирующих исследованиях. Низкая чувствительность некоторых радионуклидных методов визуализации может объясняться относительной нехваткой гранул хранения или уменьшенной экспрессией мембранного норадреналина или везикулярных моноаминных транспортеров [80]. Поэтому сцинтиграфия с 123 I-MIBG (метайодбензилгуанидином) часто не обнаруживает VHL-связанные надпочечниковые феохромоцитомы [81, 82]. ПЭТ с 6-18F-фтордопамином более чувствительный метод [36, 83]. Злокачественные феохромоцитомы с метастазами в легких, печени, костях, лимфоузлах редко встречаются при синдроме VHL [37, 74, 84—87]. Метастазы выявляются менее чем в 7% случаев [37]. К сожалению, в настоящее время нет четких признаков, позволяющих надежно отличить доброкачественную от злокачественной феохромоцитомы, хотя уже известно, что герминальная мутация гена SDHB, является в этом отношении точным маркером [86—88]. Выявление феохромоцитомы у пациентов с синдромом VHL особенно важно, учитывая высокую вероятность хирургических вмешательств по поводу других опухолей (гемангиобластом ЦНС и др.). Невыявленная феохромоцитома при других вмешательствах может привести к опасным для жизни гипертоническим кризам. Более 70% феохромоцитом у детей являются VHL-ассоциированными. Каждому пациенту с VHL-синдромом и подтвержденной феохромоцитомой до оперативного лечения необходимо проводить ПЭТ с 6-18F-фтордопамином или сцинтиграфию с 123 I-MIBG для выявления вненадпочечниковой феохромоцитомы или метастазов [89]. Лечение феохромоцитомы оперативное. В то же время 6-месячная терапия ингибиторами тирозинкиназы приводит к уменьшению опухоли на 21% и сокращению уровня норметанефринов и хромогранина А в плазме [90] (рис. 4). Рисунок 4. Двусторонняя феохромоцитома и поликистоз почек.
Цистаденомы эпидидимуса — доброкачественные опухоли, которые могут быть двусторонними [48]. Чаще они имеют около 2 см в диаметре, могут распространяться на семенной канатик, приводя к бесплодию. Хирургическое лечение обычно не проводят; необходимо наблюдение [92].
У 35—75% пациентов с синдромом VHL имеются доброкачественные кисты и микрокистозные аденомы поджелудочной железы [93—96]. По данным КТ, у 17% пациентов выявляются панкреатические нейроэндокринные опухоли [95, 97, 98]. Из 633 пациентов с VHL-синдромом у 108 (17%) обнаруживались панкреатические эндокринные опухоли, и у 9 (8%) были выявлены метастазы [99]. Метастазирование более вероятно, если размеры опухоли превышают 3 см. У 78% пациентов с метастазами (7 из 9) мутация локализовалась в экзоне 3 гена VHL, и удвоение массы опухоли в среднем происходило за 337 дней. Таким пациентам необходимо оперативное лечение. Если размеры опухоли меньше 3 см, она растет медленно, а мутация локализована не в экзоне 3, то можно ограничиться наблюдением за пациентом. Средний возраст диагностики нейроэндокринных опухолей поджелудочной железы — 35 лет. Панкреатические кисты встречаются у больных начиная с 15 лет и чаще являются бессимптомными. В зависимости от размера и местоположения клинические симптомы могут быть вызваны обструкцией желчных путей и/или ферментной недостаточностью. В таких случаях устанавливают желчный стент и/или назначают ферментные препараты.
Гормонально-активные нейроэндокринные опухоли встречаются редко [95, 100]. По данным последних исследований, смертность при панкреатических эндокринных опухолях составляет 6%. В 60% случаев при сцинтиграфии обнаруживают рецепторы соматостатина; злокачественные опухоли выявлялись в 58% случаев [101].
Опухоли внутреннего уха располагаются в лабиринте, под твердой мозговой оболочкой на задней поверхности пирамиды височной кости [102—104]. Эти опухоли практически не метастазируют [105]. Симптомы включают потерю слуха, звон в ушах, головокружение и/или парез лицевого нерва [106, 107]. Таким образом, пациентам с мутацией VHL абсолютно показаны аудиологический осмотр, а также КТ или МРТ внутреннего уха с высокой разрешающей способностью, так как раннее хирургическое вмешательство способно сохранить слух. У пациентов с двусторонними опухолями, приводящими к глухоте, слух может быть восстановлен кохлеарным имплантом [108].
У 90% носителей мутации VHL к 60-летнему возрасту имеются те или иные клинические проявления синдрома [45]. На долгосрочный прогноз и смертность обычно влияет наличие гемангиобластом сетчатки и ЦНС, а также карциномы почки на поздних стадиях [31—33]. Таким образом, своевременное обследование и выявление патологии, ассоциированной с VHL-синдромом, является залогом успешного лечения и увеличения продолжительности жизни пациента (табл. 4).
ИСТИНА
Болезнь Гиппеля-Линдау (Von Hippel-Lindau disease) доклад на конференции
- Авторы: Симонян А.С., Сперанская Е.В., Есин А.И., Новикова А.Ю., Петров Р.В., Аджемян Н.А., Егоров В.И.
- Всероссийская Конференция : III-я Научно-Практическая конференция «Актуальные вопросы современной Неврологии»
- Даты проведения конференции: 12 октября 2017
- Дата доклада: 12 октября 2017
- Тип доклада: Устный
- Докладчик: не указан
- Место проведения: Москва, Россия
- Аннотация доклада:
Болезнь Гиппеля-Линдау (VHL) классифицируется как одно из более 7000 редких заболеваний, выявленных на сегодняшний день. В 1927г Шведский патологоанатом Арвид Линдау впервые описал гемангиобластомы в головном и в спинном мозге. Данный научный труд привлек внимание одного из основоположников современной Нейрохирургии – Харви Уильямса Кушинга, который предложил назвать заболевание в честь Арвида Линдау. Долгое время заболевание именовалось в честь Арвида Линдау, но в 1964г научное мировое сообщество обратило внимание на труды Немецкого офтальмолога Эжена фон Гиппеля в которых в 1904г он впервые описал гемангибластомы глаз. Оба ученых в своих работах описывали разные проявления одного и того же патологического состояния, и сегодня с учетом заслуг Эжена фон Гиппеля и Арвида Линдау данная нозология именуется «болезнью Гиппеля - Линдау» В 20% случаев заболевание носит спорадический характер (Мозаичный кариотип - как правило следствие мутаций на ранних стадиях эмбриогенеза). У некоторых пациентов данной группы генетическое тестирование не выявило мутаций в половых клетках, несмотря на то, что в соматических клетках мутации были подтверждены и как следствие у данных людей рождается здоровое потомство. Нельзя исключить также появление подобных случаев вследствие мозаицизма гонад у родителей (Как правило, в результате мутаций на поздних стадиях эмбриогенеза). У данных пациентов вероятность передачи мутантного гена потомству составляет 50% Ген VHL расположен в 3-й хромосом (регион 3p25.3.), содержит 213 аминокислот и способствует выроботке белка pVHL, который является в целом ингибитором опухолевого роста (Ингибитором ангиогенеза). Белок pHVL взаимодействует с 3-мя белками: Elongin B, Elongin C, Cullin 2 (CUL2) и играет ключевую роль в инактивации HIF-1a. Также pVHL снижает продукцию таких факторов ангиогенеза как VEGF. Мутации в гене VHL приводят к нарушению синтеза данного белка, и отсутствие его ингибирующего влияния запускает каскад патологических процессов, что в итоге приводит к различным соматическим проявлениям. Частота заболеваемости VHL одинакова среди женского и мужского населения. Манифестация заболевания может происходить практически в любом возрасте. Заболевание может манифестировать с поражения любого из «органов мишеней». Следует отметить что VHL длительное время может протекать бессимптомно и время постановки диагноза не может считаться временем манифестации у большинства пациентов. Клиническая картина заболевания зависит от количества и степени вовлеченности «органов мишеней», и зачастую сильно варьирует даже у пациентов из одной семьи. Диагностические мероприятия при подазрении на VHL Генетическое тестирование Нейровизуализация (МРТ головного мозга, в том числе с фокусировкой внимания на область внутреннего уха, МРТ шейного, грудного, пояснично-крестцового отделов позвоночника) Флюоресцентная ангиография, офтальмоскопия (Можно также провести дополнительные методы обследования: УЗИ, ЦДК, ОКТ) КТ брюшной полости с контрастным усилением Тест на фракционированные метанефрины, в частности выявление свободного норметанефрина в плазме крови или в анализе суточной мочи. Патогенетического лечения болезни Гиппеля-Линдау на данный момент нет. Наблюдением и лечением пациентов должна заниматься мультидисциплинарная команда специалистов, со знанием особенностей течения данного заболевания.
Читайте также: