Диагностика пантотенаткиназа-ассоциированной нейродегенерации по КТ, МРТ
Добавил пользователь Алексей Ф. Обновлено: 30.01.2026
Является наиболее распространенным типом нейродегенерации с накоплением железа в головном мозге (NBIA) - редкое нейродегенеративное расстройство, характеризующееся прогрессированием экстрапирамидной дисфункции (дистония, ригидность, хореоатетоз), накоплением железа в мозге и наличием сферических аксонов в центральной нервной системе.
Распространенность оценивается в 1-2 /1000000. Наследуется аутосомно-рецессивно.
Вызывается мутациями гена PANK2 (20p13-p12.3).
Классическая форма пантотенаткиназа-ассоциированной нейродегенерации (75% случаев) характеризуется ранним началом, как правило, до шести лет и быстрым прогрессированием. Атипичная форма (25% случаев) имеет позднее начало, от 13 до 14 лет, и более медленное прогрессирование. При классическом варианте синдрома, пациенты обращаются с нарушениями походки и частыми падениями, которые обусловлены дистонией, ригидностью, нарушениями равновесия и спастичностью, обычно они теряют способность самостоятельно передвигаться через 10-15 лет после начала заболевания. Эпизоды быстрого прогрессирования заболевания могут включать в себя status dystonicus, перемежаются с более длительными периодами относительной стабильности. У пациентов наблюдается задержка развития, в первую очередь двигательная. Часто развивается пигментная дегенерация сетчатки и дизартрия. Позже присоединются общие нарушения, которые включают дисфагию, желудочно-пищеводный рефлюкс, хронический запор, аспирационную пневмонию и недоедание. При атипичном варианте болезни Галлервордена-Шпатца пациенты обращаются с затрудненной речью, мягкими нарушениями походки, рядом психиатрических симптомов, которые могут включать в себя депрессию, эмоциональную лабильность, импульсивность или вспышки насилия, пигментной дегенерацией сетчатки (реже, чем при классической форме) и вербальным и двигательным туреттизмом. Потеря подвижности происходит в течение 15-40 лет от начала заболевания. HARP синдром (гиперпребатилопопротеинемия, акантоцитоз и пигментный ретинит) также относится к спектру пантотенаткиназа-ассоциированной нейродегенерации.
Болезнь Галлервордена-Шпатца обычно подозревают при обнаружении на МРТ классического симптома "глаз тигра", гиперинтенсивности в центральной области, окруженной обоком гипоинтенсивности на корональных или поперечных Т2-взвешенных изображениях бледного шара. Генетический анализ необходим для подтверждения диагноза.
Пренатальная диагностика возможна, если обе болезнетворные мутации были выявлены у заболевшего члена семьи.
Дифференциальный диагноз включает болезнь Вильсона и другие виды нейродегенерации с накоплением железа, которые могут быть дифференцированы благодаря МРТ и генетическому тестированию.
Лечение направлено на облегчение симптомов, используют баклофен (перорально или с помощью интратекального насоса), тригексифенидил для снижениия дистонии и спастичности, ботулинический токсин в случаях, когда можно повысить качество жизни путем инъекций на ограниченных участках тела. Пациенты обычно не реагируют на применение леводопы. Глубокая стимуляция мозга может облегчить течение некоторых симптомов. Диета, кормление через гастростому и удаление зубов (в случаях с тяжелой оробукколингвальной дистонии) также могут потребоваться пациентам с пантотенаткиназа-ассоциированной нейродегенерацией.
Болезнь Галлервордена-Шпатца является прогрессирующим заболевание, потерянные навыки, как правило, не восстанавливаются. Скорость прогрессирования коррелирует с возрастом начала заболеевания. Продолжительность жизни разнится, но часто наступает преждевременно.
Детская нейроаксональная дистрофия
Синонимы: инфантильная нейроаксональная дистрофия, болезнь Зейтельбергера (Seitelberger)
Определение и общие сведения
Детская нейроаксональная дистрофия является одним из видов нейродегенерации с накоплением железа в мозге, характеризуется задержкой и регрессом психомоторного развития с признаками вовлечения симметричного пирамидного тракта и спастической тетраплегии. Выделяют классическую и атипичную форму детской нейроаксональной дистрофии.
Распространенность неизвестна, более 150 случаев было описано, большинство из которых являются классическим вариантом детской нейроаксональной дистрофией.
Этиология и патогенез
Детская нейроаксональная дистрофия вызывается мутациями гена PLA2G6 (22q13.1). Мутации изменяют метаболизм фосфолипидов и часто приводят к патологическому накопление железа в базальных ганглиях.
Клинические проявления
Классическая форма обычно манифестирует в возрасте от шести месяцев до трех лет задержкой и регрессом психомоторного развития, поздним началом ходьбы или нарушением походки. Характеризуется ранней стволовой гипотонией прогрессирующей в тетрапарез (как правило, спастический, но может быть арефелекторный) и слабоумием. Также наблюдаются косоглазие, маятниковый нистагм, несогласованные движения глаз, атрофия зрительного нерва и отсутствии зрения. Редко возникают судороги.
Манифестация атипичной формы детской нейроаксональной дистрофии, как правило, происходит в раннем детстве, но может возникать в подростковом возрасте, развитие ее протекает более медленно. Наблюдаются задержка речи и нейроповеденческие нарушения, в виде импульсивности, плохой концентрации внимания и эмоциональной лабильности. Тетрапарез возникает поздно и ему не обязательно предшествует клиника стволовой гипотонии, пациенты, как правило, демонстрируют прогрессирующую дистонию и дизартрию. Атрофия зрительного нерва, нистагм и судороги соответствуют картине классической формы заболевания.
Диагностика
У большинства пациентов с обеими формами болезни Зительбергера развивается мозжечковая атрофия, заметная на МРТ уже в относительно раннем возрасте. Это, плюс высокое содержание железа в головном мозге или атрофия зрительного нерва, предполагает диагноз. Генетическое тестирование его подтверждает.
Дифференциальный диагноз
Дифференциальный диагноз включает инфантильный нейрональный цероидный липофусциноз, атаксию-телеангиэктазию и наследственную атаксию (мозжечковая атрофия обычно проявляется позже у этих расстройств), пантотенаткиназа-ассоциированную нейродегенерацию (характерный "глаз тигра" знак не наблюдается при младенческой нейроаксональной дистрофии), инфантильный GM2 ганглиозидоз, болезнь Нимана-Пика типа C, аутизм и болезнь Менкеса.
Лечение паллиативное и включает в себя терапию спастичности и судорог, баклофен внутрь или интратекально при значительной дистонии, физиотерапевтическое лечение, кормление через желудочный зонд и трахеостомию для предотвращения аспирационной пневмонии. Хелатотерапия препаратами железа настоящее время не рекомендуется.
Прогрессирование классической детской нейроаксональной дистрофии происходит, как правило, быстро и многие дети никогда не научаются ходить. Тяжелая спастичность, прогрессивное снижение когнитивных функций и нарушение зрения может привести в развитию вегетативного состояния у ребенка. Многие пациенты не доживают до 10 лет, но некоторые доживают до подросткового возраста и старше. Продолжительность жизни при атипичной форме как правило выше.
Диагностика пантотенаткиназа-ассоциированной нейродегенерации по КТ, МРТ
Пантотенаткиназа-ассоциированная нейродегенерация (ПКАН) на МРТ
а) Терминология:
• Пантотенаткиназа-ассоциированная нейродегенерация (ПКАН):
о Наиболее частая форма нейродегенерации с отложением железа в головном мозге (НОЖМ)
о Вызывается мутацией гена пантотенаткиназы 2 (PANK2)
б) Визуализацияпантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Лучший диагностический критерий: симптом «глаза тигра» = диффузное снижение интенсивности сигнала на Т2-ВИ от бледных шаров с гиперинтенсивными очагами в их медиальных отделах:
о Очень характерен для ПКАН
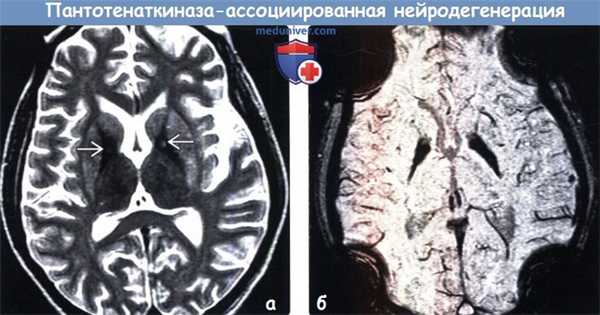
(а) МРТ, Т2-ВИ, аксиальный срез: у пятилетнего ребенка с диагнозом детский церебральный паралич определяется характерный для пантотенаткиназа-ассоциированной нейродегенерации (ПКАН) симптом «глаза тигра»: симметричные гиперинтенсивные очаги в медиальных отделах гипоинтенсивных бледных шаров.
(б) МРТ-исследование того же пациента четыре года спустя по поводу дистонии, Т2-ВИ, аксиальный срез: отмечается уменьшение интенсивности сигнала от «глаз» и их размеров на фоне снижения интенсивности сигнала от окружающих отделов бледных шаров. На момент исследования очевидная потеря объема мозговой ткани, особенно в лобных долях. (а) МРТ, Т2-ВИ, корональный срез: у этого же пациента в возрасте 9 лет определяется аномальное снижение интенсивности сигнала от бледных шаров и черной субстанции.
(б) МРТ, Т2* GRE, аксиальный срез: у того же пациента в возрасте девяти лет определяется эффект «выцветания» изображения в виде гипотензивного сигнала от бледных шаров, что обусловлено парамагнитными свойствами железа. Выявленные изменения характерны для эволюции классического пантотенаткиназа-ассоциированной нейродегенерации (ПКАН): уменьшение калибра «глаз», усиление степени снижения интенсивности сигнала от окружающих отделов бледных шаров и прогрессирование потери объема мозговой ткани.
в) Дифференциальная диагностика:
• Нарушения с ↑ интенсивности сигнала на Т2-ВИ от бледных шаров:
о Метаболического характера: метилмалоновая ацидемия, синдром Кернс-Сейр, L-2-гидроксиглутаровая ацидурия, болезнь Канавана, нейроферритинопатия
о Ишемического/токсического характера: аноксическая энцефалопатия, отравление монооксидом углерода/цианидом, ядерная желтуха
г) Клиническая картина пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Классическая форма пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
о Дистония, дизартрия, мышечная ригидность, хореоатетоз у ребенка младшего возраста
• Атипичная форма пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
о Психические, речевые, пирамидные/экстрапирамидные нарушения у детей старшего возраста/подростка
• Эпидемиология:
о Редко; встречаемость неизвестна
• Прогноз:
о Классическая форма ПКАН: фатальный прогноз; средняя продолжительность жизни после начала симптомов составляет 11 лет
о Атипичная форма ПКАН: возможны тяжелые нарушения/ле-тальный исхол
• Эффективное лечение отсутствует
Диагностика нейродегенерации с отложением железа в головном мозге (НОЖГМ) по КТ, МРТ
а) Терминология:
1. Сокращения:
• Пантотенаткиназа-ассоциированная нейродегенерация (ПКАН)
• Инфантильная нейроаксональная дистрофия (ИНАД)
• Нейродегенерация с отложением железа в головном мозге (НОЖГМ)
2. Определение:
• Группа нейродегенеративных нарушений, характеризующаяся дистонией, паркинсонизмом и мышечной спастичностью:
о Вызывается мутациями в гене L-ферритина FTL1
о Все мутации приводят к аномальному накоплению Fe в базальных ганглиях
о Включает ПКАН, ИНАД, ацерулоплазминемию и т.д.:
- Тельца Леви, набухание аксонов, гиперфосфорилированные тау-белки при некоторых подтипах
б) Визуализация:
1. Общие характеристики нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Лучший диагностический критерий:
о Снижение интенсивности сигнала на Т2-ВИ от бледных шаров (БШ)
• Локализация:
о ПКАН и ИНАД:
- БШ, черная субстанция (ЧС) ± зубчатые ядра (ЗЯ)
о Нейроферритинопатия и ацерулоплазминемия:
- БШ, ЧС, ЗЯ, кора, полосатое тело и таламус
2. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТс Т2-ВИ* (градиентноеэхо) или изображения, взвешенные по восприимчивости магнитного поля (SWI)
3. КТ признаки нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Бесконтрастная КТ:
о Атрофия больших полушарий и мозжечка при PANK2-негативной нейродегенерации с отложением железа в головном мозге (НОЖГМ)
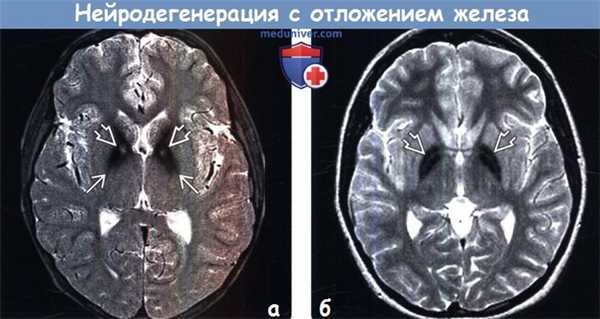
(а) МРТ, Т2-ВИ, режим подавления сигнала от жира: определяется классическая визуализационная картина при ПКАН (синдром Галлервордена-Шпатца), которая входит в спектр нейродегенерации с отложением железа в головном мозге (НОЖГМ). Обратите внимание на снижение интенсивности сигнала от бледных шаров, обусловленное отложением в них железа, на фоне чего визуализируются центральные гиперинтенсивные очаги — симптом «глаза тигра», очень специфичный для мутации PANK2.
(б) МРТ, Т2-ВИ, аксиальный срез: определяется симметричное снижение интенсивности сигнала от бледных шаров. Симптом «глаза тигра» отсутствует, вследствие чего данное состояние не относится к ПКАН. Выявленные изменения характерны для нейродегенерации с отложением железа в головном мозге (НОЖГМ).
в) Дифференциальная диагностика нейродегенерации с отложением железа в головном мозге (НОЖГМ):
1. Нормальные процессы отложения железа:
• Визуализируется на ЗТ
• Наблюдается при нормальном процессе старения
2. Болезни Паркинсона и Альцгеймера:
• Симптом «глаза тигра» отсутствует; пожилые пациенты
3. Рассеянный склероз (PC):
• Накопление железа в базальных ганглиях связано с PC
• Должны быть другие классические демиелинизирующие поражения
4. Поверхностный сидероз:
• Перегрузка железом вследствие переливания крови или рецидивирующих кровоизлияний в ЦНС
5. Гемохроматоз:
• Поражение печени и селезенки обычно происходит до поражения ЦНС
г) Патология:
1. Общие характеристики нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Генетика:
о Аутосомно-рецессивные мутации генов PANK2, PLA2G6 и СР
о Аутосомно-доминантная мутация гена FTL
2. Макроскопические и хирургические особенности:
• Накопление железа, пигментация цвета ржавчины
3. Микроскопия:
• Накопление железа в БШ и ЧС, потеря нейронов, набухание аксонов (сфероиды)
• Симптом «глаза тигра» может отражать кистозную дегенерацию
• Наличие нейрофибриллярных сплетений и тел Леви предполагает схожий с болезнями Альцгеймера и Паркинсона патогенез
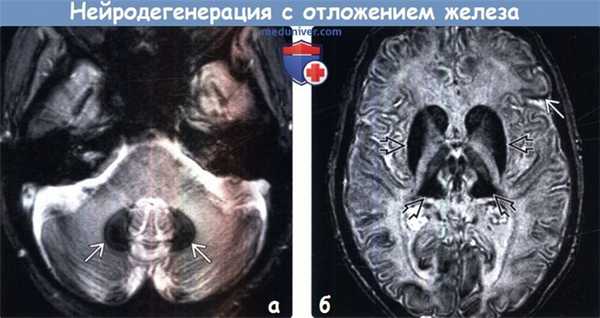
(а) МРТ, Т2* GRE, аксиальный срез: у пациента с ацерулоплазминемией определяется выраженное снижение интенсивности сигнала от зубчатых ядер мозжечка вследствие отложения в них железа. Ацерулоплазминемия и нейроферритинопатия встречаются у взрослых и имеют аналогичные визуализационные признаки.
(б) МРТ, Т2*SWI, аксиальный срез: у того же пациента определяется диффузно распространенное линейное снижение интенсивности сигнала от коры с выраженным эффектом «выцветания» изображения в базальных ганглиях и таламусах, что обусловлено отложением железа. Также типично вовлечение в процесс и черной субстанции.
д) Клиническая картина:
1. Проявления нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Наиболее частые признаки/симптомы:
о Атаксия, дизартрия, дистония
о Дегенерация сетчатки и атрофия зрительных нервов
• Другие признаки/симптомы:
о Ацерулоплазминемия: дебют во взрослом возрасте с триадой, включающей диабет, дегенерацию сетчатки и двигательные нарушения
о Нейроферритинопатия: дебют во взрослом возрасте с хореей или дистонией
2. Демография:
• Возраст:
о ПКАН: классический вариант- возраст < 6 лет, дебют в подростковом возрасте не характерен
о ИНАД: классический вариант-возраст < 2 лет, дебют в возрасте 4-6 лет не характерен
о НОЖГМ с дебютом во взрослом возрасте: средний возраст = 40 лет
2. Течение и прогноз:
• ПКАН и ИНАД: причины летального исхода разнятся, обычно вторичные причины, такие как как истощение и аспирация
• НОЖГМ с дебютом во взрослом возрасте: прогрессирующее ухудшение двигательного статуса
3. Лечение нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Симптоматическое лечение: лекарственные препараты и глубокая стимуляция головного мозга
• Ацерулоплазминемия: хелатная терапия дефероксамином и церулоплазмином из свежезамороженной плазмы
а) Терминология:
1. Сокращения:
• Пантотенаткиназа-ассоциированная нейродегенерация (ПКАН)
2. Синонимы:
• Нейродегенерация с отложением железа в головном мозге 1 типа (НОЖГМ-1)
• Синдром Галлервордена-Шпатца:
о ПКАН и НОЖГМ-1 = предпочтительные термины
3. Определение:
• Нейродегенерация с отложением железа в головном мозге (НОЖГМ) = зонтичный термин, который охватывает нейродегене-ративные расстройства, характеризующиеся накоплением железа в головном мозге:
о К известным причинам ее развития относятся ПКАН (наиболее частое нарушение), ацерулоплазминемия, нейроферритинопатия и инфантильная нейроаксональная дистрофия
• ПКАН вызывается мутацией гена пантотенаткиназы 2 (PANK2)
1. Общие характеристики пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Лучший диагностический критерий: симптом глаза тигра = диффузное снижение интенсивности сигнала на Т2-ВИ от бледных шаров с гиперинтенсивными очагами в их медиальных отделах:
о Очень характерен для ПКАН
о Гиперинтенсивный сигнал «глаз» может предшествовать снижению интенсивности сигнала от окружающих бледных шаров
о По мере прогрессирования заболевания ↓ калибр «глаз» и интенсивность сигнала от них
о По мере прогрессирования заболевания интенсивность сигнала от бледных шаров снижается
о Симптом «глаз тигра» также описан при нейроферритинопатии
• Вариабельное i интенсивности сигнала на Т2-ВИ от черной субстанции > > зубчатых ядер
• По мере прогрессирования заболевания развивается атрофия мозговой ткани
• Локализация: бледные шары (БШ), черная субстанция (ЧС), зубчатые ядра (ЗЯ)
• Морфология: изменения сигнальных характеристик бледных шаров напоминают глаза тигра
• Отложение железа (в ферритине) является причиной снижения интенсивности сигнала от мозговых структур на Т2-ВИ
2. КТ признаки пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Бесконтрастная КТ: вариабельный характер изменений; гипо-, гиперденсные или нормальные БШ
• КТ с контрастированием: патологическое накопление контраста отсутствует
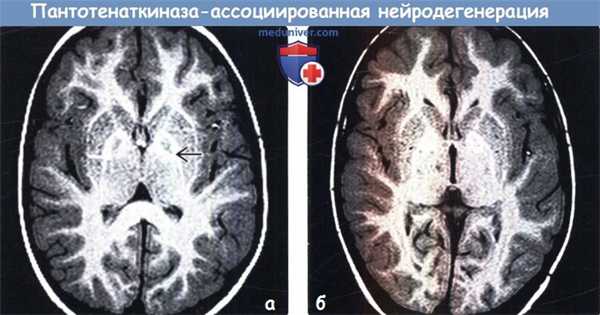
(а) МРТ, Т1 -ВИ, аксиальный срез: у пятилетнего ребенка с классической формой пантотенаткиназа-ассоциированной нейродегенерации (ПКАН) определяется, что «глаза тигра» гипоинтенсивен и окружен несколькими точечными участками гиперинтенсивного сигнала.
(б) МРТ, Т1-ВИ, аксиальный срез: у этого же пациента в возрасте девяти лет определяется, что «глаза» имеют преимущественно гиперинтенсивный сигнал. Выраженность «глаз» при симптоме «глаза тигра» вариабельна и зависит от стадии заболевания. Прогрессирующее по мере развития заболевания отложение железа в бледных шарах, вероятно, отражается в укорочения времени Т1.
4. Радионуклидная диагностика:
• ОФЭКТ с Tc-99m: ↑ активности в медиальных отделах БШ
о Возможна хелатация Тс-99m с помощью цистеина в бледных шарах
5. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ
• Советы по протоколу исследования:
о Рассмотрите возможность получения SWI или Т2* GRE изображений для оценки минерализации
о Снижение интенсивности сигнала на Т2-ВИ более заметно на спин-эхо последовательностях (по сравнению с быстрыми спин-эхо последовательностями) и при использовании магнитов с большей напряженностью поля
в) Дифференциальная диагностика пантотенаткиназа-ассоциированной нейродегенерации (ПКАН). Нарушения с ↑ интенсивности сигнала на Т2-ВИ от бледных шаров:
• Метаболического характера:
о Метилмалоновая ацидемия (ММА): ↑ интенсивности сигнала на Т2-ВИ от БШ ± перивентрикулярного белого вещества (БВ) о Синдром Кернс-Сейр/L:-2-гидроксиглутаровая ацидурия: Т интенсивности сигнала на Т2-ВИ от БШ (в > степени по сравнению с другими глубокими серыми структурами) и периферического БВ
о Болезнь Канавана: ↑ интенсивности сигнала на Т2-ВИ от БШ (в > степени по сравнению с другими глубокими серыми структурами) и субкортикального БВ; макроцефалия; ↑ пика NAA
о Нейроферритинопатия: гиперинтенсивные на Т2-ВИ очаги вариабельных размеров в БШ, скорлупе, головках хвостатых ядер со ↑ интенсивности сигнала на Т2-ВИ от ЧС, ЗЯ; заболевание взрослых
о Недостаточность гуанидиноацетатметилтрансферазы (нарушение синтеза креатина)
• Ишемического/токсического характера:
о Аноксическая энцефалопатия: ↑ интенсивности сигнала на Т2-ВИ от БШ (и других глубоких серых структур), а также коры о Отравление монооксидом углерода: ↑ интенсивности сигнала на Т2-ВИ от БШ (± других глубоких серых структур, коры, БВ)
о Отравление цианидом: ↑ интенсивности сигнала на Т2-ВИ от базальных ганглиев, за которым следует геморрагический некроз
о Ядерная желтуха: ↑ интенсивности сигнала на Т2-/Т1-ВИ от бледных шаров у новорожденных
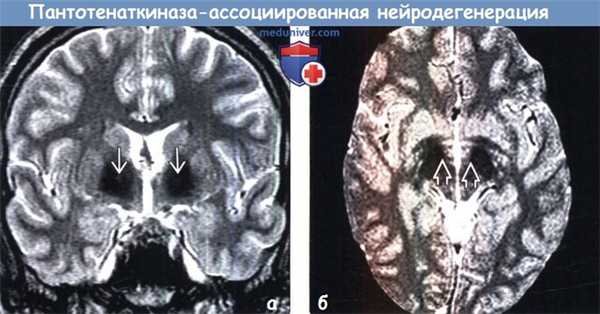
(а) МРТ, Т2-ВИ, корональный срез: у пациента с классической формой пантотенаткиназа-ассоциированной нейродегенерации (ПКАН) определяется классический симптом «глаза тигра» с мелкими гиперинтенсивными очагами, расположенными в медиальных отделах бледных шаров и окруженными аномальным гипоинтенсивным сигналом от последних.
(б) МРТ, Т2* GRE, аксиальный срез: у пациента с классической формой пантотенаткиназа-ассоциированной нейродегенерации (ПКАН) определяется эффект «выцветания» в виде гипоинтен-сивного сигнала от нижних отделов бледных шаров и черной субстанции. При диагностической визуализации аномальное отложение железа в черной субстанции становится более заметным по мере прогрессирования заболевания.
1. Общие характеристики пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Отложение железа при ПКАН, вероятно, является вторичным процессом:
о При динамическом MPT-наблюдении за пациентами с ПКАН было выявлено, что появление гиперинтенсивных очагов в БВ предшествовало снижению интенсивности сигнала от последних
• Эмбриология, анатомия:
о Встречается прогрессирующее физиологическое отложение железа в головном мозге в БШ, ЧС > красных и зубчатых ядрах о ↓ интенсивности сигнала на Т2-ВИ от БШ в норме выявляется у большинства пациентов в возрасте >25 лет, но никогда у детей до 10 лет
• Генетика:
о Аутосомно-рецессивное наследование (в 50% случаев спорадический характер)
о Идентифицировано > 100 мутаций гена PANK2 в локусе 20р 12.3-р13:
- Симптом «глаза тигра», выявляемый при МРТ, сильно коррелирует с мутациями гена PANK2
- Ген PANK2 кодирует митохондриальнотаргетированную пантотенаткиназу 2, ключевой фермент в биосинтезе коэнзима А (КоА):
Помимо других его функций КоА необходим для энергетического обмена и метаболизма жирной кислоты
- Нулевые мутации чаще встречаются при раннем дебюте заболевания и быстро прогрессирующем его течении
- Миссенс-мутации чаще встречаются при позднем дебюте заболевания и более медленно прогрессирующем его течении
о Предполагают остаточную активность пантотенаткиназы 2 при заболевании с поздним дебютом (менее тяжелая форма)
о Синдром HARP: (гипопребеталипопротеинемия, акантоцитоз, пигментная дегенерация сетчатки и дегенерация бледных шаров:
- Аллелен ПКАН
- Выраженная орофациальная дистония; ранний дебют паркинсонизма
• Этиология:
о Ведущая теория:
- Сутация гена PANK2 → недостаточность КоА → дисгомеостаз энергии и липидов → образование свободных радикалов кислорода → разрушение фосфолипидных мембран
- Базальные ганглии и сетчатка уязвимы для окислительного поражения, поскольку имеют высокие метаболические потребности
о Дополнительные факторы:
- Накопление цистеина в БШ, обусловленное ↓ фосфопантотената вызывает хелатирование железа и пероксидативное поражение клеточных мембран
- Аксональные сфероиды еще больше компрометируют функцию глии и нейронное
2. Макроскопические и хирургические особенности:
• Симметричная пигментация БШ ржаво-коричневого цвета (медиальных > латеральных его отделов), и ретикулярной части ЧС: о В дополнение к накоплению железа пигментацию также обусловливает интра-/экстранейрональное отложение цероид-липофусцина и меланина
• Вариабельная выраженность атрофии
3. Микроскопия:
• Классические признаки:
о ↑ железа в медиальных отделах БШ и ретикулярной части ЧС
- Железо, расположенное в астроцитах, микроглиальных клетках, нейронах и вокруг сосудов
о Потеря нейронов, глиоз и глиальные включения в первую очередь развиваются во внутренних отделах БШ и ретикулярной части ЧС
о Округлые или овальные безядерные набухания аксонов («сфероиды») в БШ, ЧС, коре и головном мозге
• «Рыхлая» ткань (состоящая из реактивных астроцитов, дистрофических аксонов и вакуолей в передне-медиальных отделах БШ) соответствует «глазу» в симптоме «глаз тигра» на МР-томограммах
• Вариабельное количество акантоцитов (в мазке крови)
(а) МРТ по поводу паллидотомии у ребенка 12 лет с поздней стадией классической формы ПКАН, Т2-ВИ, аксиальный срез: гипоинтенсивные и атрофичные бледные шары со слабо выраженным симптомом «глаза тигра». Обратите внимание на диффузную потерю объема мозговой ткани.
(б) МРТ, SWI, аксиальный срез: у этого же пациента определяется эффект «выцветания» в виде гипотензивного сигнала от бледных шаров. Симптом «глаза тигра» больше не наблюдается, поскольку «затмевается» вышеупомянутым эффектом. SWI — более чувствительная по сравнению с Т2* GRE последовательность вследствие взвешенности по магнитной восприимчивости.
г) Клиническая картина:
1. Проявления пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Клиническая классификация на классическую и атипичную форму:
о Классическая форма ПКАН: ранний дебют, более быстрое прогрессирование, единообразный фенотип
о Атипичная форма ПКАН: поздний дебют, более медленное прогрессирование, гетерогенный фенотип
• Наиболее частые признаки/симптомы:
о Классическая форма ПКАН: дистония:
- Другие экстрапирамидные признаки/симптомы: дизартрия, мышечная ригидность, хореоатетоз
- Часто наблюдаются признаки/симптомы поражения верхних моторных нейронов и когнитивные нарушения
- Пигментная ретинопатия (66%)
о Атипичная форма ПКАН: психические и речевые нарушения:
- Другие признаки/симптомы: пирамидные/экстрапирамид-ные нарушения (включая феномен «застывания»), деменция
• Клинический профиль:
о Классическая форма ПКАН: ребенок младшего возраста с нарушениями походки, постуральной недостаточностью о Атипичная форма ПКАН: подросток с речевыми, психическими нарушениями
• Нормальный уровень железа в сыворотке и СМЖ
• При подозрении на ПКАН всегда следует проводить подтверждающий анализ на мутацию PANK2
2. Демография:
• Возраст:
о Классическая форма ПКАН: у большинства заболевание проявляется до возраста 6 лет
о Атипичная форма ПКАН: средний возраст при дебюте - 13 лет
• Эпидемиология: редкое заболевание; встречаемость неизвестна
3. Течение и прогноз:
• Анамнез:
о Классическая форма ПКАН: быстрое нелинейное прогрессирование (с периодами ухудшения, сменяющимися периодами стабилизации), ведущее к раннему наступлению летального исхода во взрослом возрасте
о Атипичная форма ПКАН: более медленнее прогрессирование с потерей способности ходить через 15-40 лет после дебюта заболевания
• Прогноз:
о Классическая форма ПКАН: фатальный; средняя продолжительность жизни после начала симптомов составляет 11 лет
о Атипичная форма ПКАН: возможны тяжелые нарушения ± летальный исход во взрослом возрасте
4. Лечение пантотенаткиназа-ассоциированной нейродегенерации (ПКАН):
• Эффективное лечение отсутствует; хелатирование железа неэффективно
• Паллиативная терапия:
о Баклофен, тригексифенидил часто неэффективны
о Стереотаксическая паллидотомия
о Перспективны первые результаты глубокой стимуляции бледных шаров
д) Диагностическая памятка. Советы по интерпретации изображений:
• Симптом «глаза тигра» очень характерен для ПКАН
• У подростка/взросых физиологическое снижение интенсивности сигнала от БШ трудно отличить от патологического
Болезнь Галлервордена-Шпатца ( Пантотенаткиназа-ассоциированная нейродегенерация )
Болезнь Галлервордена-Шпатца — нейродегенеративная наследственная патология, обусловленная отложением железа в базальных ганглиях головного мозга. Проявляется синдромом паркинсонизма, нарушениями интеллектуальной сферы и психики, гиперкинезами, зрительными расстройствами. Основное диагностическое значение имеет обнаружение рисунка «глаз тигра» в зоне бледного шара при проведении МРТ церебральных структур. Лечение симптоматическое: агонисты дофамина, вальпроаты, антиконвульсанты, нейролептики, антидепрессанты. Прогноз неблагоприятный.
МКБ-10
Общие сведения
Болезнь Галлервордена-Шпатца описана в 1922 г. немецкими морфологами, в честь которых и получила свое название. К наиболее типичным клиническим маркерам данной патологии относят гиперкинезы, синдром паркинсонизма, интеллектуальное снижение, атрофию зрительных нервов, пигментную ретинопатию. Болезнь Галлервордена-Шпатца встречается крайне редко. В зависимости от времени ее манифестации различают детскую, ювенильную (подростковую) и взрослую формы. Ранее заболевание диагностировалось лишь посмертно по данным аутопсии. После внедрения в практическую неврологию МРТ стала возможна прижизненная постановка диагноза. Прорыв в изучении этиологии был сделан в 2001 г., когда было установлено, что в основе заболевания лежит генетический дефект, обуславливающий нарушения в синтезе фермента пантотенаткиназы. После этого болезнь Галлервордена-Шпатца была официально переименована в пантотенаткиназа-ассоциированную нейродегенерацию.
Причины
Болезнь Галлервордена-Шпатца является генетической патологией, носящей как семейный, так и спорадический характер, передающейся по наследству аутосомно-рецессивным путем. Генетическим субстратом выступают аберрации в гене пантотенаткиназы (локус 20р12.3–р13 20-й хромосомы). Всего известно более 50 мутаций. Результатом генетического дефекта является уменьшение продукции пантотенаткиназы, что ведет к аккумуляции в базальных структурах цистеина. Последний образует устойчивые химические соединения с ионами железа, которые неблагоприятно воздействуют на белки и запускают процесс перекисного окисления, приводящий к апоптозу нейронов. На месте некротизированных нейронов происходит разрастание глиальной ткани.
Описанный патологический процесс затрагивает преимущественно бледный шар и черное вещество (субстанцию nigra), где морфологически обнаруживаются внеклеточные отложения железа, имеющие коричневую пигментацию. Кроме этого, имеют место сфероидные периаксональные образования, расположенные в белом церебральном веществе, коре мозга, спинном мозге и периферических нервных стволах.
Симптомы болезни Галлервордена-Шпатца
Классическим вариантом болезни Галлервордена-Шпатца считается ранняя детская форма с клинической манифестацией в период от 4 до 10 лет (обычно после 5-летнего возраста). В 90% случаев первым признаком заболевания выступает торсионная дистония, затрагивающая мышцы ног. Ведущей жалобой является затруднение ходьбы. Затем, как правило, происходит генерализация процесса с его распространением на мышцы глотки, лица, туловища. Наряду с генерализованными вариантами могут отмечаться мультифокальный или сегментарный тип дистонии. Наиболее часто наблюдается писчий спазм, блефароспазм, лицевой параспазм, спастическая кривошея. У трети пациентов отмечаются признаки паркинсонизма: мышечная ригидность и гипокинезия. В ряде случаев имеют место эпилептические приступы.
Болезнь Галлервордена-Шпатца характеризуется когнитивными расстройствами в виде снижения внимательности и памяти с постепенным развитием олигофрении; психическими изменениями с преобладанием агрессивности и асоциального поведения. Отмечается дизартрия. У большинства больных имеются нарушения остроты зрения. В 68% случаев они обусловлены атрофией зрительных нервов, в 29% случаев — пигментной ретинопатией. Для детской формы болезни Галлервордена-Шпатца типично быстрое прогрессирование с полной потерей в течение 10-15 лет способности к передвижению.
Подростковый вариант болезни Галлервордена-Шпатца проявляется в возрасте от 10 до 18 лет и характеризуется более замедленным течением. Дебютирует проявлениями фокальной торсионной дистонии, наиболее часто в мышцах конечностей или ортомандибулярной области. Сопровождается психическими, интеллектуальными и поведенческими расстройствами.
Диагностика
Благодаря полиморфизму симптоматики, постановка диагноза болезни Галлервордена-Шпатца представляет трудную задачу для неврологов. Основными критериями заболевания считаются дебют в возрасте до 30 лет, экстрапирамидные расстройства, неуклонное прогрессирование симптомов, наличие типичной МРТ-картины. К дополнительным признакам отнесены наличие пирамидных знаков, прогрессирующее интеллектуальное снижение, эпиприступы, атрофия зрительных нервов, пигментная атрофия сетчатки, аутосомное наследование по рецессивному типу.
В диагностике опираются на данные неврологического статуса и электроэнцефалографии. При нарушении зрения проводят консультацию офтальмолога, визиометрию, офтальмоскопию. Определение типа наследования осуществляет генетик путем составления генеалогического древа. Возможна ДНК-диагностика (поиск мутаций в гене пантотенаткиназы). При проведении ПЭТ головного мозга удается выявить сниженный метаболизм в зоне паллидума. Основанием для исключения болезни Галлервордена-Шпатца является наличие симптомов другой патологии, в рамки которой может укладываться имеющаяся клиническая картина: болезни Вильсона, хореи Гентингтона, нейроакантоцитоза, болезни Мачадо-Джозефа.
Основополагающим методом диагностики болезни Галлервордена-Шпатца выступает МРТ. Во всех типичных вариантах патологии в режиме Т2 на МРТ головного мозга определяется расположенная в области бледного шара гиперинтенсивная зона овальной формы, окруженная еще большей гипоинтенсивной зоной. Подобная МРТ-картина является патогномоничной и получила название «глаз тигра». Гипоинтенсивная зона представляет собой «глаз», а гиперинтенсивная — его «зрачок». Время появления этого признака на томограммах пока дискутируется. По мнению одних авторов «глаз тигра» может появляться еще до клинической манифестации болезни, по мнению других — через несколько лет от дебюта клинических симптомов.
Лечение болезни Галлервордена-Шпатца
В настоящее время болезнь Галлервордена-Шпатца не имеет эффективных методов лечения. Попытки терапии препятствующими накоплению железа хелатными соединениями (дефероксамином) и антиоксидантами не имели успеха. В связи с этим применяется симптоматическое лечение. Синдром паркинсонизма служит показанием к назначению дофаминовых агонистов (пирибедила, прамипексола) или производных амантадина. Однако при данном заболевании он, как правило, резистентен к проводимому лечению.
При гиперкинезах применяют вальпроаты, бензодиазепины (диазепам, клоназепам). При спастике рекомендованы миорелаксанты (баклофен, толперизона гидрохлорид), при эпиприступах — топирамат или вальпроаты, при когнитивных расстройствах — ипидакрин и холина альфосцерат, при психических отклонениях — нейролептики (рисперидон, кветиапин, клоназепам), антидепрессанты 3-го поколения (венлафаксин, циталопрам, дапоксетин).
Симптоматическая терапия болезни Галлервордена-Шпатца позволяет уменьшить проявленность клинических симптомов, продлить способность пациентов к самообслуживанию. Вместе с тем продолжается разработка новых способов лечения. Исследуется эффективность применения пантотеновой кислоты. Получены данные о положительном влиянии на течение заболевания магнитной стимуляции бледного шара.
Прогноз
Прогноз зависит от формы заболевания. Наиболее неблагоприятное течение имеет ранняя форма, при которой полная инвалидизация наступает в промежутке от 10 до 15 лет с момента дебюта симптомов. Более благоприятен взрослый вариант, особенно в случаях, когда деменция слабо выражена. Его средняя продолжительность составляет более 20 лет.
Читайте также:
- Рабдомиосаркома - стадии, механизмы развития
- Синдром Февра-Лангепен - клиника, диагностика
- Диагностика бронхолегочного аспергиллеза. Лечение бронхолегочного аспергиллеза
- Рентгенограмма, КТ при переломе диафизов большеберцовой и малоберцовой костей
- Паклитаксел. Применение и побочные эффекты паклитаксела