Диагностика подострого склерозирующего панэнцефалита по КТ, МРТ
Добавил пользователь Владимир З. Обновлено: 24.01.2026
Подострый склерозирующий панэнцефалит — медленная инфекция, вызванная вирусом кори. Латентный период составляет до 8 лет. Подострый коревой панэнцефалит дебютирует расстройствами личности и поведения, затем появляются прогрессирующие мышечно-тонические, двигательные, зрительные, когнитивные нарушения, пароксизмальные состояния. Основные методы диагностики: электроэнцефалография, КТ/МРТ головного мозга, анализ цереброспинальной жидкости, выявление антител к кори в крови, ликворе. Этиотропная терапия осуществляется антивирусными фармпрепаратами, симптоматическое лечение проводится противоэпилептическими средствами.
МКБ-10
Общие сведения
Подострый склерозирующий панэнцефалит (ПСП) впервые был описан в 1933 году американским патоморфологом Дж. Доусоном под названием «энцефалит с включениями». В 1945 году в Бельгии невролог Ван Богарт дополнил имеющиеся сведения о заболевании, указал на процессы демиелинизации с преимущественным поражением белого церебрального вещества и предложил новый термин — «подострый склерозирующий лейкоэнцефалит». В честь упомянутых исследователей в современной зарубежной литературе по неврологии ПСП упоминается под названиями «энцефалит Доусона», «лейкоэнцефалит Ван-Богарта». Подострый склерозирующий панэнцефалит встречается с частотой 1 случай на 1 млн. населения, 85% заболевших относятся к возрастной категории 5-15 лет, мальчики болеют в 3 раза чаще. Известны отдельные случаи заболевания у детей младшего возраста и у взрослых старше 30 лет.
Причины ПСП
Патология является классической медленной инфекцией ЦНС. Инфекционным агентом выступает вирус кори, сохраняющийся в организме в персистирующем состоянии после перенесённой естественной коревой инфекции. В большинстве случаев ПСП наблюдается у детей, переболевших корью в возрасте до двух лет. Латентный период продолжается 6-8 лет, по истечении которых развивается быстро прогрессирующий коревой панэнцефалит. Факторы, обуславливающие вирусную персистенцию, остаются неизвестными. Отдельные авторы предполагают, что в основе лежит изменённая иммунологическая реактивность организма, обуславливающая неполную элиминацию вируса.
Патогенез
Этиопатогенетические механизмы не изучены, вызывающие активацию вируса триггеры не определены. После многолетнего латентного периода персистирующие в церебральных клетках вирусы кори начинают активную репликацию, что обуславливает тотальные воспалительные изменения тканей головного мозга — подострый панэнцефалит. В воспалительный процесс вовлекаются белое вещество, подкорковые ганглии, кора, мозговые оболочки. Очаги поражения распределяются неравномерно, в них происходит гибель нейронов, компенсаторно разрастается глиальная ткань. На поздних стадиях склерозирующий панэнцефалит характеризуется диффузной демиелинизацией в сочетании с глиозом. Микроскопически в поражённых нейронах обнаруживаются специфические включения. Исследование при помощи меченых антител выявляет в них наличие антигенов коревого вируса.
Классификация
Клиническая симптоматика ПСП весьма вариабельна, однако у всех пациентов прослеживается стадийное течение. Понимание фазы заболевания имеет клиническое и прогностическое значение, в связи с чем подострый склерозирующий панэнцефалит классифицируют на 4 основные стадии:
- I стадия (начальная, психотическая) характеризуется нарастающими изменениями в характере, поведении, интеллектуальных способностях больного. До появления мышечно-тонических нарушений часто ошибочно диагностируется как психиатрическая патология. Продолжается 2-12 месяцев.
- II стадия начинается с появления двигательных расстройств (гиперкинезов), пароксизмальных эпизодов (судорожных приступов, абсансов, атонических падений). В дальнейшем присоединяется различная неврологическая симптоматика. Стадия продолжается 6-12 месяцев.
- III стадия протекает с быстрым прогрессированием деменции, нарастанием мышечной ригидности, ослаблением судорожного синдрома. Длится несколько месяцев.
- IV стадия (терминальная, коматозная) — полный распад психических функций, децеребрационная ригидность, кахексия. Пациенты впадают в кому с последующим летальным исходом.
Симптомы ПСП
Подострый склерозирующий панэнцефалит дебютирует постепенно усугубляющимися отклонениями в поведении: больной становится неряшлив, упрям, агрессивен, раздражителен, равнодушен к окружающим. Начинают преобладать примитивные качества: жадность, эгоизм. Появляются психопатоподобные реакции, нарушения сна, у школьников возникают сложности в обучении. К концу начального периода наблюдаются прогрессирующие мнестические расстройства, интеллектуальное снижение, нарушения речи (дизартрия, афазия).
Затем присоединяется экстрапирамидная симптоматика: непроизвольные движения в форме атетоза, тремора, торсионной дистонии, гемибаллизма, миоклонии, смешанные расстройства мышечного тонуса. Наблюдаются генерализованные судорожные приступы, эпизоды «отключения» сознания (абсансы), атонические пароксизмы. На фоне развёрнутой клиники возникают пирамидные нарушения (парезы, патологические рефлексы), мозжечковая атаксия. Прогрессирует когнитивная дисфункция, характеризующаяся апраксией, амнезией, аграфией, алексией, агнозией. В половине случаев выявляются нарушения зрения, обусловленные развитием коревого хориоретинита, поражением зрительного нерва, коры затылочной доли.
Постепенно гиперкинезы сменяются симптоматикой паркинсонизма, смешанные изменения тонуса переходят в тотальную ригидность. Экстрапирамидные расстройства сочетаются с вегетативной симптоматикой: повышенной сальностью кожи, гипергидрозом, лабильностью давления, гиперсаливацией. Нарастающая децеребрационная ригидность приводит к постепенному исчезновению судорожных пароксизмов. Отмечается полный распад личности, насильственный смех/плач, гипертермические кризы, расстройства глотания, дыхания. В терминальной стадии конечности пациента согнуты, имеются сгибательные контрактуры, продуктивный контакт отсутствует, угнетение сознания доходит до стадии комы, возникают трофические изменения тканей.
Осложнения
Прогрессирование зрительных нарушений приводит к амаврозу. Лежачее положение больного на последних стадиях ПСП может вызвать образование пролежней. Инфицирование последних ведёт к местным воспалительным изменениям, попадание инфекции в кровоток — к возникновению сепсиса. Обездвиженность пациента, дыхательные расстройства центрального генеза способствуют возникновению застойной пневмонии. Дисфагия опасна попаданием пищи в дыхательные пути с развитием аспирационной пневмонии. Перечисленные инфекционные осложнения выступают наиболее частыми причинами гибели больных.
Диагностика
Полиморфизм, неспецифичность симптоматики, изолированные психотические проявления начального периода, редкая встречаемость обуславливают позднее диагностирование заболевания. В ходе диагностики невролог опирается на анамнестические сведения о перенесённой в детстве кори, изменения неврологического статуса, данные ЭЭГ и нейровизуализации, результаты исследований на противокоревые антитела. Биопсия головного мозга не показательна, поскольку мозаицизм церебрального поражения делает возможным забор материала из неизменённого участка мозговых тканей. Перечень необходимых дополнительных обследований включает:
- Электроэнцефалографию. Типична картина повышенной медленноволновой активности, возникающая с интервалом 6-8 с и чередующаяся с периодами сниженной биоэлектрической активности. Регистрируемые комплексы носят двусторонний характер, симметричны, синхронны.
- Нейровизуализацию. Проводится методами компьютерной или магнитно-резонансной томографии. МРТ, КТ головного мозга диагностируют диффузное множественное поражение церебральных тканей, атрофические изменения коры, в ряде случаев — гидроцефалию. Изменения в белом веществе наблюдаются спустя 4 месяца от дебюта ПСП. В 50% случаев поражения базальных ганглиев определяются уже на 2-ой стадии.
- Исследование цереброспинальной жидкости. Ликвор для анализа получают путём люмбальной пункции. Исследование выявляет умеренное повышение концентрации белка со значительным нарастанием относительного содержания гамма-глобулинов, наличие специфических олигоклональных IgG. Резкое нарастание титра антикоревых антител свидетельствует о коревой этиологии поражения.
- Анализ крови на противокоревые антитела. Определяет повышение антител в сыворотке до 1:4 — 1:128. В норме титр составляет 1:200 – 1:500. Выявление повышенного титра в крови и цереброспинальной жидкости является важнейшим диагностическим признаком.
Выделение вируса кори из поражённых тканей мозга редко применяется в клинической практике по причине сложности выполнения. В начальной стадии подострый склерозирующий панэнцефалит необходимо дифференцировать от психических расстройств: неврастении, шизофрении, истерии. В последующем дифдиагноз осуществляется с опухолью головного мозга, прогрессирующим краснушным панэнцефалитом, клещевым энцефалитом, болезнью Крейтцфельдта-Якоба.
Лечение ПСП
Специфическая терапия не разработана. Большое значение имеет правильный уход за больным, предупреждение инфекционных осложнений. Проводится этиотропное лечение противовирусными средствами (рибавирин, инозин пранобекс), препаратами интерферона, но оно малорезультативно. В качестве симптоматической терапии назначают антиконвульсанты, эффективные в отношении миоклоний (диазепам, производные вальпроевой кислоты). Для снятия спастического гипертонуса применяют миорелаксанты (толперизон, баклофен). Нарушения дыхания на заключительных стадиях заболевания являются показанием к переводу пациентов на ИВЛ. Тщательный уход и симптоматическое лечение позволяют продлить жизнь больного.
Прогноз и профилактика
В 80% случаев подострый склерозирующий панэнцефалит имеет длительность 1-3 года и приводит к летальному исходу. У 10% пациентов наблюдается молниеносное течение. В 10% случаев удаётся увеличить продолжительность жизни больного, иногда до 10 лет. Наилучшими превентивными мерами, позволяющими предупредить возникновение ПСП, являются мероприятия по предупреждению заболевания корью. Специфическая профилактика проводится путём плановой вакцинации против кори детей в возрасте 12 месяцев.
Диагностика подострого склерозирующего панэнцефалита по КТ, МРТ
а) Терминология:
1. Сокращения:
• Подострый склерозирующий панэнцефалит (ПСПЭ)
2. Синоним:
• Энцефалит Доусона
3. Определение:
• Прогрессирующий энцефалит, обусловленный вирусом кори
б) Визуализация:
1. Общие характеристики подострого склерозирующего панэнцефалита:
• Лучший диагностический критерий:
о Зоны повышения интенсивности сигнала на Т2-ВИ в перивентрикулярном, глубоком или субкортикальном белом веществе (БВ)
• Локализация:
о Лобные > теменные > затылочные доли
2. КТ признаки подострого склерозирующего панэнцефалита:
• Бесконтрастная КТ:
о Часто изначально наблюдается нормальная визуализационная картина; отек коры, снижение плотности коры/субкортикального белого вещества
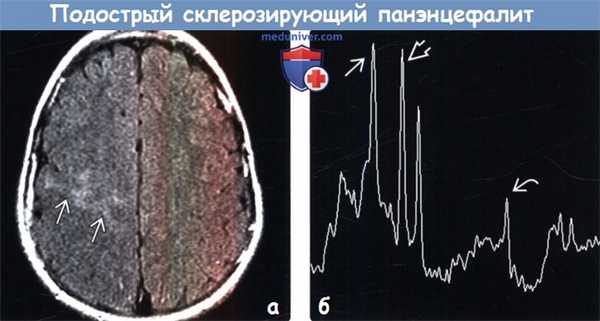
(а) МРТ, FLAIR, аксиальный срез: у семилетнего мальчика с прогрессирующим нарушением походки и миоклоническими судорогами в области коры и субкортикального белого вещества заднего отдела правой лобной доли определяется зона слабого повышения интенсивности сигнала.
(б) Одновоксельная протонная МР-спектроскопия (ТЕ = 35 мс), исследование субкортикального белого вещества правой теменной доли: у того же пациента определяется повышение пиков холина и миоинозитола и выраженное снижение пика NAA. При этих метаболических изменениях наблюдались лишь умеренные изменения сигнала на Т2-ВИ и FLAIR.
3. МРТ признаки подострого склерозирующего панэнцефалита:
• Т1-ВИ:
о Зоны ↓ интенсивности сигнала в БВ, мозолистом теле
• Т2-ВИ:
о Зоны ↑ интенсивности сигнала в БВ лобных > теменных > затылочных долях, обычно симметричные и, в конечном счете, прогрессирующие до диффузной атрофии
• Постконтрастные Т1-ВИ:
о Отсутствие накопления контраста
• МР-спектроскопия
о ↑ пиков холина и миоинозитола; ↓ пика NAA (может предшествовать изменениям на МРТ)
4. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ
• Советы по протоколу исследования:
о МРТ + в/в введение контрастного вещества + МР-спектроскопия
в) Дифференциальная диагностика:
1. Острый диссеминированный энцефаломиелит:
• Перенесенное вирусное заболевание, периферические зоны ↑ интенсивности сигнала на Т2-ВИ
2. Псевдотуморозная форма рассеянного склероза:
• Очаги по типу объемных образований, расположенные в перивентрикулряном БВ, контрастирование по периметру
3. Вирус иммунодефицита человека:
• Атрофия, зоны Т интенсивности сигнала на Т2-ВИ с нечеткими контурами, расположенные в БВ
• Повышение плотности базальных ганглиев (бесконтрастная КТ)
4. Прогрессирующая мультифокальная лейкоэнцефалопатия:
• Зоны ↑ интенсивности сигнала на Т2-ВИ, иммунокомпрометированные пациенты (СПИД, рак)
г) Патология:
1. Общие характеристики:
• Этиология:
о Постинфекционный прогрессирующий энцефалит, возникающий спустя годы после перенесенной кори
2. Макроскопические и хирургические особенности:
• Диффузный отек большого мозга на раннем этапе заболевания; субкортикальное и перивентрикулярное белое вещество, базальные ганглии
• Быстро прогрессирующая диффузная кортикальная атрофия
3. Микроскопия:
• Лептоменингеальное, периваскулярное воспаление, воспаление мозговой паренхимы
• Реактивный глиоз с демиелинизацией белого вещества, внутриядерными включениями в нейронах и клетках олигодендроглии
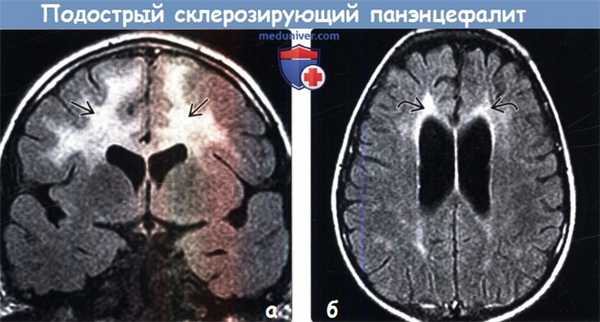
(а) МРТ, FLAIR, корональный срез: у семилетнего мальчика спустя шесть месяцев после первого появления симптомов (после подтверждения кори на основании положительного теста фиксации комплемента) в белом веществе лобной доли определяется зона повышения интенсивности сигнала с прогрессирующей динамикой.
(б) МРТ, FLAIR, аксиальный срез: у этого же пациента (спустя 10 месяцев после появления симптомов на фоне прогрессирующих ухудшения когнитивных функций и клонических судорог) отмечается потеря объема белого вещества обеих лобных долей и повышение интенсивности сигнала от него (демиелинизация и глиоз).
д) Клиническая картина:
1. Проявления подострого склерозирующего панэнцефалита:
• Наиболее частые признаки/симптомы:
о Поведенческие изменения, ухудшение психического состояния, атаксия, миоклонические судороги и нарушения зрения:
• Клинический профиль
о Положительный результат теста фиксации комплемента ликвора, плазмы крови на вирус кори
о ЭЭГ: периодические остроконечные комплексы, высоковольтаж-ные медленные волны
2. Демография:
• Возраст:
о Дети, молодые подростки; редко встречается у взрослых
• Эпидемиология:
о У большинства пациентов перенесенная в возрасте до двух лет корь → увеличение риска развития подострого склерозирующего панэнцефалита (ПСПЭ) в 16 раз
3. Течение и прогноз:
• Легкие поведенческие изменения; моторные нарушения (миоклонические судороги); смерть (спустя 1-3 года после появления симптоматики)
4. Лечение:
• Лечения не существует, но существует определенная польза от внутрижелудочкового введения а-интерферона и изопринозина
е) Диагностическая памятка:
1. Обратите внимание:
• Предполагайте СПСЭ у детей из семей иммигрантов с поведенческими изменениями и мультифокальным поражением БВ
2. Совет по интерпретации изображений:
• Обнаруживаемые на МРТ изменения слабо коррелируют с клинической картиной
Лейкоэнцефалит Шильдера
Лейкоэнцефалит Шильдера — дегенеративно-демиелинизирующее поражение головного мозга, сопровождающееся образованием крупных или сливных зон демиелинизации. Имеет неуклонно прогрессирующее течение с неспецифичной и полиморфной клинической картиной, которая может включать психические нарушения, пирамидный и экстрапирамидный синдромы, когнитивный дефицит, поражение черепно-мозговых нервов, эписиндром. Диагностируется лейкоэнцефалит Шильдера по клиническим критериям и результатам МРТ после исключения другой патологии с подобными проявлениями. Терапия осуществляется глюкокортикостероидами, антиконвульсантами, миорелаксантами и психотропными средствами. Однако лечение малоэффективно.
Лейкоэнцефалит Шильдера впервые был рассмотрен в качестве самостоятельной нозологии в 1912 г. психоневрологом, имя которого прочно закрепилось в названии заболевания, хотя сам автор обозначил описанную им патологию термином «периаксиальный диффузный лейкоэнцефалит». Позже различными исследователями были представлены описания других клинических форм лейкоэнцефалита: в 1941 г. - геморрагического лейкоэнцефалита, в 1945 г. - подострого склерозирующего лейкоэнцефалита. Поскольку основной патоморфологический субстрат болезни составляют диффузные зоны демиелинизации белого вещества, лейкоэнцефалит Шильдера входит в группу демиелинизирующих заболеваний.
Преимущественный возраст манифестации болезни Шильдера до сих пор остается спорным вопросом. Зарубежные специалисты в области неврологии считают характерным дебют в возрастном периоде от 7 до 12 лет, а отдельные авторы предлагают относить заболевание к детской форме рассеянного склероза. Наблюдения отечественных неврологов, напротив, свидетельствуют о равной степени поражения лиц различной возрастной категории.
Причины лейкоэнцефалита Шильдера
Этиопатогенез болезни Шильдера находится в стадии изучения. Из названия заболевания видно, что первоначально подразумевалась воспалительная этиология церебрального поражения, т. е. энцефалит. Предполагается вирусная теория заболевания по типу медленных инфекций. Среди возможных инфекционных агентов дискутируется роль кори, герпетической инфекции, миксовирусов, которые, возможно, запускают процесс аутоиммунного церебрального воспаления. Однако безуспешные попытки выделения возбудителя привели к возникновению иной этиопатогенетической теории. Последняя предполагает связь лейкоэнцефалита Шильдера с дисфункцией регуляторных механизмов липидного обмена, что сближает заболевание с наследственными лейкодистрофиями.
Морфологические изменения заключаются в образовании в белом церебральном веществе полушарий значительных зон демиелинизации, имеющих четкие заостренные очертания и зачастую асимметрично расположенных. В ряде случаев подобные очаги формируются в мозжечке и мозговом стволе. У пациентов, заболевших в пубертатном периоде и во взрослом возрасте, описаны случаи, когда наряду с зонами обширной демиелинизации наблюдаются округлые бляшковидные очаги, напоминающие бляшки рассеянного склероза.
Симптомы лейкоэнцефалита Шильдера
Заболевание отличается наличием неспецифичной и полиморфной симптоматики. Может манифестировать исподволь развивающимися психическими расстройствами: лабильностью настроения, апатией, нарушением поведения, эпизодами возбуждения с галлюцинаторным синдромом. Интеллектуальное снижение прогрессирует вплоть до деменции. Наблюдаются аграфия, акалькулия, алексия, агнозия, апраксия. Вследствие демиелинизации черепных нервов возникает неврит зрительного нерва, офтальмоплегия, тугоухость, снижение зрения, бульбарные расстройства. При поражении мозжечка появляется мозжечковая атаксия, скандированная речь, интенционный тремор. Поражение зрительной зоны коры проводит к гемианопсии, корковому амаврозу. Возможны экстрапирамидные нарушения в виде гиперкинезов, торсионной дистонии и т. п. Пирамидные расстройства обычно наблюдаются на поздних этапах лейкоэнцефалита в виде моно-, геми- и тетрапарезов. Зачастую присутствует судорожный синдром (по типу джексоновкой эпилепсии или с генерализованными эпиприступами), характеризующийся отсутствием специфической ЭЭГ-картины.
Вариативность сочетаний различных симптомокомплексов настолько выражена, что не позволяет выделить типичный вариант течения болезни Шильдера. В ряде случаев клиника сходна с прогредиентным вариантом рассеянного склероза, в других - имеет псевдотуморозный характер, в третьих — напоминает психиатрическую патологию. В последнем случае пациенты могут проходить лечение у психиатра вплоть до развития явной неврологической симптоматики.
Диагностика лейкоэнцефалита Шильдера
Прижизненно диагностировать лейкоэнцефалит Шильдера весьма затруднительно. Эта задача требует от невролога тщательного сопоставления анамнестических, клинических и томографических данных, внимательного проведения дифдиагностики со схожими заболеваниями. С целью обследования зрительного и слухового анализаторов к консультациям могут привлекаться офтальмолог и отоларинголог.
Электроэнцефалография выявляет признаки диффузного церебрального поражения: снижение альфа-активности и дезорганизацию ритма; зачастую определяется эпилептиформная активность. При исследовании цереброспинальной жидкости обнаруживается повышение уровня гамма-глобулина на фоне снижения удельного веса альбуминовой фракции. Наиболее информативным способом инструментальной диагностики выступает МРТ головного мозга. Болезнь Шильдера подтверждает наличие как минимум одного большого или пары сливных очагов демиелинизации в белом церебральном веществе.
Для установления окончательного диагноза многие неврологи руководствуются критериями C.M. Poser 1985 г.: наличие по данным МРТ 1-2 округлых зон демиелинизации величиной не менее 2х3 см; отсутствие патологии надпочечников; исключение любой иной церебральной патологии (внутримозговой опухоли, рассеянного энцефаломиелита, инсульта и пр.); соответствие норме уровня жирных кислот в сыворотке крови; выявление на аутопсии зон диффузного хронического склероза. В некоторых случаях отличить лейкоэнцефалит Шильдера от лейкодистрофии позволяют лишь гистологические исследования церебральных тканей пораженной зоны.
Лечение и прогноз лейкоэнцефалита Шильдера
Отсутствие ясных представлений об этиопатогенезе болезни Шильдера пока не позволило разработать более или менее эффективные методы ее лечения. Отмечен некоторый эффект глюкокортикостероидной терапии, в связи с чем многим пациентам назначают метилпреднизолон, вначале парентерально в ударной дозе, а затем внутрь с постепенным снижением дозы. Параллельно проводится курс нейропротекторной, антиоксидантной и сосудистой терапии, при необходимости назначаются антиконвульсантное лечение (карбамазепин, диазепам), миорелаксанты (амантадин, толперизон, амидин), противоотечные мероприятия (фуросемид, ацетазоламид, магния сульфат), психотропные фармпрепараты.
Своевременно начатое лечение способно лишь несколько задержать прогрессирование патологии. Однако, не смотря на его проведение, все пациенты погибают. Время наступления летального исхода варьирует от нескольких месяцев до 3 лет с момента дебюта лейкоэнцефалита.
Диагностика энцефалита Расмуссена по КТ, МРТ
а) Терминология:
1. Сокращения:
• Энцефалит Расмуссена (ЭР)
2. Синонимы:
• Хронический фокальный (локализованный) энцефалит
3. Определение:
• Хроническое прогрессирующее одностороннее воспаление мозговой ткани неясной этиологии
• Характеризуется рефрактерной фокальной эпилепсией, прогрессирующей гемиплегией, когнитивными нарушениями
2. КТ признаки энцефалита Расмуссена:
• Бесконтрастная КТ:
о Изначально изменения отсутствуют → атрофия
• КТ с контрастированием:
о Контрастирование обычно не наблюдается
о Редко преходящее контрастирование мягкой мозговой оболочки и/или коры
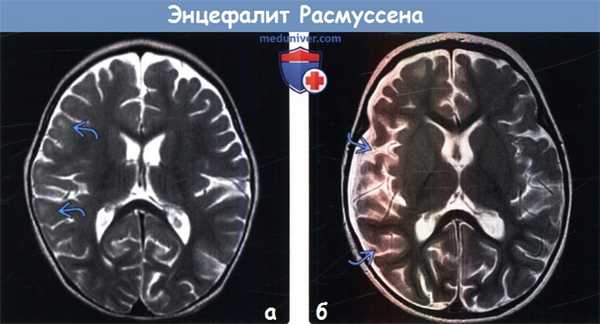
(а) МРТ, Т2-ВИ, аксиальный срез: у годовалого ребенка с фокальными судорожными приступами, устойчивыми к лекарственной терапии, и прогрессирующим левосторонним гемипарезом в субкортикальном белом веществе правого полушария головного мозга определяется легкое повышение интенсивности сигнала.
(б) МРТ, Т2-ВИ, аксиальный срез: у этого же пациента через месяц наблюдается выраженная потеря объема мозговой ткани правого большого полушария и углубление борозд. На серии МР-томограмм при энцефалите Расмуссена прослеживается прогрессирующая атрофия головного мозга в сочетании с изменениями сигнальных характеристик коры/субкортикального белого вещества.
4. Радионуклидная диагностика:
• Сцинтиграфия с Тс-99m ГМПАО: ↓ перфузии даже при нормальной МР-картине
• ПЭТ/ОФЭКТ:
о Диффузное ↓ метаболизма/перфузии в полушарии
о Перекрестный мозжечковый диашиз
о Преходящий гиперметаболизм может быть связан с недавно перенесенным судорожным приступом (редко):
- Мультифокальный гиперметаболизм поданным исследования с С-11-метионином
5. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ + оценка клинических симптомов + соответствующие признаки на ЭЭГ
• Советы по протоколу исследования:
о МРТ с контрастным усилением ± ПЭТ (ФДГ)
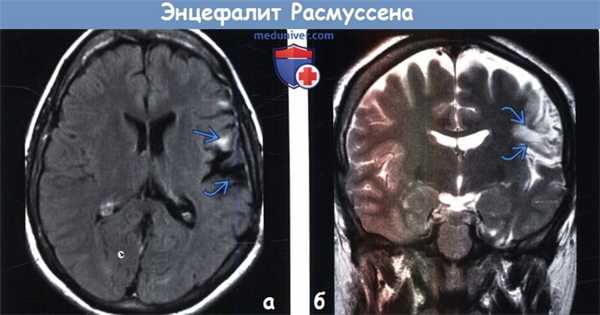
(а) МРТ, FLAIR, аксиальный срез: у пациента с энцефалитом Расмуссена (подтвержден по данным биопсии) определяется расширение левой сильвиевой борозды, обусловленное потерей объема мозговой ткани и сочетающееся с субкортикальным глиозом лобной покрышки.
(б) МРТ, Т2-ВИ, корональный срез: у того же пациента определяется потеря объема мозговой ткани и глиоз лобной покрышки слева.
в) Дифференциальная диагностика энцефалита Расмуссена:
1. Синдром Стерджа-Вебера:
• Пламенеющий невус лица и контрастируемая ангиома мягкой мозговой оболочки
• Прогрессирующая полушарная атрофия
• Кальцификация коры
2. Синдром MELAS (митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобные эпизоды):
• Острый: может вызывать повышение интенсивности сигнала от коры (наиболее часто в теменно-затылочной области) + ДВИ
• Хронический: атрофия коры, формирование лакун (базальные ганглии, таламус)
3. Синдром Дайка-Давыдова-Массона (полушарный инфаркт в пренатальном/неонатальном периоде):
• Односторонняя церебральная атрофия
• Компенсаторное утолщение костей черепа
• Приподнятость каменистого гребня височной кости и гипераэрация околоносовых пазух
• Развивается после инфаркта во внутриутробном или перинатальном периоде
4. Фокальная кортикальная дисплазия:
• Может вызывать полушарный эпилептический статус, что приводит к односторонней потере объема мозговой ткани
5. Другие аутоиммунные воспалительные заболевания:
• Односторонний васкулит головного мозга, паранеопластические синдромы, онконейрональные антитела
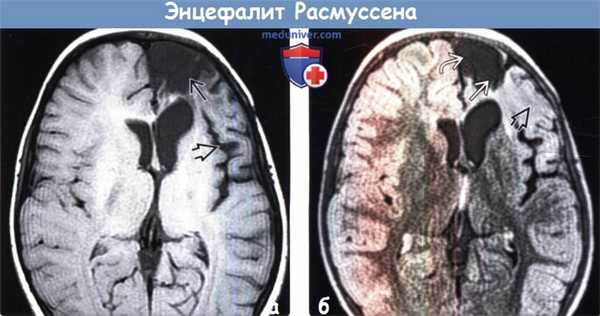
(а) МРТ, Т1-ВИ, аксиальный срез: у четырехлетней девочки с хроническим локализованным энцефалитом Расмуссена определяется выраженная атрофия лобной доли. Обратите внимание на расширение лобного рога левого бокового желудочка заместительного генеза и расширение круговой борозды островка что является признаком потери объема островковой доли.
(б) МРТ, FLAIR, аксиальный срез: у того же пациента определяется атрофия верхней и средней лобных извилин Атрофия и субкортикальный глиоз распространяются на нижнюю лобную извилину слева.
1. Общие характеристики энцефалита Расмуссена:
• Этиология:
о Этиология и патогенез энцефалита Расмуссена (ЭР) остаются неизвестными
о Накапливаются данные о том, что заболевание носит аутоиммунный характер:
- Антитело-опосредованная, Т-клеточная цитотоксичность и индуцированная микроглией денерация
• Генетика:
о Возможно существование вирусного триггера генетически обусловленной иммунодисфункции
• Ассоциированные аномалии:
о Сочетание трех потенциальных факторов может инициировать/закреплять процессы, ведущие к поражению мозговой ткани:
- Вирусная инфекция
- Аутоиммунные антитела
- Аутоиммунные цитотоксические Т-лимфоциты
2. Стадирование и классификация:
• Три стадии течения энцефалита Расмуссена (ЭР):
о Продромальная стадия: неспецифическая симптоматика, низкая частота судорожных приступов и легкая гемиплегия
о Острая стадия: частые судорожные приступы, часто парциальная непрерывная эпилепсия, прогрессирующий гемипарез, когнитивные нарушения
о Стадия резидуальных изменений: перманентная и стабильная неврологическая симптоматика и сохраняющиеся судорожные приступы
• Классификация и стадирование: МРТ (Т2-ВИ):
о Стадия 1: отек/гиперинтенсивный сигнал
о Стадия 2: нормальный объем мозговой ткани/гиперинтенсив-ный сигнал
о Стадия 3: атрофия/гиперинтенсивный сигнал
о Стадия 4: прогрессирующая атрофия и нормальные сигнальные характеристики
3. Макроскопические и хирургические особенности:
• Полушарная атрофия коры
• Зона поражения часто окружена неизменной корой или зоной легкого воспаления:
о Данные при биопсии могут вводить в заблуждение
4. Микроскопия:
• Классификация Робитэйла описывает воспаление коры, потерю нейронов и глиоз в пределах 1 полушария:
о Группа 1 (патологически активная форма): активный воспалительный процесс:
- Микроглиальные узелки ± нейронофагия, округлые пери-васкулярные клетки
о Группа 2 (активная и отдаленная форма): обострение на фоне хронического течения:
- Вышеизложенное + некроз и формирование полостей, затрагивающие всю толщину коры в ≥ 1 гиральном сегменте
о Группа 3 (менее активная «отдаленная» форма):
- Потеря нейронов/глиоз и меньшее количество микроглиальных узелков
о Группа 4 («выгорание»):
- Формирование неспецифического рубца со слабо активным воспалением
1. Проявления энцефалита Расмуссена:
• Наиболее частые признаки/симптомы:
о Рефрактерная эпилепсия, клонические судороги
о Прогрессирует до парциальной непрерывной эпилепсии
о Другие: гемипарез, зрительные и сенсорные нарушения, дизартрия, дисфазия, изменения личности
• Клинический профиль:
о Дети младшего возраста с прогрессирующей парциальной эпилепсией, некупируемой лекарственными средствами
• Клиническое течение:
о Парциальные комплексные судорожные приступы с увеличивающейся частотой возникновения
о У 20% пациентов проявляется эпилептическим статусом
о Вслед за этим наступает рост тяжести судорожных приступов, прогрессирующий гемипарез, нарушение когнитивной функции, смерть
• ЭЭГ: изначально картина от нормальной до персистирующего одностороннего замедления ± эпилептогенная активность
• СМЖ: ± олигоклональные полосы
2. Демография:
• Возраст:
о Обычно начинается в детстве (6-8 лет)
о В 10% случаев развивается у подростков или взрослых
• Пол:
о М = Ж
• Этническая принадлежность:
о Отсутствует
• Эпидемиология:
о Наличие предшествующего эпизода воспаления (50% случаев):
- Тонзиллит, инфекция верхних дыхательных путей, средний отит
3. Течение и прогноз:
• В большинстве случаев гемиплегия и нарушение когнитивной функции
• Для пациентов старшего возраста характерен более длительный продромальный период и затяжное течение
• Неблагоприятный прогноз
• Развитие гемиплегии неизбежно ± лечение
4. Лечение:
• Заболевание рефрактерно к противосудорожным препаратам
• ± преходящее улучшение состояния при плазмоферезе, внутривенной терапии иммуноглобулинами, лечении стероидными препаратами, агентами, направленными на В-клетки и Т-клетки
• При ЭР полное излечение судорожных приступов достигается только за счет хирургического вмешательства:
о функциональная гемисферэктомия/центральное разобщение
о Гемисферэктомия
е) Диагностическая памятка. Советы по интерпретации изображений:
• Предполагайте энцефалит Расмуссена при:
о ↑ частоты возникновения парциальных комплексных судорожных приступов, постиктального неврологического дефицита у пациентов (1-15 лет) с изначально «нормальной» визуализационной картиной
о Некупируемой эпилепсии с прогрессирующей атрофией одного полушария в виде повышения интенсивности сигнала на Т2-ВИ
Прогрессирующий краснушный панэнцефалит
Прогрессирующий краснушный панэнцефалит — редкая медленная вирусная инфекция ЦНС, обусловленная вирусом краснухи. Манифестирует в период от 8 до 19 лет. Проявляется прогрессирующим умственным снижением, мозжечковым синдромом, спастическим тетрапарезом, нарушениями зрения, эпилептическими пароксизмами. Диагностируется на основании анамнестических и клинических данных, картины церебральной МРТ/КТ, результатов анализа спинномозговой жидкости. Лечение осуществляется симптоматическими средствами, имеет малую эффективность. Заболевание склонно к неуклонному прогрессированию.
Причины ПКП
Этиофактором заболевания выступает краснушный вирус. Медленная инфекция развивается при заражении в результате:
- Внутриутробного инфицирования. Краснуха при беременности у женщины может протекать бессимптомно. Внутриутробная инфекция плода в первом триместре приводит к множественным порокам развития, далее вероятность аномалий уменьшается. Признаки врождённой краснухи (раннего краснушного энцефалита) обычно наблюдаются в ближайшем периоде после родов. Прогрессирующий краснушный панэнцефалит развивается, если вирус не проявляется сразу, а остаётся в латентном состоянии.
- Краснухи, перенесённой в детстве. В исключительных случаях после перенесённой инфекции не происходит полная элиминация вируса, он персистирует в организме в латентной форме.
К группе риска инфицирования корью в период беременности относятся женщины с низкой концентрацией специфических противокоревых иммуноглобулинов (10-25 МЕ/мл), осуществляющих защиту организма от краснушного вируса.
Попадание вируса краснухи в организм обычно происходит внутриутробно либо в раннем детском возрасте. По неизвестным обстоятельствам вирус переходит в неактивную форму, в которой может существовать до двух десятилетий. Причины активации находящегося в латентном состоянии вируса краснухи не установлены. Малое количество клинических наблюдений не даёт возможности подробно исследовать патогенез ПКП. Морфологически отмечается образование периваскулярных лимфоцитарных инфильтратов и мелких глиальных очагов в мозговой коре, астроцитоз с преимущественным поражением белого церебрального вещества. Изменения носят диффузный характер, захватывают различные участки мозга, за что краснушный энцефалит получил название «панэнцефалит».
Симптомы ПКП
Прогрессирующий краснушный панэнцефалит дебютирует неспецифической симптоматикой. Близкие начинают замечать перепады настроения, изменения в характере, поведении больного. Ребёнок хуже учится в школе, возникают проблемы с сосредоточением внимания, запоминанием, усваиванием нового материала. Со временем умеренные когнитивные нарушения перерастают в прогрессирующий интеллектуальный дефицит с исходом в деменцию. Отличительной особенностью ПКП является тяжёлая мозжечковая атаксия, включающая шаткость в положении стоя, неустойчивость во время ходьбы, несоразмерные крупноразмашистые движения, промахивание при выполнении целенаправленных действий.
Развёрнутая стадия заболевания характеризуется эпилептическими пароксизмами, присоединением очагового неврологического дефицита. Наблюдается выраженная атаксия (дискоординация, шаткость), зрительные расстройства, парез лицевого нерва. Типичны пирамидные симптомы: слабость мышц, спастическое повышение мышечного тонуса, повышение сухожильных рефлексов, появление патологических стопных знаков. Двигательные нарушения распространяются на все конечности, развивается тетрапарез. В некоторых случаях наблюдаются миоклонии — непроизвольные сокращения отдельных мышечных групп. Возникает сенсомоторная афазия — речь становится замедленной, упрощённой, непонятной из-за перестановки слогов, нарушается понимание услышанной речи.
В терминальной стадии спастический тетрапарез приковывает пациентов к постели, отсутствует произвольный контроль тазовых органов. Отмечается выраженная деменция, полное отсутствие речи (мутизм), расстройство движений глазных яблок (офтальмоплегия). Нередко наступает кома. Заболевание оканчивается летальным исходом вследствие тотального поражения головного мозга.
Редкая встречаемость патологии обуславливает сложности постановки диагноза «краснушный панэнцефалит». Важную роль имеет выявление в анамнезе пациента данных о перенесённой внутриутробно или в раннем детстве краснухе. Перечень диагностических мероприятий включает:
- Неврологический осмотр. В ходе обследования невролог устанавливает наличие фациального пареза, пирамидной симптоматики, мозжечкового синдрома, выраженного когнитивного снижения, сенсомоторной афазии.
- Люмбальную пункцию. Процедура позволяет получить образец цереброспинальной жидкости, провести его анализ. Характерно умеренное повышение лимфоцитов (40 кл/мкл), белка (1,5-1,5 г/л) с преобладанием гаммаглобулина. Резко увеличена концентрация специфических антикраснушных антител, что свидетельствует об их продукции в тканях ЦНС.
- Электроэнцефалографию. Не обнаруживает специфических отклонений. Типично диффузное замедление основного ритма.
- Томографию. МРТ, МСКТ и КТ головного мозга выявляют гидроцефалию, генерализованную атрофию церебральных тканей, расширение борозд мозговой коры. Особенно грубо атрофирован мозжечок.
- Анализ крови на противокраснушные антитела. Осуществляется иммуноферментным методом (ИФА). Титр антител повышен, но не столь значительно, как в цереброспинальной жидкости.
Выделить краснушный вирус из полученных путём биопсии церебральных тканей, как правило, не удаётся. Необходимо дифференцировать прогрессирующий панэнцефалит краснушной этиологии от других медленных инфекций, вирусного энцефалита, рассеянного энцефаломиелита. Дифференциальный диагноз проводят с подострым склерозирующим панэнцефалитом, отличающимся нормальной картиной цереброспинальной жидкости с повышением титра противокоревых антител.
Лечение ПКП
Специфическая терапия не разработана. Попытки лечения противовирусными препаратами (амантадином), метаболическими средствами (инозином) оказались безуспешными. Проводится симптоматическая терапия (антиконвульсанты, антигипоксанты, противоотечные средства), однако она не способна приостановить прогрессирование симптомов.
Прогрессирующий краснушный панэнцефалит оканчивается летально. Продолжительность заболевания с момента манифестации симптомов не превышает 3-х лет. В связи с отсутствием эффективной терапии большое значение придаётся профилактике инфицирования краснухой беременных женщин и детей. Специфическая профилактика осуществляется путём вакцинации против краснухи детей в возрасте 1 года с последующей ревакцинацией в 6 лет. Не привитым ранее и не переболевшим краснухой 13-летним девочкам рекомендовано однократное введение вакцины. Планирующим беременность женщинам с низким титром противокраснушных антител показана вакцинация по индивидуальным показаниям. Ведение беременности у женщин из группы риска должно осуществляться с контролем титра специфических антител в крови.
Читайте также: