Дистрофия Эмери-Дрейфуса
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Эта форма мышечной дистрофии была названа в честь британского профессора Эмери и его американского коллеги Дрейфуса, которые впервые описали эту патологию примерно 40 лет назад.
Что представляет собой мышечная дистрофия Эмери-Дрейфуса?
Как и другие виды мышечной дистрофии, дистрофия Эмери-Дрейфуса – это болезнь с постепенным нарастанием слабости мышц. Обычно болезнь проявляется в детстве или юности. Характерные особенности, отличающие данную форму мышечной дистрофии от других форм, – это ранние развитие мышечных контрактур, распространение мышечной слабости. Отличительной чертой является и тот факт, что при данной мышечной дистрофии определённым образом может быть затронуто сердце.
Что такое мышечная контрактура?
Мышечная контрактура – это стягивание и сокращение определённых групп мышц так, что суставам, связанным с этими мышцами, становиться всё сложнее двигаться. Такая контрактура едина для последних стадий в большинстве заболеваний связанных с мышечной атрофией и является результатом сидячего и неподвижного образа жизни. Но при мышечной дистрофии Эмери-Дрейфуса мышечные контрактуры развиваются очень рано и прежде, чем любая мышечная слабость будет обнаружена.
Как проявляется контрактура?
При этом заболевании ограничивается способность распрямления локтевого сгиба так, что руки часто находятся в полусогнутом состоянии, обнаруживается склонность к хождению на носочках и ограничивается возможность наклона шеи.
На какие мышцы происходит воздействие?
В верхних конечностях слабость в основном поражает плечевой пояс. В нижних конечностях сначала слабость распространяется на голени. Такое распространение мышечной слабости иногда называют «scapula-humeroperoneal» (лат. лопаточно-плечеперонеальная).
В начале трудно поднимать руки над головой и поднимать тяжёлые предметы, так же появляется склонность к спотыканию о край ковра. Затем поражаются мышцы бёдер, в результате чего становится труднее подниматься по лестнице и вставать со стула без посторонней помощи.
Как заболевание влияет на сердце?
Мышечная дистрофия Эмери-Дрейфуса влияет на сердце особенным образом по сравнению с другими типами миопатий. Вместо того, чтобы затронуть сердечную мышцу, нарушается электропроводимость (её ещё называют проводящей системой сердца), которая контролирует частоту сердечных сокращений, что называется «блокадой сердца». Частота сердечных сокращение — часто аномально медленное, могут появляться учащённое сердцебиение («трепетание» в груди — нередкое ощущение для здорового человека и в одиночку не является причиной для беспокойства), а так же приступы головокружения и обмороки, нарастает усталость, также может возникнуть одышка.
Что делать, если затронуто сердце?
Если страдает сердце, однако не у каждого пациента это случается, врач может порекомендовать введение электрокардиостимулятора. Небольшое устройство внедряется под кожу чуть ниже грудной клетки. Оно предотвращает дальнейшие проблемы, благодаря этому устройству сердце бьётся в нормальном ритме.
На сколько это серьёзная болезнь?
В целом это заболевание менее тяжёлое, чем другие формы мышечной дистрофии, и, хотя продолжительность жизни может сократиться, многие могут достигнуть среднего возраста и дальше. Однако крайне важно проверяться у врача через определённый промежуток времени, к примеру, каждые 12 месяцев, чтобы убедиться, что сердце не пострадало. Однако имеются доказательства существования более тяжёлой рецессивной формы заболевания. При этой форме присутствует слабость, быстро прогрессирующая с раннего детства. Однако данная форма заболевания встречается довольно редко.
Можно ли это вылечить?
К сожалению, пока не существует лекарства или эффективного лечения, кроме внедрения кардиостимулятора, в тех случаях, когда это необходимо. Однако правильное питание и поддержка общего хорошего состояния здоровья очень важные факторы в проявлении каждой формы мышечной дистрофии.
Можно ли увеличить мышечную силу?
Полезны регулярные лёгкие упражнения, не вызывающие стресс в организме. Следует избегать тяжёлых физических упражнений (например, упражнения с отягощениями). Очень важно иметь сбалансированный рацион питания, следует включать в меню трудно перевариваемую пищу и избегать появления избыточного веса, так как это ослабит и без того ослабленные мышцы.
Может ли операция помочь?
Для ходьбы могут быть полезны надрезы пяточного сухожилия. Другие операции могут быть назначены в отдельных случаях. Следует обратиться за советом к таким специалистам как невропатолог или хирург-ортопед. Так как это может затронуть сердце, что в свою очередь может осложнить операцию, перед операцией о диагнозе следует сообщить анестезиологу.
Стану ли я обездвиженным?
Заболевание развивается очень медленно, в течение многих лет (за исключением очень редкой рецессивной форм). Возможно, что в дальнейшем на протяжении жизни может потребоваться инвалидное кресло.
Перспективы на трудоустройство
Как и при других медленно развивающихся заболеваниях мышц, на ранних стадиях можно рассматривать большинство профессий (вероятно, кроме получения водительских прав, так как потребуется медицинский осмотр). Но с возрастом физическая трудоспособность уменьшается, и в долгосрочной перспективе более предпочтительна сидячая работа.
Может ли это заболевание отразиться на будущих детях?
Данное заболевание является наследственным и поэтому может затронуть и других членов семьи.
Во многих семьях оно передаётся по наследству по сцепленному с полом (х-хромосомному) признаку, и, следовательно, поражает мужчин, а женщины являются носительницами. Все сыновья больного мужчины будут здоровыми, однако все его дочери будут носительницами мутации. Относительно потомков женщины, которая является носительницей, в среднем каждая из её дочерей имеет 50:50 шансов быть носительницей мутации, и в среднем каждый из её сыновей имеет 50:50 шансов заболеть.
Заболевание может передаваться по аутосомно-доминантному признаку. В этом случае заболевание может проявиться и у мужчин, и у женщин. Здесь генетический риск отличен. Как и во всех аутосомно-доминантных заболеваниях, в среднем на каждого сына или дочку больного родителя приходится 50:50 шансов, что ребёнок будет болен.
В рецессивных формах у здоровых родителей есть 1 шанс из 4-х, что ребёнок будет болен.
Иногда случается, что при х-сцепленных и доминантных формах ни у кого из родственников не было такой болезни. В таких случаях заболевание возникает в результате новой мутации в человеке, который позже может передать заболевание своим детям.
По этим причинам очень важно обратиться за профессиональной консультацией к специалистам по нервно-мышечным заболеваниям или генетикам, если вы обеспокоены о риске, которому могут быть подвержены ваши дети и родственники.
Чего ждать от будущего?
Сейчас ситуация более обнадёживающая, чем раньше: не только за счёт использования кардиостимуляторов при поражениях сердца, но также и потому, что были выявлены гены, ответственные и за появление аутосомно-доминантной (кодирует белок ламин А/С), и за появление х-хромосомной форм мышечной дистрофии Эмери-Дрейфуса (кодирует белок эмерин). Эти данные важны как для поиска эффективного лечения, так и для перинатальной диагностики.
Другие схожие заболевания
Синдром ригидного позвоночника
При таком заболевании, которое развивается в детстве, мышечная слабость обычно лёгкая, и основной проблемой являются контрактуры мышц шеи, локтей и коленей, при этом нет никакого влияния на сердце. Заболевание — разнотипное, так как связано с различными дистрофиями. Необходима консультация специалиста для определения точного диагноза и способа наследования в каждом конкретном случае.
Редкая доброкачественная сцепленная с полом мышечная дистрофия
Кроме мышечной дистрофии Беккера (она рассматривается в отдельной брошюре) и мышечной дистрофии Эмери-Дрейфуса, другие протекающие в лёгких формах дистрофии, связанные с полом, — очень редки и описаны в единичных семьях.
Конечностно-поясные дистрофии
На поздних этапах дистрофии Эмери-Дрейфуса мышечная слабость может напоминать конечностно-поясную дистрофию. И вновь может потребоваться консультация у специалиста, для подтверждения точного диагноза.
[Напоминаем, что статья от лица Muscular Dystrophy Campaign, поэтому помощь возможна на территории Соединённого Королевства. Однако, вполне вероятно, что эта организация помогает людям и из других стран. Прим. администраторов сайта]
Мы будем с вами на момент постановки диагноза и на всем дальнейшем пути. Мы сможем:
- дать вам самую новую и точную информацию о вашем состоянии и состоянии вашего ребёнка;
- дать вам знать о прогрессе в исследованиях, снабдим вас советами на каждый день, написанные людьми, которые знают, каково это жить с мышечной атрофией;
- познакомить с семьями, которые живут с таким же заболеванием и которые смогут поделиться с вами опытом;
- поделиться информацией, помочь вам, услугами, оборудованием и поддержкой, на которые вы имеете полное право.
Если вам бы хотелось, чтобы ваш врач имел больше информации о мышечной дистрофии Эмери-Дрейфуса, у нас есть подходящие материалы. Мы разработали онлайн-обучение для врачей, работающих со взрослыми, имеющими атрофию мышц. Свяжитесь с нами по горячей линии или по телефону, чтобы получить больше информации.
Отказ от прав: Несмотря на то, что мы делаем всё возможное, чтобы удостоверится, что информация в этом документе полна, верная и соответствует последним исследованиям, не можем это полностью гарантировать. Организация Muscular Dystrophy UK не несёт ответственности в случае причинении какого-либо вреда в результате использования данной информации. Организация Muscular Dystrophy UK не обязана поддерживать услуги, которые предоставляются организациями в списке.
Мышечная дистрофия Эмери-Дрейфуса, FHL1 м.
Ген FHL1 (FOUR-AND-A-HALF LIM DOMAINS 1) расположен на Х-хромосоме в регионе Xq26.3. Содержит 5 экзонов.
Мутации в данном гене приводят также к развитию скапулоперонеальной миопатии, миопатии с редуцированными тельцами с манифестацией в детском возрасте, миопатии с редуцированными тельцами с ранним началом и тяжелым течением, миопатии с атрофией тонических мышц и лопаточно-перонеальной миопатии. Для данного гена характерно наличие альтернативного сплайсинга, (при этом при участии одного гена могут синтезироваться несколько видов белка) в результате которого образуются: изоформа FHL1А (состоит из 5 экзонов, экспрессируется только в скелетной мускулатуре и в минимальных количествах в сердечной мышце); изоформа FHL1В (состоит из экзонов 1, 2, 3, 4, 4b, 5, экспрессируется в высоких количествах в скелетной мускулатуре и в низких количествах в сердечной мышце, толстом кишечнике, тонкой кишке, простате); изоформа FHL1С (состоит из экзонов 1, 2, 3 и 5, экспрессируется в яичках, скелетной мускулатуре и сердечной мышце).
Заболевание мышц, проявляющееся в детском или подростковом возрасте нарастающей слабостью мышц плечевого пояса и проксимальных отделов рук.
Первые признаки возникают, как правило, у взрослых. Возможна ретракции ахилловых сухожилий и заднешейных мышц, что приводит к изменению походки и появлению вынужденной позы больного с запрокидыванием головы назад. Эти симптомы, как правило, предшествуют признакам мышечной слабости и гипотрофии. Наиболее вовлеченными в процесс оказываются мышцы проксимальных отделов рук и перонеальной группы мышц, поражение которых носит симметричный характер. Типичными признаками заболевания являются контрактуры в локтевых и межпозвоночных суставах.
В ряде случаев отмечено вовлечение в патологический процесс мышц тазового и плечевого пояса, а также лицевой мускулатуры. У абсолютного большинства больных выявляются признаки кардиомиопатии с нарушением сердечной проводимости (кардиомегалия, гипертрофия левого желудочка, брадикардия, атриовентрикулярная блокада, блокада правой ножки пучка Гисса). На ранних стадиях заболевания, эти признаки достаточно хорошо выявляются при проведении 24-часового ЭКГ-мониторинга. Интеллект больных не страдает. Для заболевания характерно медленное прогрессирующее течение. Прогноз для жизни определяется степенью вовлечения в патологический процесс миокарда. Смерть, как правило, наступает от сердечной недостаточности.
Уровень активности креатинфосфокиназы обычно повышен в 4-5 раз, но может быть не изменен. В биоптатах мышечных волокон выявляются неспецифические признаки первично-мышечного поражения.
Литература
Специальной подготовки к исследованию не требуется.
Обязательны к заполнению:
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Литература
Типичная клиническая картина. Характерным признаком является возникновение симптомов у взрослых. Страдают преимущественно мужчины.
Литература
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Сердечно-сосудистые нарушения у больных Х-сцепленной прогрессирующей мышечной дистрофией Эмери—Дрейфуса
Приведены результаты клинического наблюдения 5 больных с генетически подтвержденным диагнозом Х-сцепленной формы миопатии Эмери—Дрейфуса. При полном отсутствии жалоб со стороны сердечно-сосудистой системы по данным кардиологического обследования у всех больных выявлены нарушения ритма и проводимости сердца: трепетание предсердий — у 3, неустойчивая суправентрикулярная тахикардия — у 3, экстрасистолия III и выше градации по Лауну — у 3, атриовентрикулярная блокада — у 4. Дилатация правого предсердия была патогномоничным и самым ранним признаком поражения миокарда. Обсуждаются вопросы обследования, ведения и лечения больных. Обосновываются показания к имплантации электрокардиостимулятора (имплантирован у 2 больных) и преимущества монофокальных моделей при данной нозологии.
Ключевые слова
Об авторах
к.м.н., в.н.с. отделения патологии сердечно-сосудистой системы
к.м.н., в.н.с. отделения психоневрологии и эпилептологии
Научно-исследовательский клинический институт педиатрии
Россия
к.м.н., зав. отделения психоневрологии и эпилептологии
д.м.н., в.н.с. научно-консультативного отдела
Список литературы
1. Astejada M.N., Goto K., Nagano A. et al. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol 2007; 26: 3: 159—164.
2. Грознова О.С., Новиков П.В., Белозеров Ю.М. и др.. Диагностка и тактика лечения поражения сердца при аутосомно-доминантной прогрессирующей мышечной дистрофии Эмери-Дрейфуса. Рос вестн перинатол и педиат 2007; 3; 42—47. (Groznova O.S., Novikov P.V., Belozerov Ju.M., Rudenskaja G.E., Tverskaja S.A. Cardiac lesion in Emery-Dreifus autosome-dominant progressive musculad dystrophy: treatment and policy. Ros vestn perinatol i pediat 2007; 3; 42—47.)
3. Puckelwartz M., McNally E.M. Emery–Dreifuss muscular dystrophy. Handb Clin Neurol 2011; 101: 155—166.
4. Perrot A., Spuler S., Geier C. et al. Cardiac manifestations of muscular dystrophies. Z Kardiol 2005; 94: 5: 312—320.
5. Finsterer J., Stöllberger C. Primary myopathies and the heart. Scand Cardiovasc J 2008; 42: 1: 9—24.
6. Wożakowska-Kapłon B., Bąkowski D. Atrial paralysis due to progression of cardiac disease in a patient with Emery– Dreifuss muscular dystrophy. Cardiol J 2011; 18: 2: 189—193.
7. Грознова О.С., Новиков П.В. Ранняя диагностика поражения сердца при Х-сцепленной форме мышечной дистрофии Эмери–Дрейфусса у детей. Рос вестн перинатол и педиат 2011; 1; 27—32. (Groznova O.S., Novikov P.V. Early diagnosis of cardiac lesion in X-linked Emery-Dreifus muscular dystrophy in children. Ros vestn perinatol i pediat 2011; 1; 27—32.)
8. Cestan R., LeJonne N.I. Une myopathie avec retractions familiales. Nouv. Iconogr Salpetr 1902; 15: 38—52.
9. Emery A.E.H. X-linked muscular dystrophy with early contractures and cardiomyopathy (Emery–Dreifuss type). Clin Genet 1987; 32: 360—367.
10. Yates J.R.W., Warner J.P., Smith J.A. et al. Emery–Dreifuss muscular dystrophy: linkage to markers in distal Xq28. J Med Genet 1993; 30: 108—111.
11. Bonne G., Leturcq F., Ben Yaou R. Emery–Dreifuss Muscular Dystrophy. R.A. Pagon, T.D. Bird, C.R. Dolan et. al. (Eds). Source GeneReviews™ [Internet]. Seattle (WA): University of Washington. Seattle 2013; 427.
12. Carboni N., Mura M., Mercuri E. et al. Cardiac and muscle imaging findings in a family with X-linked Emery–Dreifuss muscular dystrophy. Neuromuscul Disord 2012; 22: 2: 152—158.
13. Finsterer J., Stöllberger C., Keller H. Arrhythmia-related workup in hereditary myopathies. J Electrocardiol 2012; 45: 4: 376—384.
14. Parmar M.S., Parmar K.S. Emery–Dreifuss humeroperoneal muscular dystrophy: cardiac manifestations. Can J Cardiol 2012; 28: 4: 516. e1—3.
15. Nigro G., Russo V., Ventriglia V.M. et al. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery–Dreifuss muscular dystrophy. Neuromuscul Disord 2010; 20: 3: 174—177.
16. Ishikawa K., Mimuro M., Tanaka T. Ventricular arrhythmia in X-linked Emery–Dreifuss muscular dystrophy: a lesson from an autopsy case. Intern Med 2011; 50: 5: 459—462.
17. Zaim S., Bach J., Michaels J. Sudden death in an Emery– Dreifuss muscular dystrophy patient with an implantable defibrillator. Am J Phys Med Rehabil 2008; 87: 4: 325—329.
18. Golzio P.G., Chiribiri A., Gaita F. Unexpected sudden death avoided by implantable cardioverter defibrillator in Emery Dreifuss patient. Europace 2007; 9: 12: 1158—1160.
19. Buckley A.E., Dean J., Mahy I.R. Cardiac involvement in Emery Dreifuss muscular dystrophy: a case series. Heart 1999; 82: 1: 105—108.
20. Dickey R.P., Ziter F.A., Smith R.A. Emery–Dreifuss muscular dystrophy. J Pediat 1984; 104: 4: 555—559.
21. Russo V., Rago A., Palladino A. et al. P-wave duration and dispersion in patients with Emery–Dreifuss muscular dystrophy. J Investig Med 2011; 59: 7: 1151—1154.
22. Russo V., Rago A., Politano L. et al. Increased dispersion of ventricular repolarization in emery dreifuss muscular dystrophy patients. Med Sci Monit 2012; 18: 11: 643—647.
23. Грознова О.С., Чечуро В.В. Лечение кардиомиопатии у больных прогрессирующими мышечными дистрофиями. Рос вестн перинатол и педиат 2011; 2; 58—62. (Groznova O.S., Chechuro V.V. Treatment for cardiomyopathies in patients with progressive muscular dystrophies. Ros vestn perinatol i pediat 2011; 2; 58—62.)
Клинический случай поздней диагностики мышечной дистрофии Эмери-Дрейфуса, ассоциированной с мутацией в гене LMNA
В статье представлен клинический случай мышечной дистрофии, манифестирующей в 5-летнем возрасте слабостью в мышцах верхних и нижних конечностей. На основании клинической симптоматики, данных электромиограммы, активности креатинфосфокиназы и «мягкой формы» течения заболевания был установлен диагноз мышечной дистрофии Беккера. Сердечная недостаточность манифестировала в 40-летнем возрасте пациента в виде нарушения сердечного ритма и проводимости с дилатацией желудочков и систолической дисфункцией левого желудочка, что указывало на ламин-ассоциированный генез мышечной дистрофии Эмери-Дрейфуса (МДЭД). Анализ гена ламина А/С (LMNA) выявил у пациента гетерозиготное носительство мутации с.1247С>G ( Thr528Arg, rs57629361, NM_001257374.2) в экзоне 9. У родственников 1-й степени родства мутации обнаружено не было. Изучение родословной показало, что МДЭД, как и другие типы мышечной дистрофии, ранее не были характерны для семьи пациента.
Ключевые слова
Об авторах
Список литературы
1. Benedetti S., Menditto I., Degano M., et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients // Neurology - 2007. - Vol. 69. № 12. - P. 1285-1292
2. Bonne G., Di Barletta M. R., Varnous S., et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy // Nat Genet. - 1999. - Vol. 21. № 3. - P. 285-288
3. Bonne G., Mercuri E., Muchir A., et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene // Ann Neurol. - 2000. - Vol.48. № 2. - P. 170-180
4. Di Barletta M. R., Ricci E, Galluzzi G, et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive emery-dreifuss muscular dystrophy // Am J Hum Genet. - 2000. - Vol.66. № 4. - Р. 1407-1412
5. Dreifuss F.E., Hogan G.R. Survival in X-chromosomal muscular dystrophy // Neurology. - 1961. - Vol. 11. - P.734-737
7. Hermans M.C.E., Pinto Y.M., Merkies I.S.J. et al. Hereditary muscular dystrophies and the heart // Neuromuscular Disorders. - 2010. - Vol. 20. - P. 479-492
8. Meinke P., Nguyen T.D., Wehnert M.S. The LINC complex and human disease // Biochem Soc Trans. - 2011. - V. 39 - P. 1693-1697
9. Sanna T., Dello Russo A., Toniolo D., et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations // Eur Heart J. - 2003. - Vol. 24. № 24. - P. 2227-2236
10. Vytopil M., Benedetti S., Ricci E, et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes // J Med Genet. - 2003. - Vol. 40. № 12. - e132
11. Zhang l., Shen H., Zhao Z., Bing Q., Hu J. Cardiac effects of the c.1583 C>G LMNA mutation in two families with Emery-Dreifuss muscular dystrophy // Molecular Medicine Reports. - 2015. - Vol.12. № 4. - P. 5065-5071
«Спина растекалась, как мороженое» Избавить Настю от постоянной боли может срочная операция. Девочке нужна ваша помощь

Насте Редько 14 лет, она живет в Москве с родителями и младшей сестрой Лизой. У Насти тяжелое генетическое заболевание — прогрессирующая мышечная дистрофия Эмери — Дрейфуса. Мышцы постепенно слабеют, позвоночник, лишенный поддержки, скручивается дугой, органы грудной клетки сдавлены, нарастает сердечная недостаточность, а легкие работают только на 40 процентов. Настя страдает от сильной боли в спине и груди, ей тяжело дышать. Избавить девочку от мучений поможет сложная операция на позвоночнике — установка фиксирующей металлоконструкции. Но такая операция стоит больше полутора миллионов рублей. Для семьи сумма неподъемная.
У Насти тяжелое заболевание, вызывающее атрофию мышц. Девочка страдает от невыносимой боли в спине и груди, ей тяжело дышать. Избавиться от мучений ей поможет операция на позвоночнике с установкой металлоконструкции, но стоит она очень дорого. Нужна ваша помощь.
Стоимость операции 1 591 645 рублей.
Всего собрано 1 663 925 рублей.
Сбор средств успешно завершен.
Друзья, всем спасибо! Вместе мы сделали доброе дело.
Первые признаки болезни у Насти появились в три года, когда ей стало трудно подниматься по ступенькам и вставать с пола, где девочка устраивалась играть в куклы. «Мама, спинка болит!» — плакала Настя.
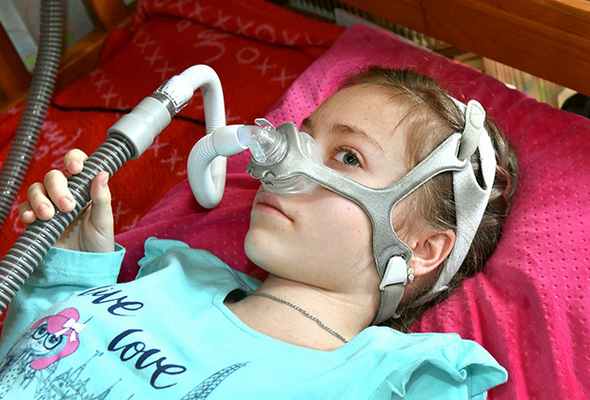

Врачи назначали массаж и физиотерапию, но состояние девочки только ухудшалось. Суставы рук и ног стягивали контрактуры, сгибать и разгибать ноги Настя уже не могла — и к пяти годам передвигалась только в коляске.
— Дочка ослабела, не могла стоять, ходить, жаловалась на боль, — рассказывает Татьяна. — А потом как-то резко сгорбилась, врачи поставили еще один диагноз: лордоз. В 2011 году Насте оформили инвалидность.
Больше всего на свете девочка боялась операции. Она готова была терпеть уколы, боль в спине и ногах, лишь бы не операция. Однако за год до школы Настя твердо заявила: «В школу пойду только на своих ногах».
В 2012 году девочке провели коррекцию контрактур тазобедренных суставов с обеих сторон — сделали надрезы на сухожилиях.
Через боль, преодолевая слабость, Настя заново училась стоять с опорой, потом передвигаться в ходунках. На школьной линейке она просто сияла от счастья. Никто из детей даже не догадывался, чего ей стоило стоять на ногах и улыбаться.
Однако радость была недолгой. Настя все больше слабела, позвоночник искривлялся все заметнее. В 2014 году девочка снова села в коляску. На вторую операцию она согласилась без лишних уговоров: «Я буду ходить? Тогда делайте что хотите».


Через год Насте провели повторную операцию. На этот раз ей удлиняли мышцы спины. После операции назначили массаж, физиопроцедуры, лечебную гимнастику.
Настя старалась изо всех сил, но так и не смогла держать спину, не было опоры на ноги. Стоять девочка могла только в вертикализаторе, из-за страшной боли в спине она не могла нормально спать.
— Дочке назначили носить жесткий ортопедический корсет, — вспоминает Татьяна. — Он помогал поддерживать спину, но стоило его снять, и спина растекалась, как растаявшее мороженое.
Настю направили на генетический анализ, по результатам которого поставили страшный диагноз: прогрессирующая мышечная дистрофия Эмери — Дрейфуса. Врачи объяснили, что это тяжелое и неизлечимое заболевание, которое нарушает работу практически всех мышц организма, включая дыхательные и сердечные.
В 2018 году Настя сильно выросла. Она уже не могла стоять даже в вертикализаторе. Позвоночник неумолимо искривлялся и принял форму буквы С. Насте было тяжело дышать и глотать, появилась аритмия. Проверка функции внешнего дыхания показала, что легкие работают лишь на 40 процентов. Теперь каждую ночь девочку подключают к аппарату искусственной вентиляции легких (ИВЛ), который помогает ей дышать во сне.
В прошлом году Настю осмотрел спинальный хирург Ильинской больницы (деревня Глухово, Московская область) Андрей Бакланов.
— Заболевание неизлечимо, но ребенку можно помочь, — сказал хирург. — Насте необходима операция на позвоночнике с установкой специальной металлоконструкции, которая позволит устранить деформацию и избавит девочку от боли. После операции спина выпрямится, внутренние органы встанут на место, Насте будет легче дышать, восстановится баланс туловища. Но операцию нужно делать срочно!
— На физкультуре я пишу тесты, — говорит Настя. — Я не знаю, что такое баскетбол и волейбол. Зато хорошо знаю, что такое вертикализатор, мешок Амбу, туторы и ИВЛ.
Эти приспособления помогают облегчить девочке жизнь. Если мы поможем оплатить операцию на позвоночнике, Насте будет намного легче дышать — и боли в ее жизни станет меньше.
Читайте также: