ЭхоКГ при аритмогенной правожелудочковой кардиомиопатии
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
Аритмогенная дисплазия правого желудочка (АДПЖ) относится к наследственным заболеваниям миокарда, при котором отмечаются структурные и функциональные нарушения в миокарде правого желудочка, вызывающие нарушения ритма и проводимости, в том числе фатальные желудочковые аритмии. АДПЖ считается одной из наиболее частых причин внезапной сердечной смерти у молодых людей и лиц, занимающихся спортом. Однако в практике встречаются случаи данного заболевания и у лиц более старшей возрастной категории. Диагностика АДПЖ по-прежнему вызывает трудности в связи с возможным длительным бессимптомным течением заболевания. В статье описан клинический случай АДПЖ у мужчины 48 лет.
Ключевые слова
Для цитирования:
For citation:
Аритмогенная дисплазия правого желудочка (АДПЖ), или аритмогенная кардиомиопатия правого желудочка, — прогрессирующее наследственное заболевание сердечной мышцы. Характеризуется очаговым фиброзно-жировым перерождением миокарда правого желудочка в пределах так называемого треугольника дисплазии, находящегося между выносящим, приносящим путём правого желудочка и верхушкой сердца, клинически проявляется желудочковыми нарушениями ритма, в том числе пароксизмальной желудочковой тахикардией (ЖТХ) [1, 2]. Однако в настоящее время жировая инфильтрация миокарда ПЖ не является патогномоничным признаком, поскольку она может встречаться у пожилых людей без заболевания. На фоне атрофии мышечной ткани продолжается разрушение кардиомиоцитов: наличие остаточных клеток — менее 60% (большой критерий), с фиброзным или жировым замещением или без него [3]. АДПЖ относится к редко встречающимся болезням, однако сопровождается высоким риском внезапной сердечной смерти (ВСС). В целом ряде исследований показана высокая инфицированность больных кардиотропными вирусами, такими как энтеровирус, парвовирус [3, 4].
Впервые клинику описал итальянский врачанатом Джованни-Мария Ланчизи. АДПЖ была описана как наследственное заболевание, наблюдающееся в четырех поколениях у всех членов одной семьи. Несмотря на то, что заболевание считается наследственным, генетическая причина развития патологии встречается менее чем в половине случаев [4].
У 60% пациентов с АДПЖ причиной заболевания являются мутации в генах, кодирующих десмосомные белки. В настоящее время известно более 100 генов и мутаций, принимающих участие в различных формах заболеваний сердца (гипертрофическая, дилатационная и аритмогенная кардиомиопатия) [4, 5]. В результате многочисленных исследований показано, что более 10% носителей мутаций генов десмосомных белков имеют высокий риск развития сложных аритмий и ВСС [6, 7].
Клинически АДПЖ характеризуется желудочковой тахикардией, блокадой проводящей системы сердца, дилатацией правого желудочка и сердечной недостаточностью.
В дебюте заболевание длительно может протекать бессимптомно, однако и в отсутствие специфической симптоматики высок риск жизнеугрожаемых аритмий.
При прогрессировании АДПЖ на первый план выходит ряд симптомов: перебои в работе сердца (>50% случаев), кардиалгии (46% случаев), сердцебиения (60% случаев), синкопальные состояния (40% случаев), ВСС (26% случаев) [8,9]. В клинике заболевания могут присутствовать такие признаки, как повышенная утомляемость, головокружение, одышка, снижение толерантности к физическим нагрузкам, указывающие на присутствие или прогрессирование сердечной недостаточности (СН).
Развитие АДПЖ имеет несколько стадий (латентную, электрических нарушений, правожелудочковой СН, бивентрикулярной СН).
Во время латентной стадии ВСC может быть первым и единственным проявлением, возникновение которой может провоцироваться активной физической нагрузкой. При развитии стадии электрических нарушений регистрируются нарушения ритма сердца и морфологические изменения правого желудочка. При прогрессировании процесса развивается диффузное поражение миокарда, которое может приводить к развитию бивентрикулярной сердечной недостаточности (СН), осложненной различными нарушениями ритма сердца (включая фибрилляцию предсердий). Финальная стадия процесса, как правило, представлена клиникой дилатационной кардиомиопатии [10].
В молодом возрасте АДПЖ является второй по частоте причиной внезапной смерти после ишемической болезни сердца [8].
Учитывая бессимптомное течение болезни или развитие ВСС как первого проявления, эпидемиологические данные остаются спорными. В среднем, заболевание встречается с частотой 6 на 10 тыс. жителей. По другим данным, АДПЖ является одной из самых распространенных кардиомиопатий и встречается, как минимум, у 200 пациентов в городе с населением 1 млн человек [8]. Отмечается, что болезнь чаще диагностируется у мужчин (около 80%) среднего возраста (до 40 лет) [5]. Считается, что у пациентов, страдающих АДПЖ, встречается примерно 17% случаев ВСС. Пристальное внимание стали уделять ранней диагностике заболевания у лиц, занимающихся спортом профессионально, после изучения причин ВВС у 16 молодых спортсменов E. Larsson и соавт. в 1999 г. В результате проведенного исследования выяснилось, что АДПЖ была обнаружена у каждого 4-го пациента. Однако возможно, что в данном случае частота встречаемости заболевания обусловлена более тщательным обследованием лиц данной категории. Начало тахикардии может быть отсрочено на многие годы, пока ПЖ значительно не увеличен, а размер аритмогенного субстрата недостаточно велик, чтобы вызвать постоянную желудочковую тахиаритмию.
Учитывая возможность длительного латентного течения АДПЖ, процесс диагностики остаётся сложным. Кроме стандартных методов обследования у пациентов с подозрением на АДПЖ в некоторых ситуациях показано проведение магниторезонансной томографии (МРТ), контрастной вентрикулографии сердца и эндомиокардиальной биопсии (ЭМБ). Для оценки структуры миокарда, а также электрических и функциональных изменений ПЖ проводится 3D-анатомическое картирование [10].
Прогноз заболевания напрямую зависит от своевременной профилактики внезапной сердечной смерти.
Представляем собственное наблюдение.
Пациент В., 48 лет, служащий, поступил в кардиологическое отделение Дорожной больницы на станции Нижний Новгород с жалобами на приступы сердцебиения, которые сопровождаются резкой слабостью, головокружением, чувством нехватки воздуха. С 45 лет отмечает перебои в работе сердца, за медицинской помощью не обращался. Ухудшение в течение трёх месяцев, когда появились вышеописанные приступы сердцебиения. Накануне госпитализации во время быстрой ходьбы возник подобный приступ, впервые была кратковременная потеря сознания.
В анамнезе редкие острые респираторные заболевания. Служил в армии. Физическая активность — умеренная, спортом не занимался. Не курит, алкоголь употребляет умеренно. Отец умер внезапно в 47 лет, причина неизвестна.
Результаты осмотра. Правильного телосложения, избыточного питания (индекс массы тела — 29,6 кг/м 2 ). Кожные покровы чистые, обычной окраски. При аускультации легких дыхание везикулярное, хрипов нет. Частота дыхания — 16 в минуту. Тоны сердца ритмичные, приглушены, частота сердечных сокращений 78 уд/мин. Артериальное давление — 115/85 мм рт.ст. Печень не увеличена, периферических отёков нет.
По данным проведенных лабораторных исследований, клинические и биохимические анализы без патологии, NT-proВNP – 125 пг/мл (верхняя граница нормы).
Данные инструментальных методов обследования. ЭКГ: ритм синусовый, регулярный. ЧСС 76 уд/мин. Горизонтальное положение ЭОС. Нарушение внутрижелудочковой проводимости. Инверсия зубца Т в V1-V5. Эпсилон волна в V1-V3 (рис.1).
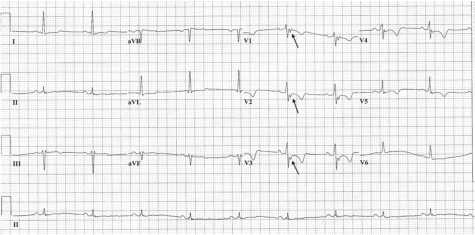
Рисунок 1. Электрокардиограмма больного В.: нарушение внутрижелудочковой проводимости, инверсия зубца Т V1-V5, эпсилон-волна V1-V3.
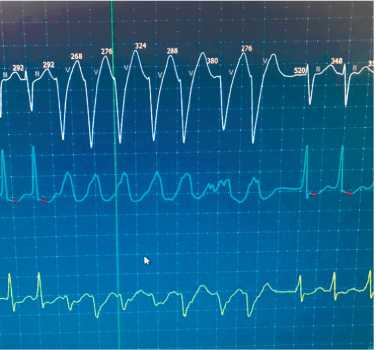
Рисунок 2. Холтеровское мониторирование больного В.: пароксизм неустойчивой ЖТ с частотой 214 уд/мин.
Рентгенография органов грудной клетки: воспалительных и очаговых изменений не выявлено. Плевральные синусы свободны. Легочный рисунок несколько усилен. Корни легких не изменены. Сердце расположено нормально, размеры не увеличены. Сосудистый пучок не изменен. Аорта без особенностей. Верхняя полая вена не расширена. Диафрагма без особенностей.
По данным трансторакальной эхокардиографии (ЭХО-КГ, рис. 3), выявлено расширение полости правого желудочка (ПЖ) со снижением его сократительной способности (фракция выброса ПЖ составила 39%), увеличение полости правого предсердия до 6,1 × 4,6 см и левого предсердия до 5,6 × 5,0 см. КДО ПЖ — 164 мл; КСО ПЖ — 102 мл. При изучении левого желудочка (ЛЖ) патологии выявлено не было, ФВ ЛЖ – 51%. Нижняя полая вена не расширена, диаметр — 2,0 см, коллабировала на вдохе более 50%. Данных о врождённом пороке сердца не выявлено.
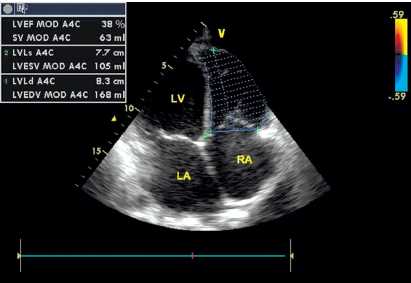
Рисунок 3. Эхокардиограмма больного В. Расширение предсердий и правого желудочка.
Примечание: ЛП — левое предсердие; ПП — правое предсердие; ЛЖ — левый желудочек; ПЖ — правый желудочек; КСР — конечно-систолический размер; КДР — конечно-диастолический размер; КСО — конечно-систолический объем; КДО — конечно-диастолический объем; ФВ — фракция выброса.
Диагностическая коронарография: данных о гемодинамически значимых стенозах коронарных артерий не получено.
Для уточнения состояния правых отделов сердца и исключения врождённых пороков была проведена магнитно-резонансная томография (МРТ) сердца с контрастным усилением гадолинием.
Рисунок 4. Магнитно-резонансная томограмма больного В. Дискинез передней стенки правого желудочка, микроаневризмы передней стенки правого желудочка, фаза диастолы.
Магнитно-резонансная томография с контрастным усилением. Сердце расположено типично. ПП 60× 45 мм (увеличено), левое предсердие 48 × 50 мм (увеличено). Увеличение правого желудочка: КДО — 166 мл, нормализованный показатель КДО (КДО/ площади поверхности тела (ППТ)) — 111 мл/м 2 , КСО ПЖ — 104 мл. Фракция выброса ПЖ — 39%. Отмечается гипокинез стенок ПЖ с мелкими участками истончения и дискинеза до 3–4 мм (микроаневризмы). Жировая инфильтрация миокарда ПЖ. ЛЖ: КДР ЛЖ — 45 мм, КСР ЛЖ — 34 мм, КДО ЛЖ — 155 мл, КСО ЛЖ — 68 мл, ФВ — 52%. Клапаны не изменены. Заключение: дилатация полостей правых отделов сердца, снижение сократительной способности ПЖ (ФВ — 39%); гипокинез с участками неравномерного истончения стенок ПЖ; жировая инфильтрация миокарда ПЖ.
На основании клинической картины, результатов лабораторных и инструментальных методов исследования поставлен диагноз: Аритмогенная дисплазия правого желудочка. Синкопальное состояние от 13.04.2020 г.
В настоящем случае отсутствует поражение левого желудочка. По данным литературы, бивентрикулярное поражение отмечается в 16% случаев и прогноз у этих пациентов хуже [9]. Для постановки диагноза АДПЖ, безусловно, наиболее важными были данные, полученные при МРТ сердца.
Для более точной диагностики заболевания в 2010 г. рабочей группой по заболеваниям миокарда и перикарда Европейского общества кардиологов были скорректированы диагностические критерии, которые включают в себя большие и малые критерии из следующих 6 различных категорий:
- региональная дисфункция и структурные изменения по данным Эхо-КГ, МРТ и/или ангиографии ПЖ;
- характеристика стенки по данным эндомиокардиальной биопсии;
- семейный анамнез (ВСС до 35 лет от АДПЖ);
- нарушения реполяризации по данным ЭКГ;
- нарушения деполяризации/проводимости по данным ЭКГ и/или сигналам усредненной ЭКГ аритмии по данным холтеровского мониторирования ЭКГ.
Точный диагноз определяется как присутствие 2 больших критериев, или 1 большого и 2 малых критериев, или 4 малых критериев.
Ниже приведены критерии, которые присутствуют у нашего пациента.
- региональный дискинез правого желудочка в сочетании со сниженной фракцией выброса (ФВПЖ - 39%);
- инвертированный Т-зубец в правых грудных отведениях V1–V3;
- эпсилон-волна в правых грудных отведениях V1–V3.
Малые критерии — желудочковые аритмии, ЖТХ с морфологией блокады ЛНПГ (ось сердца неопределенная) и/или ЖЭС ≥ 500/сут.
Таким образом, у пациента 3 больших критерия и 1 малый, что соответствует определенному диагнозу АДПЖ [11].
Жировая инфильтрация миокарда правого желудочка, определяемая у пациента, более не считается типичным признаком АДПЖ. Более специфичны нарушения кинетики правого желудочка. При эндомиокардиальной биопсии возможно выявление разрушающихся кардиомиоцитов с фиброзным и жировым замещением, атрофии мышечной ткани, но отсутствие этих изменений не отрицает возможность наличия АДПЖ, так как процесс носит очаговый характер. Эндомиокардиальная биопсия не является рутинным методом обследования из-за частичного повреждения миокарда, что приводит к низкой диагностической ценности.
Поскольку АДПЖ часто является причиной ВСС, её предупреждение чрезвычайно актуально у этих пациентов. К предикторам ВСС относятся следующие:
- индуцированная во время электрофизиологического исследования ЖТХ;
- неустойчивая ЖТХ, зафиксированная во время неинвазивной оценки/мониторинга;
- значительная дилатация или обширное вовлечение ПЖ;
- предшествующая остановка сердца в анамнезе;
- необъяснимые обмороки;
- вовлечение левого желудочка;
- мужской пол;
- раннее начало заболевания (ранее 5 лет).
В нашем случае присутствуют такие предикторы ВСС, как индуцированная во время электрофизиологического исследования желудочковая тахикардия; неустойчивая желудочковая тахикардия, зафиксированная во время мониторинга ЭКГ; необъяснимые обмороки, мужской пол. Безусловно, внезапная потеря сознания у больного носила аритмогенный характер и была обусловлена пароксизмом гемодинамически значимой желудочковой тахикардии, что, согласно современным рекомендациям по ВСС, является показанием к имплантации кардиовертера-дефибриллятора [12], что и было произведено. Инвазивное электрофизиологическое исследование может рассматриваться с целью стратификации риска ВСС [12]. Эффективность интервенционного лечения желудочковых тахикардий у пациентов с АДПЖ — низкая из-за очагового и прогрессирующего характера поражения, что не улучшает прогноз. С целью вторичной профилактики ВСС больному был имплантирован автоматический кардиовертердефибриллятор Medtronic GEM III VR 7231 Сх. За 6 месяцев наблюдения не отмечалось его срабатывания. Для уменьшения симптомов, обусловленных аритмией, и их частоты был назначен бетаадреноблокатор метопролол-сукцинат 100 мг/ сут. При повторном ЭКГ-мониторировании отмечалось некоторое снижение ЖЭС с 3412 до 2792, но субъективная переносимость их улучшилась. «Пробежки» ЖТХ не регистрировались. Пациенту рекомендовано избегать активных физических нагрузок и воздержаться от участия в соревновательных видах спорта, поскольку это может спровоцировать фатальные желудочковые аритмии.
Заключение
Представленный случай демонстрирует возможность достаточно позднего дебюта АДПЖ. Для улучшения прогноза заболевания необходима своевременная диагностика с комплексным использованием современных методов и адекватное лечение, направленное, прежде всего, на профилактику внезапной смерти.
Список литературы
2. Бокерия О.Л., Ле Т.Г. Аритмогенная дисплазия правого желудочка. Анналы аритмологии. 2015;12(2):89-99. DOI: 10.15275/annaritmol.2015.2.4
3. Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, DeMazumder D, et al. Central role for GSK3P in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;1(5):e85923. DOI: 10.1172/jci.insight.85923.
6. Bosman LP, Sammani A, James CA, Cadrin-Tourigny J, Calkins H, van Tintelen JP, et al. Predicting arrhythmic risk in arrhythmogenic right ventricular cardiomyopathy: A systematic review and meta-analysis. Heart Rhythm. 2018;15(7):1097-1107. DOI: 10.1016/j.hrthm.2018.01.031
7. Brignole M, Moya A, de Lange FJ, Deharo JC, Elliott PM, Fanciulli A, et al. 2018 ESC Guidelines for the diagnosis and management of syncope. Eur Heart J. 2018;39(21):1883-1948. DOI: 10.1093/eurheartj/ehy037.
9. Gilotra NA, Bhonsale A, James CA, Te Riele ASJ, Murray B, Tichnell C, et al. Heart Failure Is Common and UnderRecognized in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circ Heart Fail. 2017;10(9):e003819. DOI: 10.1161/CIRCHEARTFAILURE.116.003819.
11. Focardi M., Cameli M., Carbone S.F., Massoni A., De Vito R., Lisi M. et al. Traditional and innovative echocardiographic parameters for the analysis of right ventricular performance incomparison with cardiac magnetic resonance. Eur. Heart J. Cardiovasc. Imaging. 2015;6 (1):47-52. DOI: 10.1093/ehjci/jeu156.
12. Silvia P.G., Blomstrom-Lundqvist C., Mazzanti+ A., Bloma N., Borggrefe M., Camm J., et al. Рекомендации ESC по лечению пациентов с желудочковыми нарушениями ритма и профилактике внезапной сердечной смерти 2015. Российский кардиологический журнал. 2016;(7):5-86. DOI: 10.15829/1560-4071-2016-7-5-86
Об авторах
Соловьева Елена Витальевна - кандидат медицинских наук, доцент, кафедра терапии и кардиологии.
Попова Наталия Алексеевна - кандидат медицинских наук, доцент, кафедра терапии и кардиологии.
Власова Татьяна Владимировна - кандидат медицинских наук, доцент, кафедра терапии и кардиологии.
Горбунова Марина Леонидовна - кандидат медицинских наук, доцент, врач кардиолог.
ГБУЗ НО Специализированная кардиохирургическая клиническая больница им. академика Б.А. Королева
Россия
Анцыгина Людмила Николаевна - кандидат медицинских наук, врач кардиолог.
Рецензия
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Аритмогенна кардіоміопатія правого шлуночка
Аритмогенна кардіоміопатія правого шлуночка — захворювання м’яза серця, що характеризується частковим або повним прогресуючим фіброзно-жировим заміщенням міокарда ПШ, пізніше — залученням у процес ЛШ з відносною інтактністю перегородки.
Епідеміологія
Захворювання недавно ідентифіковане та важко діагностується, тому його поширеність точно невідома, але вважається, що може варіювати в межах від 1:3000 до 1:10 000, співвідношення чоловіки : жінки становить 2,5:1. Перші клінічні прояви можуть виникнути в юнацькому віці, рідко — старше 40 років.
Етіологія
Точна причина захворювання невідома, однак у деяких родинах існують безсумнівні докази його успадковування. У більшості родин, де більше одного хворіючого, найбільш імовірним типом успадкування є аутосомно-домінантний. Описаний також один добре відомий варіант аритмогенної кардіоміопатії ПШ, що успадковується за аутосомно-рецесивним типом.
Генетичними дослідженнями ідентифіковано 7 локусів генів, відповідальних за розвиток захворювання. З аритмогенною кардіоміопатією ПШ також асоціюються мутації генів, що кодують білки вставних дисків (десмоплакін, плакоглобін зі специфічним фенотипом, плакофілін, десмоглеїн, десмоколін). Ознаки захворювання можуть варіювати навіть у членів однієї родини, і патологія може виявитися через покоління. Вважається, що заняття спортом не можуть викликати аритмогенну кардіоміопатію ПШ, проте захворювання частіше реєструють серед спортсменів. Мутації генів ріанодинових рецепторів серця (RyR2) асоціюються з поліморфною шлуночковою тахікардією, викликаною фізичними навантаженнями і ювенільною раптовою смертю.
Патологічна анатомія
При морфологічному дослідженні серця залученим виявляється частіше ПШ, що має плямистий вигляд: змінені ділянки можуть бути оточені нормальними тканинами. Залучення ПШ може бути регіонарним (20%) або дифузним (80%). Міокард ПШ прогресивно редукується, заміщаючись жировою і фіброзною тканиною, що відрізняється від нефіброзної жирової інфільтрації, що виникає в ПШ з віком. На ранніх стадіях захворювання стінки правих відділів серця товщають, але надалі через нагромадження жирової тканини та появи ділянок дилатації вони стають тоншими (рис. 12.1а, б).
Рис. 12.1. Аритмогенна кардіоміопатія ПШ: а) ділянки жирової тканини призводять до послаблення та вибухання м’язової стінки; б) ПШ збільшується
Жирове переродження міокарда поширюється частіше від епікардіальних шарів до ендокарда. Міокард уражується переважно в області виносного тракту, верхівки та субтрикуспідальної зони, які розглядаються як «трикутник дисплазії».
При аритмогенній кардіоміопатії ПШ ліпоматоз супроводжується переважно дилатацією виносного тракту, ПШ або генералізованою дилатацією. Фіброліпоматоз характеризується наявністю фокальної аневризми ПШ і випинанням в області верхівки, нижньої стінки, субтрикуспідальної та інфундибулярної зони.
У міру прогресування фіброзно-жирова дистрофія уражує також ЛШ та передсердя.
Патогенез
Серед молекулярних механізмів аритмогенної кардіоміопатії ПШ розглядаються генетично детермінуючі мутації в десмосомальних протеїнах, а також інгібування сигнальних шляхів. Стрес-індукований розрив десмосомальних зв’язків клітин може запускати процес апоптоза, викликати атрофію міокарда і заміщення його жировою тканиною.
Вогнища жирового переродження і інтерстиціального фіброзу при аритмогенній кардіоміопатії ПШ не проводять електричні імпульси, внаслідок чого дезорганізована структура серця зумовлює виникнення безладної електричної активності, електричні імпульси можуть ставати невпорядкованими, внаслідок чого, крім порушень ритму серця, можуть виникати порушення його скоротності (рис. 12.2а, б).
Рис. 12.2. Серце при аритмогенній кардіоміопатії ПШ: а) до скорочення; б) після скорочення
Клінічна картина
Основними клінічними симптомами аритмогенної кардіоміопатії ПШ є:
- відчуття серцебиття, перебоїв у роботі серця, напади шлуночкової тахікардії;
- підвищена стомлюваність, запаморочення, непритомність;
- симптоми СН;
- раптова зупинка кровообігу.
Описані 4 клінічні стадії захворювання:
- субклінічна, незначні шлуночкові аритмії можуть відзначатися або бути відсутні;
- стадія явних електричних порушень, правошлуночкові аритмії і ризик зупинки серця пов’язані з морфофункціональними змінами ПШ;
- стадія правошлуночкової недостатності з прогресуючим залученням ПШ і наступною його глобальною систолічною дисфункцією;
- стадія кінцевої бівентрикулярної СН.
Діагностика
На ЕКГ визначаються:
- спонтанні шлуночкові тахікардії зі зміною комплексу QRS по типу блокади лівої ніжки пучка Гіса;
- негативні зубці Т у відведеннях V1—4 на фоні синусового ритму;
- розширення комплексу QRS;
- неповна блокада правої ніжки пучка Гіса;
- ектопічні тяжкі аритмії: шлуночкова екстрасистолія, фібриляція шлуночків, передсердна тахікардія, фібриляція передсердь.
Приблизно у ⅓ пацієнтів реєструється характерна епсілон-хвиля і ППШ.
Методом холтерівського моніторингу можна діагностувати епізоди шлуночкової тахіаритмії. Для оцінки прогресування захворювання важливо проводити реєстрацію ЕКГ у динаміці.
При ехоКГ-обстеженні виявляються:
- дилатація ПШ і порушення його скоротності (асинергія, дифузна гіпокінезія, зниження ФВ);
- локальна аневризма ПШ;
- підвищена трабекулярність;
- трикуспідальна регургітація;
- емболія ЛА;
- збільшення правого передсердя;
- ліві відділи серця частіше не змінені.
За допомогою допплєр-ехоКГ визначається порушення діастолічної функції ПШ і трикуспідальна регургітація. Для більш точної візуалізації ПШ застосовують контрастну ехоКГ міокарда.
Методом МРТ візуалізують ділянки заміщення міокарда жировою тканиною, фокальне стоншення стінки та локальні аневризми. Продемонстровано гарну кореляцію між результатами цього методу і результатами морфологічного дослідження міокарда.
Для підтвердження діагнозу використовують рентгенконтрастну вентрикулографію, при якій виявляють дилатацію ПШ з сегментарними порушеннями його скорочення, випинання контуру в ділянці дисплазії і підвищення трабекулярності.
При ендоміокардіальній біопсії визначають фіброзно-жирову інфільтрацію міокарда ПШ.
Через труднощі та ризик проведення біопсії для підтвердження діагнозу «аритмогенна кардіоміопатія ПШ», а також неточностей в оцінці структури і функції ПШ за допомогою неінвазивних тестів Європейським кардіологічним товариством та Міжнародним товариством і кардіологічною федерацією розроблені критерії, згідно з якими діагноз встановлюють при наявності 2 великих або 1 великого + 2 малих або 4 малих діагностичних критеріїв (Corrado D. et al., 2000).
Великі діагностичні критерії:
- сімейний характер захворювання, підтверджений даними аутопсії або при хірургічному втручанні;
- епсилон-хвиля або локалізоване розширення комплексу QRS (>110 мс) у правих грудних відведеннях (V1–V3);
- фіброліпоматозне заміщення міокарда за даним ендоміокардіальної біопсії;
- значна дилатація і зниження ФВ ПШ при відсутності або мінімальному залученні ЛШ;
- локалізована аневризма ПШ;
- виражена сегментарна дилатація ПШ.
Малі діагностичні критерії:
- наявність у сімейному анамнезі випадків передчасної раптової смерті (у осіб віком молодше 35 років) внаслідок передбачуваної аритмогенної кардіоміопатії ПШ;
- ППШ на усередненій ЕКГ;
- інвертований зубець Т у правих грудних відведеннях у осіб віком старше 12 років при відсутності блокади правої ніжки пучка Гіса;
- шлуночкова тахікардія з ознаками блокади лівої ніжки пучка Гіса, документована за даними ЕКГ або результатами холтерівського моніторування або під час навантажувального тесту;
- часті шлуночкові екстрасистоли (>1000/24 год при холтерівському моніторуванні ЕКГ);
- помірна глобальна дилатація або зниження ФВ ПШ при незміненому ЛШ;
- помірна сегментарна дилатація ПШ;
- регіонарна гіпокінезія ПШ.
Лікування
Для вибору антиаритмічної терапії необхідне проведення інвазивного ЕФД і проб з дозованим фізичним навантаженням. Серед антиаритмічних засобів ефективні аміодарон і соталол. Дигоксин застосовують при тахісистолічній формі фібриляції передсердь для зниження ЧСС. Для відновлення синусового ритму проводять кардіоверсію.
Діуретики застосовують при СН у хворих із затримкою рідини.
З хірургічних методів лікування застосовують абляцію, якщо джерело порушеної електричної активності ідентифіковано за допомогою електрофізіологічних тестів. У випадках, якщо аритмія не контролюється за допомогою лікарських засобів або абляції (велике ураження або наявність множинних аритмогенних вогнищ), імплантують кардіовертер-дефібрилятор, у деяких випадках потрібна імплантація водія ритму. Трансплантацію серця застосовують рідко, якщо неможливе проведення контролю ритму іншими методами.
Прогноз
Результати нещодавно проведеного дослідження, яке включало 130 пацієнтів з аритмогенною кардіоміопатією ПШ, показали, що серцево-судинна смертність становила 16% (n=24), найбільш частою причиною була раптова смерть (29%) і СН (59%). Аналіз чинників ризику виявив найбільш несприятливі — наявність дисфункції ПШ або ЛШ і шлуночкову тахікардію.
Правожелудочковая аритмогенная кардиомиопатия
Кардиомиопатии – заболевания миокарда неизвестной этиологии. Выделяют 4 вида кардиомиопатий: дилатационная, гипертрофическая, рестриктивная и правожелудочковая аритмогенная. Если гипертрофическая и дилатационная кардиомиопатии относительно известны кардиологам, то диагноз правожелудочковая аритмогенная кардиомиопатия (ПКМП) встречается крайне редко и не всегда правильно трактуется. Иногда ПКМП диагностируется у больных с дилатацией полости правого желудочка (ПЖ) после предшествующих длительных нарушений ритма. Это принципиально неправильно, так как нарушается причинно-следственная связь: при ПКМП первичным является перерождение миокарда с замещением его жировой и соединительной тканью. Гетерогенность миокарда правого желудочка (наличие мышечной, жировой, соединительной ткани) с различными электрофизиологическими характеристиками является основанием для развития в последующем правожелудочковых аритмий.
Согласно определению McKenna, «правожелудочковая аритмогенная кардиомиопатия – заболевание, характеризующееся прогрессивным замещением миокарда правого желудочка соединительной или жировой тканью, с редким вовлечением в процесс миокарда левого желудочка, как правило, не поражает межжелудочковую перегородку». Термин «аритмогенная дисплазия правого желудочка» предложен G. Fontaine в 1977 г., поэтому это заболевание часто называют болезнью Фонтейна. В 1982 г. Marcus предложил термин «аритмогенная правожелудочковая кардиомиопатия, или аритмогенная болезнь правого желудочка». Многие авторы рассматривают ПКМП как миокардиальный феномен, однако, по мнению Fontaine, ПКМП является проявлением дисплазии. D. Corrado и соавт. считают, что в 76% случаев ПКМП в процесс вовлекается и левый желудочек (ЛЖ). А по данным C. McRae и соавт., ЛЖ поражается в 50% случаев ПКМП, что сопровождается наличием выраженной дилатационной кардиомиопатии.
ПКМП – относительно редкое заболевание и по данным Perry встречается с частотой 1:5000. Выделены отдельные регионы, где ее распространенность выше, например Греция (о. Наксос) и Италия (Венеция). Ранняя диагностика ПКМП позволяет предупредить возникновение летальных аритмий и внезапную сердечную смерть. Трагичность заболевания заключается в его бессимптомном начале и неясной причине возникновения.
Несмотря на то что ПКМП описана более 30 лет назад, в отечественной литературе практически нет посвященных ей работ. В значительной степени это связано с относительно редкой встречаемостью ПКМП, отсутствием специфической клинической картины, необходимостью использования дорогостоящих, не всегда доступных методов инструментального обследования, а также с недостаточной настороженностью и знаниями врачей об этом заболевании. Судить о распространенности ПКМП в Украине достаточно сложно, так как заболевание практически не диагностируется.
Известно, что заболевание чаще манифестирует в молодом (до 40 лет) возрасте, преимущественно у мужчин (соотношение 4:1). Иногда первым проявлением заболевания является внезапная смерть. Наряду с гипертрофической кардиомиопатией, ПКМП рассматривается как основная причина внезапной смерти у молодых спортсменов – 22%.
Генетические основы ПКМП
Этиология правожелудочковой аритмогенной дисплазии в настоящее время изучена недостаточно. Чаще всего заболевание носит идиопатический или наследственный характер. Показано, что с генетической точки зрения когорта больных достаточно гетерогенна, выявлены как аутосомно-доминантные, так и рецессивные типы наследования. Кроме того, идентифицировано 6 генов и 9 независимых локусов, ответственных за развитие правожелудочковой дисплазии/кардиомиопатии. Мутантные гены, ассоциированные с правожелудочковой кардиомиопатией, выявлены в 14 [q23-24] и 17, 12, 18 [q21] хромосомах. Они включают промежуточные филаменты, десмоплакин, плакофиллин, плакоглобин, ядро – факторы защиты миокарда от воздействия механического стресса на клеточном уровне. Помимо этого, десмосомы входят в структуру вставочного сердечного диска и участвуют во внутриклеточных сигнальных сетях, которые сейчас прицельно изучаются in vitro. Проявлением данных мутаций является нарушение функции сократительных белков и их взаимодействия.
Кроме того, выделяют и другие варианты ПКМП:
1. Врожденная аномалия развития миокарда ПЖ с клиническим проявлением – внезапной смертью.
2. Следствие дисплазии, обусловленной метаболическими нарушениями, поражающими ПЖ и вызывающими прогрессирующее замещение миоцитов.
3. Воспалительного генеза: дисплазия как результат миокардита, когда инфекция не оставляет следов первичного воспаления. По данным F. Calabrese и соавт., в случаях ПКМП часто обнаруживали миокардит, в связи с чем рассматривают этиологический агент заболевания в виде группы кардиотропных вирусов. Е. Нурмухаметова считает самой частой причиной миокардита поражение вирусом Коксаки группы В. При этом возможно вовлечение как проводящей системы сердца, так и непосредственно миокарда. Но Fontaine придерживается другой точки зрения: пациенты с ПКМП склонны к возникновению инфекционных миокардитов, то есть изменена интерпретация причинно-следственной связи. По мнению Peters, острый/хронический миокардит приводит к вовлечению в процесс левые отделы сердца, что является прогностически неблагоприятным признаком. Ввиду противоречивых данных роль инфекционного миокардита при ПКМП требует дальнейшего изучения.
4. По мнению Turrini, Corrado, ПКМП является следствием дистрофии миокарда с уменьшением массы миокарда, его дисфункцией, электрической нестабильностью и сердечной недостаточностью.
5. Morgera и соавт. отметили ассоциации блокады левой ножки пучка Гиса с аритмиями и идиопатическими желудочковыми тахиаритмиями.
6. По мнению Folino, существует корреляционная зависимость между снижением вагусного воздействия и степенью тяжести болезни.
Классификация ПКМП
Группа ученых во главе с G. Fontaine на протяжении 23 лет исследовала более 250 пациентов не только во Франции, но и за ее пределами, включая Японию, США, Австралию. На основании наблюдения ими была предложена классификация ПКМП.
Дисплазия с вовлечением ЛЖ
1. Бивентрикулярная дисплазия характеризуется поражением обоих желудочков. Типичная гистологическая структура ЛЖ: замещение жировой тканью, фиброзным ограничением. Это состояние приводит к сердечной недостаточности в связи с чрезмерным уменьшением миокарда ЛЖ и может быть ошибочно диагностировано как идиопатическая дилатационная кардиомиопатия. Дифференциально-диагностическим критерием является наличие жировой инфильтрации миокарда.
2. Дисплазия, осложненная миокардитом – в таком случае вовлекаются оба желудочка, прогноз неблагоприятный. В большинстве случаев в структурной основе ПКМП миокардит генетически предопределен. При миокардите с вовлечением обоих желудочков возникает сердечная недостаточность, приводящая к смертельному исходу, уносящая жизни 1% пациентов в год.
Сложной является постановка диагноза в случаях неаритмогенных форм, осложненных миокардитом.
Диагностика
Диагноз ПКМП ставится на основании структурных, гистологических, ЭКГ, генетических факторов. Определить функциональные и структурные повреждения позволяют исследования: ЭхоКГ, ангиография, ядерно-магнитный резонанс с томографией или радионуклидное исследование.
Диагностика правожелудочковой кардиомиопатии достаточно сложна, так как нет характерных специфических или клинических жалоб либо клинических проявлений. В начале течение заболевания может быть бессимптомным или со стертой симптоматикой. Основные жалобы связаны с нарушениями ритма, и их проявления зависят от тяжести аритмии. Единичные правожелудочковые экстрасистолы больные могут субъективно не ощущать, в то время как наличие желудочковой экстрасистолии высоких градаций или желудочковой тахикардии проявляется пресинкопальные и синкопальные состояния, общая слабость.
Большое значение в диагностике правожелудочковой кардиомиопатии имеет ЭКГ, изменения на которой относительно специфичны. При проведении ЭКГ часто отмечаются отрицательные зубцы Т в отведениях V1-V2, а при вовлечении ЛЖ – также в V4. При этом продолжительность комплекса QRS в правых грудных отведениях превышает 110 мс при его неизмененной ширине в отведении V6. Большая продолжительность комплекса QRS в правых грудных отведениях по сравнению с левыми сохраняется и в случаях блокады правой ножки пучка Гиса. Такая более чем полная блокада обусловлена сопутствующей париетальной блокадой проводящей системы ПЖ.
Весьма характерны различные эктопические желудочковые аритмии, вплоть до стойкой желудочковой тахикардии, при которой желудочковые комплексы обычно имеют вид блокады левой ножки пучка Гиса, а электрическая ось сердца может быть отклонена как вправо, так и влево. Пароксизмальная желудочковая тахикардия в большинстве случаев возникает в ПЖ и легко индуцируется при электрофизиологическом исследовании. У таких больных зачастую выражена дисперсия интервала QT в различных отведениях, а на сигнал-усредненной ЭКГ обнаруживаются поздние желудочковые потенциалы. По данным Corrado, в 14% случаев внезапной смерти у молодых пациентов в Венеции на ЭКГ зафиксирована элевация сегмента ST, аутопсия подтвердила ПКМП.
При ЭхоКГ определяется дилатация ПЖ, сокращения которого в типичных случаях носят асинергичный характер. У небольшой части больных наблюдается диффузная гипокинезия ПЖ. Определяется снижение фракции выброса (ФВ) ПЖ до 28%. Левые отделы сердца чаще не изменены. При сопутствующем миокардите характерно вовлечение ЛЖ со снижением его ФВ. По мнению Y. Juilliere, ФВ обоих желудочков является предиктором выживаемости при развитии идиопатической дилатационной кардиомиопатии. Согласно данным D. Mehta, наиболее чувствительным методом диагностики является 2-режимное эхокардиографическое исследование.
Сопоставляя результаты ЭКГ в 12 отведениях, ЭхоКГ (увеличение полости ПЖ) и эндомиокардиальной биопсии, можно говорить о степени тяжести дисфункции ПЖ.
Контрастная селективная правожелудочковая вентрикулография является золотым стандартом с условием соответствующего обзора и при избежании появления преждевременных желудочковых экстрасистолий [32, 48]. При этом характерна дилатация ПЖ в сочетании с сегментарными нарушениями его сокращения, выпячиваниями контура в областях дисплазии и увеличением трабекулярности. Это отличает аритмогенную кардиомиопатию ПЖ от правожелудочковой дилатационной кардиомиопатии и «чистого» миокардита, при которых гипокинезия ПЖ, а также ЛЖ носят диффузный характер.
Радионуклидная ангиография определяет снижение ФВ ПЖ с нормальной или относительно сохраненной функцией ЛЖ. Такие факторы, как наличие QRS-дисперсии, синкопальных эпизодов в анамнезе и подтвержденные радионуклидной ангиографией изменения ПЖ и/или ЛЖ, являются независимыми неинвазивными предикторами внезапной смерти.
Электронная микроскопия позволяет определить ремоделирование вставочных дисков кардиомиоцитов.
Магнитно-резонансная томография позволяет производить запись замещения миокарда фиброзно-жировой тканью, выявлять очаги диаметром от нескольких миллиметров, производить расчет содержания жировой ткани.
Стандартом диагностики является гистологическое подтверждение трансмурального фиброзно-жирового замещения миокарда ПЖ. Патологический процесс носит очаговый характер. Лишь в поздних стадиях слияние отдельных очагов может создавать впечатление диффузного поражения ПЖ. Тем не менее, биопсия не является чувствительным методом исследования из-за сегментного (островкового) повреждения, а также в связи с тем, что забор материала производят из области межжелудочковой перегородки, тогда как при данной патологии жировое перерождение распространяется из эпикарда в эндокард. При осмотре макропрепарата сердца ПЖ дилатирован, истончен и покрыт жировой тканью. Часто определяются аневризмы книзу от трехстворчатого клапана и в области верхушки. В ряде случаев дисплазия распространяется на часть ЛЖ. W.J. McKenna предложил диагностические критерии аритмогенной дисплазии ПЖ, среди которых выделяют большие и малые (табл.). О наличии ПКМП свидетельствует выявление 2 больших, либо 1 большого и 2 малых, либо 4 малых критериев.
Прогноз
Неинвазивными предикторами неблагоприятного прогноза при ПКМП являются преклонный возраст, синкопы, сердечная недостаточность, вовлечение в процесс ЛЖ и желудочковые аритмии. Дисперсия QRS ≥40 мс рассматривается как независимый предиктор синдрома внезапной сердечной смерти при ПКМП. Течение заболевания может быть осложнено развитием сердечной недостаточности. Сердечная недостаточность может быть как изолированной правожелудочковой, так и бивентрикулярной с развитием систолической недостаточности в течение 4-8 лет с момента возникновения полной блокады правой ножки пучка Гиса.
Лечение
При ПКМП применяют медикаментозное, неинвазивное и оперативное лечение.
Медикаментозное лечение проводится только как симптоматическая терапия и предусматривает устранение и предотвращение жизнеугрожающих аритмий, реже – проявлений застойной сердечной недостаточности. Лучшие результаты получены при использовании соталола (83%) в сравнении с верапамилом, эффективность которого составила 50%, амиодароном (25%) и бета-блокаторами (29%). В тяжелых случаях при хорошей переносимости с соблюдением мер предосторожности можно использовать комбинации препаратов, например амиодарона с бета-адреноблокаторами или амиодарона с флекаинидом или другими антиаритмическими средствами класса 1С. В первом случае учитывается положительное фармакодинамическое, а во втором – фармакокинетическое взаимодействие комбинируемых лекарственных средств. Флекаинид можно сочетать также с бета-адреноблокаторами. При недостаточной эффективности, оцениваемой с использованием данных холтеровского мониторирования ЭКГ, подбор методов антиаритмической терапии целесообразно проводить с помощью электрофизиологического исследования.
Лечение застойной сердечной недостаточности проводят общепринятыми методами. Особенно эффективны карведилол и ингибиторы АПФ.
При брадикардии, в том числе и индуцированной антиаритмической терапией, рекомендуется установка электрокардиостимулятора.
В случаях рефрактерности к терапии и при высоком риске развития синдрома внезапной сердечной смерти прибегают к неинвазивным методам лечения: имплантации дефибриллятора-кардиовертера или радиочастотной абляции. По данным Gatzoulis, на о. Наксос двум пациентам со злокачественными желудочковыми аритмиями имплантированы автоматические дефибрилляторы. По мнению S. Peter, абляция проводится только при ангиографическом подтверждении фокальной дисплазии. По данным Masedo, при липоматозной инфильтрации ПЖ≥6 мм (согласно результатам магнитно-резонансного исследования) без локальной или распространенной дисфункции ПЖ следует осторожно проводить имплантацию кардиовертерадефибриллятора и использовать лекарственные препараты. Имплантация кардиовертера-дефибриллятора, как правило, переносится без осложнений и позволяет снизить смертность.
У больных с упорными потенциально фатальными желудочковыми аритмиями, особенно в сочетании с дисфункцией ЛЖ и застойной сердечной недостаточностью, эффективно хирургическое лечение – вентрикулотомия, обеспечивающая прерывание циркуляции патологической волны возбуждения в ПЖ.
Среди методов оперативного вмешательства самым эффективным является трансплантация сердца. Однако, ввиду многих причин, она производится крайне редко, и данные по этому поводу в литературе встречаются нечасто.
Наряду с тем что в изучении ПКМП достигнуты определенные результаты, присутствие «белых пятен» в этиологии этого заболевания свидетельствует о необходимости проведения дальнейших исследований по данной теме.
Список литературы находится в редакции.
СТАТТІ ЗА ТЕМОЮ
Компанія Berlin Chemie глибоко засмучена трагічною ситуацією в Україні, руйнуванням і жертвами серед мирного населення. Ми категорично засуджуємо всі акти агресії в Україні та світі, докладаємо значних зусиль для допомоги в розв’язанні гуманітарної кризи та забезпеченні доступу пацієнтів до лікарських засобів. .
Система гемостазу є складною, адже фізіологічно вона призначена для захисту людини від кровотечі. Ця система задіюється одразу після ушкодження ендотелію судин з одночасною синергічною взаємодією тромбоцитів і факторів згортання. Водночас у здорових людей цей процес здійснюється так, щоб уникнути надлишкового утворення та відкладення фібрину всередині кровоносних судин, з одного боку, і бути готовим зупинити кровотечу – з іншого. Для досягнення цієї найважливішої мети потрібне тонке регулювання її діяльності. Інакше кажучи, всі дії системи гемостазу перебувають під постійним контролем для забезпечення ідеального балансу на відстані від Сцилли (кровотеча) та Харибди (тромбоз). Ця система є динамічною і дуже залежить від віку. Існують значні відмінності між системою згортання в новонароджених порівняно з дітьми та дорослими. Так само як Одіссею та аргонавтам для того, щоб вижити, потрібно було пропливти через вузьку протоку між Сциллою та Харибдою, так і новонародженому в перші години та дні життя необхідно лавірувати між ризиком тромбозу та ризиком геморагії. .
При складанні плану ведення пацієнта з хронічним болем старшої вікової групи слід розглянути можливість комбінованого застосування фармакологічного та нефармакологічного втручань. Вибір лікарських засобів для фармакологічного контролю болю в осіб старших вікових груп потребує суворого персоніфікованого підходу.
Сьогодні в рамках оцінки чинників ризику серцево-судинних (СС) захворювань дедалі більший інтерес наукового світу прикутий до ендотелію та його функції. Ендотелій уражається насамперед унаслідок артеріальної гіпертензії (АГ), це проявляється розвитком його дисфункції та морфологічною перебудовою артеріальної стінки. Добре відомо, що з віком підвищується судинна жорсткість, що пов’язують зі зниженням в артеріальній стінці вмісту еластину та підвищенням кількості колагену 3. У разі АГ перебіг цих процесів значно прискорюється – судинний вік починає випереджати хронологічний (паспортний). Тому дедалі більше науковців для опису функціонального та морфологічного стану серцево-судинної системи використовують термін «синдром раннього старіння судин», або EVA‑синдром (early vascular aging), як модель старіння судин, яка більш точно відображає структурні та функціональні зміни, що відбуваються в організмі в міру його старіння [1].
Аритмогенная дисплазия правого желудочка. Аритмогенная правожелудочковая кардиомиопатия
Аритмогенная (вызывающая аритмию) дисплазия/кардиомиопатия правого желудочка (АДПЖ, АКМП ПЖ) – столь громоздким названием означается опасное кардиологическое заболевание, известное с ХVIII века, но до сих пор в ряде аспектов изученное недостаточно.
Прославленный врач-анатом и мыслитель-материалист своего времени, автор ряда выдающихся трудов о малярии, чуме сельскохозяйственных животных, сифилисе, строении мозга, – итальянец Джованни Мария Ланчизи (1654-1720) занимался исследованиями, в том числе, сердечнососудистой системы. Однако одна из главных его работ, – «De Motu Cordis et Aneurysmatibus», – в которой дано первое клиническое описание аритмогенной дисплазии ПЖ, была опубликована лишь посмертно, в 1728 году. Ланчизи описал семейную болезнь, появлявшуюся в четырех поколениях и результировавшую неожиданно и фатально (спустя два с половиной века такой исход станут называть внезапной сердечной смертью).
Современные нозологические определения «аритмогенная дисплазия», «аритмогенная кардиомиопатия правого желудочка» появились сравнительно недавно, в конце 1970 - начале 80-х годов, и в МКБ-10 они были отнесены к рубрике «Другие болезни сердца / Кардиомиопатии»). Суть данной патологии повторяет закономерности очень многих фиброзов, однако обладает и выраженными отличительными особенностями: паренхиматозная мышечная ткань сердца, состоящая из функциональных клеток-кардиомиоцитов, перерождается и вытесняется отчасти жировой, отчасти соединительной (рубцовой) тканями, что неизбежно сказывается на функционировании миокарда в целом, – вызывая, в частности, характерные сбои сердечного ритма, которые и дали название болезни. Специфика заключается в том, что фиброзирующий процесс и жировая дегенерация почти всегда локализованы в т.н. «диспластическом треугольнике» правого желудочка сердечной мышцы, т.е. в зоне, ограниченной верхушкой сердца, входными и выводными путями желудочка.
АКМП ПЖ считается редким заболеванием, однако же установлено, что оно выступает второй из наиболее распространенных причин внезапной сердечной смерти в молодом возрасте. В особенности это касается спортсменов, среди которых такой исход нередко становится первым, единственным и последним проявлением болезни. В целом, доля АКМП ПЖ как причины внезапной сердечной смерти составляет, по разным оценкам, от 20% до 25%, а в возрастных выборках младше 20 лет этот показатель достигает 26%. Около 80% случаев аритмогенной правожелудочковой кардиомиопатии диагностируется у лиц в возрасте до 40 лет.
Частота встречаемости АКМП ПЖ в пересчете на общую популяцию оценивается по-разному; обычно приводятся данные на уровне 0,05%-0,5%. В странах средиземноморского бассейна заболевание регистрируется существенно чаще: до 0,8% от общей популяции. Преобладают пациенты мужского пола.
Однако все эти данные требуют тщательной верификации и уточнения на больших выборках, что затрудняется сложностью диагностики (во многих случаях аритмогенная дисплазия ПЖ не распознается и не регистрируется как таковая) и относительной редкостью заболевания.
2. Причины
Генетический, наследственный характер заболевания распространяется лишь на 30-50% случаев, причем уже идентифицированы ответственные за его развитие гены. В региональном варианте АКМП ПЖ, известном как болезнь острова Наксос (Греция), фиброзно-жировая дегенерация и прогрессирующая сердечная недостаточность сопровождаются врожденными аномалиями кожи и волос (кератодермия, «шерстяные волосы»).
Этиопатогенез всех прочих, ненаследственных случаев остается, по сути, неизвестным. Рассматриваются и обсуждаются гипотезы, согласно которым дегенеративный процесс в правом желудочке миокарда запускается вследствие бактериального или вирусного инфекционного воспаления; идиопатическими (сугубо индивидуальными) особенностями внутриутробного развития; аномально ускоренным апоптозом (преждевременной гибелью кардиомиоцитов после значительно меньшего, чем в норме, числа клеточных делений); спонтанным перерождением кардиомиоцитов в клетки других типов (трансдифференциация). Однако даже если какое-либо из этих предположений подтвердится статистически, оно мало чего будет стоить без понимания внутренних причин: почему у одних людей аритмогенная дисплазия запускается и развивается, а у других нет.
3. Симптомы и диагностика
Клиническая картина АКМП ПЖ требует дифференциации с множеством иных заболеваний и состояний, при которых постепенно прогрессирующая сердечная недостаточность сочетается с эпизодами аритмии. Как правило, пациенты начинают обращать внимание на усиленное сердцебиение, перебои сердечного ритма, одышку, затем появляются боли в сердце, головокружения, внезапные синкопальные состояния с потерей сознания (прогностически неблагоприятный признак, который считается одним из возможных предвестников внезапной сердечной смерти). Нарастает утомляемость, пациент все хуже переносит физические нагрузки.
Однако во многих случаях прогрессирование фиброзно-жировой дегенерации в течение длительного времени не сопровождается сколько-нибудь значимыми субъективными ощущениями или клиническими симптомами.
Учитывая диагностическую сложность АКМП ПЖ, международные кардиологические ассоциации предпринимали попытки не только унифицировать терминологию, но и разработать надежные протоколы распознавания и дифференциальной диагностики. Так, необходимым для установления диагноза считается присутствие не менее 3% соединительной и не менее 40% жировой ткани в гистологической структуре правого желудочка; применяются также электрокардиографические, аускультативные и другие диагностические критерии. В силу очевидных причин требуется изучение семейного анамнеза.
По мере необходимости назначают ЭЭГ, эхокардиографию, нагрузочные тесты, холтеровское мониторирование, рентгенографию, МРТ, а если все эти методы не позволяют однозначно определиться с диагнозом – приходится прибегать к инвазивной эндомиокардиальной биопсии, отбирая тканевые образцы для гистологического анализа.
4. Лечение
Этиопатогенетической терапии в настоящее время не существует, и она не появится до окончательного прояснения всех вопросов, связанных с причинами и патогенезом аритмогенной правожелудочковой дисплазии. Риск внезапной фибрилляции и остановки сердца снижают бета-адреноблокаторами и другими антиаритмическими средствами; симптоматически назначают сердечные гликозиды, мочегонные, ингибиторы АТФ и т.д. Эффективность таких тактических схем требует дополнительных исследований. Обязательно вносятся коррективы в образ жизни, категорически исключаются любые факторы риска и факторы-провокаторы. В качестве лечения некоторыми кардиоцентрами практикуется радиочастотная катетерная абляция, однако этот опыт также пока изучен недостаточно.
Высказываются мнения о том, что методом выбора следует признать радикальное вмешательство – пересадку сердца или, по меньшей мере, вживление кардиовертера-дефибриллятора, – однако другим специалистам риск возможных осложнений при транс- или имплантации представляется оправданным лишь в тех случаях, когда процесс прогрессирует быстро и проявляется заведомо угрожающей симптоматикой.
Читайте также: