Энцефалит Расмуссена как причина эпилепсии
Добавил пользователь Владимир З. Обновлено: 29.01.2026
Провели клинические и ЭЭГ-обследования 2 пациентов с острым энцефалитом, вызванным вирусом простого герпеса (ЭВВПГ): М. Б., 23-летняя женщина, получавшая лечение на дому по поводу тяжелого органического психосиндрома, постоянный уход за которой осуществляла медсестра, и Л. Й., 16-летняя пациентка, на 3-м году жизни перенесшая ЭВВПГ, который перешел в аутоагрессивную форму, напоминавшую энцефалит Расмуссена, с эпилептическим статусом в виде длительной парциальной эпилепсии. Проанализировали ранние и поздние исходы.
Данные ЭЭГ статистически обработали с использованием «спектрального анализа мощности» и трехмерного цветного картирования, позволяющего выявить топическую локализацию отдельных диапазонов частот ЭЭГ-кривых и измерить межнейронные связи в продольном и поперечном направлениях с помощью определения средней когерентности (показателя межнейронных связей).
На 9-м году жизни Л. Й. перенесла ветряную оспу с выраженными высыпаниями на коже и повышением температуры тела. При этом у нее выявили уменьшение высокоактивных изменений на ЭЭГ и клинической симптоматики, в том числе фокальной. Данное уменьшение произошло на фоне применения анестетика тиопентала и болюсного введения кортикостероидов, а также приема амантадина сульфата.
При осмотре во время самостоятельной ходьбы у Л. Й. обнаружили центральный монопарез правой нижней конечности, а также — 4–5 коротких миоклонических спазмов в суставах рук при приведении, в сочетании с непроизвольными выкриками, напоминающими крик осла, глубокими вдохами и выдохами. Больная находилась при этом в полном сознании. По данным МРТ выявили область гиперинтенсивного сигнала парасагиттально на левой стороне двигательного центра, по данным ЭЭГ — признаки эпилептогенной активности в области прилежащего рубца. По результатам сравнения последней ЭЭГ пациентки Л. Й. с ЭЭГ ее гомозиготной сестры патологической активности не выявили — записи были схожи.
Обе пациентки выжили на фоне длительного противовирусного лечения, интенсивной терапии, направленной на защиту нейронов головного мозга, эффект которой был доказан в динамике по данным классического и статистического анализа ЭЭГ.
Поздние исходы у обследованных пациенток были диаметрально противоположными. Пациентка Л. Й. окончила 9-ти летнюю среднюю школу с хорошими результатами. Пациентка М. Б. была постепенно мобилизована, несмотря на клинические признаки постэнцефалитной энцефалопатии с недостаточным ответом на нейролептики и седативные средства. После получения согласия родителей ее перевели в областную больницу.
Ключевые слова
Об авторах
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса
03659, г. Мартин, ул. Колларова, д. 2
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса, 03659, г. Мартин, ул. Колларова, д. 2;
Клиника инфектологии и медицины путешествий, медицинский факультет им. Я. Ессениуса, 03659, г. Мартин, ул. Колларова, д. 2
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса
03659, г. Мартин, ул. Колларова, д. 2
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса, 03659, г. Мартин, ул. Колларова, д. 2;
Клиника инфектологии и медицины путешествий, медицинский факультет им. Я. Ессениуса, 03659, г. Мартин, ул. Колларова, д. 2
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса
03659, г. Мартин, ул. Колларова, д. 2
Беата Дробна Саньева
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса
03659, г. Мартин, ул. Колларова, д. 2
Клиника анестезиологии и интенсивной медицины, медицинский факультет им. Я. Ессениуса
03659, г. Мартин, ул. Колларова, д. 2
Список литературы
1. Mayer V., Drobný M., Mittrová E. Pomalé vírusové neuroinfekcie. Edition, Osveta, Martin, 1983, 407p.
2. Xu F., Sternburg M., Kottiri B., McQuilan G., Lee F., Nahmias A. Berman S., Markowitz L. Trends in Herpes Simplex Virus Type1 and Type 2 Seroprevalence in the United States. AMA. 2006; 8. 296 (8): 964–973. PMID: 16926356 DOI: 10.1001/jama.296.8.964
3. Whitley R.J. Herpes simplex encephalitis: adolescents and adults. Antiviral Res. 2006; 71 (2–3): 141–148. PMID: 16675036 DOI: 10.1016/j.antiviral.2006.04.002
4. Whitley R.J., Gnann J.W. Viral encephalitis: familiar infections and emerging pathogens. Lancet. 2002; 359 (9305): 507–513. PMID: 11853816 DOI: 10.1016/S0140-6736(02)07681-X
5. Pandian J.D., Thomas S.V., Santoshkumar B., Radhakrishnan K., Sarma P.S., Joseph S., Kesavadas C. Epilepsia partialis continua-a clinical and electroencephalography study. Seizure. 2002; 11 (7): 437–441.
6. Varatharaj A., Nicoll James A.R., Pelosi E., Pinto A.A. Corticosteroid-responsive focal granulomatous herpes simplex type-1 encephalitis in adults. Practical Neurology. 2017; 17 (2): 140–144. PMID: 28153849 DOI: 10.1136/practneurol-2016-001474
7. Sániová B., — Drobný M., Lehotský J., Šulaj M., Schudichová J: Biochemical and clinical improvement of cytotoxic state by amantadine sulphate In: Cellular and Molecular Neurobiology. 2006; 26 (7–8): 1473–1480. PMID: 16710757 DOI: 10.1007/s10571-006-9033-0
8. Saniova B., Drobny M., Kneslova L., Minarik M. The outcome of patients with severe head injuries treated with amantadine sulphate. J Neural Transm. 2004; 111 (4): 511–514. PMID: 15057520 DOI: 10.1007/s00702-004-0112-4
9. Wang Y., Qin Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010; 15: 1382–1402. DOI: 10.1007/s10495-010-0481-0
10. Bien C.G., Bauer J., Deckwerth T.L., Wiendl H., Deckert M., Wiestler O.D., Schramm J., Elger C.E., Lassmann H. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol. 2002; 51 (3): 11–18. DOI: 10.1002/ana.10100
11. O’Rourke D.J., Bergin A., Rotenberg A., Peters J., Gorman M., Poduri A., Cryan J., Lidov H., Madsen J., Harini C. Rasmussen’s encephalitis presenting as focal cortical dysplasia. Epilepsy Behav Case Rep 2014; 2, 86–89. PMID: 25667877 PMCID: PMC4307873 DOI: 10.1016/j.ebcr.2014.01.009
12. Frigeri T., Hemb M., Paglioli E., Hoefel J.R., Silva V., Vinters H., Palmini A. Bilateral Rasmussen’s encephalitis associated with type II focal cortical dysplasia: dormant ‘second’ epileptogenic zone in contralateral disease. Epilepsy Behav Case Rep. 2013; 1: 66–68. PMID: 21216675 DOI: 10.1016/j.yebeh.2010.12.004
13. Prayson R.A. Dual pathology in Rasmussen’s encephalitis: a report of coexistent focal cortical dysplasia and review of the literature. Case Rep Pathol. 2012; 2012: 1–4. PMID: 23056977 PMCID: PMC3465884 DOI: 10.1155/2012/569170
15. Buzsáki G., Wang X. J. Mechanisms of Gamma Oscillations. Annu. Rev. Neurosci. 2012; 35: 203–225. PMID: 22443509 PMCID: PMC4049541 DOI: 10.1146/annurev-neuro-062111-150444
16. Mukamel E. A., Pirondini Elvira., Babadi B.W., Kin F. K., Pierce E. T. P., Harrell P.G., Walsh J. L., Salazar-Gomez A. F., Cash S. S., Eskandar E. N., Weiner V. S., Brown E. N., Purdon. P.L. A Transition in Brain State during Propofol-Induced Unconsciousness. Journal of Neuroscience. 2014, 34 (3) 839–845. DOI: 10.1523/JNEUROSCI.5813-12.2014
17. Cannon J., McCarthy M.M., Lee S., Lee J., Börgers Ch., Whittington M., Kopell N. Review Neurosystems; brain rhythms and cognitive processing. Eur J Neurosci. 2014; 39 (5): 705–719. PMID: 24329933 PMCID: PMC4916881 DOI: 10.1111/ejn.12453
18. Fischer M., Drobny M., Saniova B., Bakosova E., Hamzik J., Schnierer M.,Osinova D. Electrical changes in deeper cortical structures during balanced general anesthesia with the aim on inhalation anesthetics effects. Acta Medica Martiniana. 2015; 15 (3): 21–29. DOI: 10.1515/acm-2015-0014
19. Press C., Wallace A., Chapman K.E. The Janus-faced nature of Rasmussen’s encephalitis. Semin Pediatr Neurol. 2014; 21, 129–136. PMID: 25149947 DOI: 10.1016/j.spen.2014.04.018
20. Castellano J.F., Meyer J.A., Lado F.A. A Case Series of Adult-Onset Rasmussen’s Encephalitis: Diagnostic and Therapeutic Challenges, Front Neurol. 2017; 8, 564. PMID: 29118737 PMCID: PMC5660978 DOI: 10.3389/fneur.2017.00564
21. Bien C.G., Widman G., Urbach H., Sassen R., Kuczaty S., Wiestler O.D., Schramm J., Elger C.E. The natural history of Rasmussen’s encephalitis. Brain. 2002; 125 (8): 1751–1759. DOI: 10.1093/brain/awf176
22. Eggers C., Burghaus L., Fink G.R., Dohmen C. Epilepsia partialis continua responsive to intravenous levetiracetam. Seizure. 2009; 18 (10): 716–718. PMID: 19836263 DOI: 10.1016/j.seizure.2009.09.005
23. Pardo C.A., Vining E.P., Guo L., Skolasky R.L., Carson B.S., Freeman J.M. The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia. 2004; 45 (5): 516–526. DOI: 10.1111/j.0013-9580.2004.33103.x
24. Bien C.G., Granata T., Antozzi C., Cross J.H., Dulac O., Kurthen M., Lassmann H., Mantegazza R., Villemure J.-G., Spreafico R., Elger C.E. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. 2005; 128 (3): 454-471. PMID: 15689357
25. Takei H., Wilfong A., Malphrus A., Yoshor D., Hunter J.V., Armstrong D.L., Bhattacharjee M.B. Dual pathology in Rasmussen’s encephalitis: a study of seven cases and review of the literature. Neuropathology. 2009; 30: 381–391. PMID: 20051019 DOI: 10.1111/j.1440-1789.2009.01079.x
26. Cheong J.Y., Wong C., Bleasel A., Varikatt W., Ng T., Dexter M.A. Late onset Rasmussen’s encephalitis with triple pathology. J Clin Neurosci. 2009; 16: 1677–1681. PMID: 19800797 DOI: 10.1016/j.jocn.2009.02.042
28. Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Andermann F., editor. Chronic encephalitis and epilepsy Rasmussen’s syndrome. Boston: Butterworth-Heinemann; 1991: 79–110.
29. Varadkar S., Bien C.G., Kruse C.A., Jensen F.E., Pardo C.A., Vincent A., Mathern G.W., Cross J.H. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol. 2014; 13 (2): 195–205. DOI: 10.1016/S1474-4422(13)70260-6
30. Eggers C., Burghaus L., Fink G.R., Dohmen Ch. Epilepsia partialis continua responsive to intravenous levetiracetam. Seizure. 2009; 18 (10): 716–718. PMID: 19836263 DOI: 10.1016/j.seizure.2009.09.005
Энцефалит Расмуссена
Энцефалит Расмуссена — это прогрессирующий энцефалит, затрагивающий только одну гемисферу головного мозга и имеющий хроническое течение. Клинически представляет собой сочетание кожевниковской эпилепсии, когнитивного снижения и очагового сенсомоторного дефицита. В ходе диагностики сопоставляются данные анамнеза, неврологического осмотра, ЭЭГ, периметрии, томографических исследований головного мозга. Терапия включает противоэпилептическую, глюкокортикостероидную, иммуномодулирующую или иммуносупрессивную составляющие. Возможно хирургическое лечение, целью которого является «выключение» пораженной гемисферы из функционирования ЦНС.
МКБ-10
Общие сведения
Энцефалит Расмуссена — прогрессирующий очаговый энцефалит с хроническим течением, характеризующийся наличием кожевниковской эпилепсии, очагового сенсомоторного неврологического дефицита и когнитивных расстройств. Подробная информация о нем впервые была предоставлена медицинскому сообществу в 1958 году американским врачом Т. Расмуссеном. Как отдельная нозологическая единица энцефалит Расмуссена выделен относительно недавно. Заболевание является крайне редким. Сам Расмуссен в течение 35 лет наблюдал лишь 51 случай подобного энцефалита.
Заболеванию подвержены только дети. Около 85% случаев приходится на возраст до 10 лет. Пик заболеваемости попадает на возрастной период от 5 до 8 лет. Единичные варианты, когда энцефалит Расмуссена дебютирует до 1 года или после 14-летнего возраста, считаются атипичными. Характерно начало энцефалита после перенесенного инфекционного заболевания (чаще ОРВИ). В литературе по неврологии указаны случаи, когда энцефалит Расмуссена сочетался с прогрессирующей гемиатрофией лица.
Причины
В настоящее время этиопатогенез не установлен. В качестве гипотез рассматриваются 2 варианта: вирусная этиология и аутоиммунный генез заболевания.
- Приверженцы вирусной гипотезы считают энцефалит Расмуссена медленно текущей вирусной нейроинфекцией и соответственно относят его к вирусным энцефалитам. В пользу вирусной теории говорят изменения, обнаруживаемые при гистологическом исследовании пораженных церебральных тканей (периваскулярные инфильтраты, мелкоочаговый глиоз, пролиферация микроглии). Однако все попытки выделить возбудитель пока остаются безрезультатными.
- В пользу аутоиммунной теории свидетельствуют исследования 2002 года, выявившие антитела к глутаматному рецептору, в результате воздействия которых происходит раскрытие ионных каналов и возбуждение нейронов. При этом эксайтотоксичность (повреждение нервных клеток под действием возбуждающих нейротрансмиттеров) обуславливает гибель астроцитов и нейронов. Кроме того, у пациентов, имеющих энцефалит Расмуссена, отмечается высокая встречаемость антигенов НLА. Вероятно, подверженность нейронов указанным выше патологическим изменениям обусловлена иммуногенетическими факторами.
Патоморфология
Морфологически энцефалит Расмуссена отличается очаговым характером возникающих изменений. Патологические процессы распространяются на одну долю или одну гемисферу мозга. В отдельных случаях при раннем дебюте заболевания имеет место двусторонний характер морфологических изменений, однако всегда выявляется первичный очаг поражения. Варианты с распространением патологического процесса на вторую гемисферу мозга являются прогностически неблагоприятными и зачастую приводят к гибели пациентов.
Симптомы энцефалита Расмуссена
Базовым симптомокомплексом выступает кожевниковская эпилепсия — сочетание парциальных судорожных эпиприступов с миоклониями. Наиболее часто наблюдаются простые (без утраты сознания) моторные фокальные приступы, локализующиеся в лице или одной из конечностей. Они могут носить клонический или тонический характер. Возможна вторичная генерализация пароксизмов с переходом в клонико-тонический генерализованный эпиприступ. Миоклонии — фокальные мышечные подергивания — носят практически постоянный характер. В начале заболевания они могут отсутствовать, появляются через несколько месяцев или возникают вместе с очаговым неврологическим дефицитом спустя 1,5-2,5 года после дебюта эпиприступов.
Очаговая симптоматика представлена в основном центральным гемипарезом, контралатеральным пораженной гемисфере. Наблюдаются сенсорные нарушения по проводниковому типу, гемианопсия, афазия (при нарушениях в доминантном полушарии). Когнитивные расстройства, сопровождающие энцефалит Расмуссена, сводятся к прогрессирующим нарушениям памяти, внимания, праксиса и приводят к развитию олигофрении. Сочетаются с психическими отклонениями.
Стадии энцефалита Расмуссена
- Продромальный период может продолжаться несколько лет. У 70% пациентов характеризуется возникновением простых фокальных судорожных пароксизмов двигательного типа. В 20% случаев энцефалит Расмуссена манифестирует эпилептическим статусом. Возможно развитие вторично-генерализованных эпиприступов. В начальном периоде частота пароксизмов, как правило, небольшая; в дальнейшем наблюдается ее существенный рост. Уже в дебюте энцефалита возможно появление паралича Тодда — транзиторного пареза, возникающего в конечностях вслед за происходящими в них в ходе эпиприступа судорожными сокращениями.
- Активный период знаменуется присоединением очаговых неврологических симптомов (гемипареза, гемианопсии, гемигипестезии, нарушений речи), мнестических нарушений и практически постоянными миоклониями. В этой стадии у 80% пациентов наблюдаются простые фокальные моторные пароксизмы, у 28% - сложные фокальные приступы, у 40% - вторично-генерализованные, у 23% - соматосенсорные. Эпилептические гемиприступы по типу джексоновской эпилепсии, затрагивающие мышцы одной половины туловища, согласно некоторым авторам, встречаются лишь в 10% случаев. Период длительности двигательного дефицита при параличе Тодда постепенно увеличивается, затем отмечается перманентный гемипарез, со временем принимающий стойкий характер.
- Период стабилизации у большей части пациентов (около 80 %) наступает спустя не более 3-х лет от времени манифестации энцефалита. Отмечается стабилизация и даже некоторое понижение частоты судорожных приступов. Однако на этом фоне продолжают прогрессировать сенсомоторные, зрительные и когнитивные нарушения. У четверти пациентов диагностируют нейроэндокринные расстройства: преждевременное половое созревание, ожирение.
Диагностика
В продромальном периоде в связи с отсутствием очаговой симптоматики постановка точного диагноза весьма затруднительна. В активной стадии невролог при обследовании выявляет наличие центрального гемипареза с усилением рефлексов и пирамидными знаками, постоянные миоклонические сокращения, нарушения речи, повышенную психическую истощаемость, снижение памяти и внимания и пр. нарушения.
- Электроэнцефалография. В активном периоде выявляет нарушения у всех пациентов. Отмечается замедление основного ритма, возможно полное отсутствие альфа-ритма. В пораженном полушарии наблюдается пик-волновая активность.
- Офтальмологическая диагностика.Определение полей зрения выявляет гемианопсию, при офтальмоскопии изменения глазного дна зачастую отсутствуют.
- Лабораторная диагностика. Исследование цереброспинальной жидкости проводятся с целью исключения другой клинически сходной патологии ЦНС.
- Томография. Первостепенное значение в постановке диагноза имеет проведение КТ или МРТ головного мозга в динамике. Патогномоничным томографическим признаком, характеризующим энцефалит Расмуссена, выступает прогрессирующая церебральная гемиатрофия. Как правило, вначале регистрируется увеличивающееся с течением времени расширение сильвиевой щели, затем отмечаются атрофические изменения конвекситальных отделов церебральной коры. При наблюдении в динамике типично увеличение зоны корковой атрофии наподобие растекания масляного пятна по пергаменту.
Дифференциальная диагностика проводится с:
- внутримозговыми опухолями;
- церебральными кистами;
- энцефалитами другой этиологии;
- иными видами эпилепсии у детей (синдромом Леннокса-Гасто, синдромом Ландау-Клеффнера, фокальной корковой дисплазией);
- лейкодистрофиями;
- лейкоэнцефалитом Шильдера и др.
Лечение энцефалита Расмуссена
Консервативная терапия включает противоэпилептическое лечение и попытки патогенетической терапии, направленной на замедление прогрессирования атрофических изменений в мозге. Сопровождающие энцефалит Расмуссена эпилептические приступы относятся к резистентным формам эпилепсии.
Консервативная терапия
Антиконвульсантную терапию обычно начинают с назначения вальпроатов. Препаратами выбора также являются топирамат, леветирацетам и фенобарбитал. Резистентность пароксизмов заставляет врачей переходить на комбинированную терапию. Рекомендованы сочетания вальпроатов с топираматом или леветирацетамом, топирамата с леветирацетамом. У некоторых пациентов эффективно сочетание вальпроатов с карбамазепином, хотя последний противопоказан в качестве монотерапии по причине усугубления миоклонических проявлений. Отдельные авторы указывают на временный антиконвульсивный эффект внутривенных инфузий больших доз ноотропила.
Относительно методов патогенетической терапии пока не существует единого мнения. Применяется иммуносупрессивное и иммуномодулирующее лечение, назначение противовирусных фармпрепаратов и глюкокортикостероидов. Последнее время в качестве стартового лечения все чаще используют терапию иммуноглобулинами. По мере прогрессирования симптоматики к ним добавляют кортикостероиды (дексаметазон, метилпреднизолон). При отсутствии положительного результата такой терапии переходят к иммуносупрессии с использованием азатиоприна или циклофосфана. Однозначных данных о результативности противовирусного лечения с применением зидовудина, ацикловира, интерферона пока нет. Возможно проведение плазмафереза, иммуносорбции.
Хирургическая тактика
Хирургическое лечение на ранних стадиях энцефалита позволяет добиться устойчивой ремиссии у 23-52% пациентов. Оно проводится нейрохирургами и заключается в функциональной или анатомической гемисферэктомии. В первом случае производится «выключение» пораженного полушария путем пересечения всех его связей, во втором — хирургическое удаление полушария или его части.
Прогноз
В большинстве случаев энцефалит Расмуссена имеет неутешительный прогноз. За исключением единичных случаев спонтанной стабилизации заболевания, у пациентов отмечается тяжелейший двигательный и когнитивный дефицит. Летальный исход может наступить в период от 3 до 15 лет со времени дебюта энцефалита.
Энциклопедия
Этиология. Синдром (энцефалит) Кожевникова - Расмуссена (СКР) - прогрессирующее заболевание головного мозга, предположительно аутоиммунной природы. До настоящего времени этиология СКР неизвестна. Согласно современным представлениям, заболевание относится к медленным нейроинфекциям вирусной этиологии, но вирус не идентифицирован. Обсуждаются 3 основные причины патологического процесса [Granata и соавт., 2002]: хроническая вирусная инфекция; острая вирусная инфекция, приводящая к локальным иммунным изменениям; аутоиммунный механизм, не связанный с инфекцией. В последние годы появились публикации об обнаружении у больных СКР повышенного титра антител к глютаматным (GluR3) рецепторам [Hart & Andermann, 2002]. По Проекту классификации 2001 года, заболевание относится к группе симптоматической фокальной неокортикальной эпилепсии.
Эпилепсия Кожевникова (ЭК) представляет полиэтиологичное заболевание, проявляющееся определенным симптомокомплексом: облигатным наличием постоянного миоклонуса, обычно в сочетании с фокальными моторными, вторично – генерализованными эпилептическими приступами и очаговыми неврологическими симптомами. Принципиально важно, что эпилепсия Кожевникова – отдельная форма эпилепсии, но не нозологически самостоятельное заболевание. Симптомокомплекс ЭК встречается при большом количестве различных неврологических заболеваний: энцефалиты, другие инфекционные заболевания головного мозга с масс – эффектом, травматические, сосудистые поражения головного мозга, опухоли, фокальные кортикальные дисплазии [Walker & Shorvon, 1996]. В России эпилепсия Кожевникова наиболее часто встречается при клещевом энцефалите и синдроме Кожевникова – Расмуссена.
Диагностические критерии. Синдром Кожевникова - Расмуссена впервые был описан А.Я. Кожевниковым в 1894 году и идентифицирован T. Rasmussen, J. Olszewski и Lloyd - Smith в 1958 году. СКР представляет собой тяжелое заболевание головного мозга - подострый прогрессирующий очаговый энцефалит. Заболевание характеризуется клинической триадой: эпилептические приступы, двигательные нарушения (центральный гемипарез) и расстройство высших психических функций в сочетании с неуклонно прогрессирующим течением и возможным летальным исходом.
Дебют в широком возрастном диапазоне – от 1 до 14 лет, с пиком в 5-8 лет. Первым признаком СКР являются эпилептические приступы. Обычно заболевание начинается с фокальных моторных или вторично – генерализованных приступов, реже – диалептических пароксизмов. В 20% случаев эпилептический статус знаменует начало СКР. Наиболее характерны приступы, исходящие из моторной коры лобной доли: клонические судороги руки, лица, реже – ноги или гемиконвульсивные пароксизмы. Судорогам нередко предшествуют соматосенсорные ощущения (жжение, покалывание, онемение) контралатерально очагу поражения. Уже на начальных этапах заболевания типично развитие преходящего постиктального пареза (или гемипареза) в конечностях, которые были вовлечены в эпилептический приступ (парез Тодда).
Обычно через несколько месяцев после появления первых фокальных приступов, у больных присоединяются длительные (до нескольких дней), а затем постоянные, локализованные в одной половине туловища и конечностей, миоклонические пароксизмы, которые могут трансформироваться в генерализованные судороги. Данный симптомокомплекс был описан А.Я. Кожевниковым в 1894 году и носит название «Кожевниковская эпилепсия». Таким образом, эпилепсия Кожевникова - один из симптомокомплексов СКР. С течением времени эпилептический миоклонус распространяется на все конечности, лицевую мускулатуру, мышцы передней брюшной стенки и становится постоянным, не исчезая и во сне.
Bancaud и соавт. (1992) выделили 3 стадии развития СКР. 1 стадия (продромальный период) характеризуется дебютом заболевания с простых парциальных моторных приступов (которым может предшествовать соматосенсорная аура) или со вторично – генерализованных пароксизмов. Частота приступов относительно невелика. После фокальных моторных приступов начинает появляться преходящий постприступный парез Тодда. Возможно также присоединение унилатеральных миоклонических приступов. В течение нескольких недель или месяцев с момента дебюта, происходит значительное нарастание частоты приступов. Средняя продолжительность этого периода составляет около 7 мес.
2 стадия (активный период) характеризуется частыми, продолжительными приступами, статусным течением, постоянным эпилептическим миоклонусом в одной половине тела (симптомокомплекс ЭК). Продолжительность постприступных симптомов выпадения увеличивается, и постепенно развивается перманентный гемипарез. Присоединяются нарушение чувствительности по проводниковому типу, выпадение полей зрения. Постепенно нарастают расстройства высших психических функций и речи. Продолжительность этой стадии заболевания – около 8 мес.
3 стадия (период стабилизации) в 80% случаев наступает в пределах 3-х лет от начала заболевания. Этот период характеризуется некоторой стабилизацией и урежением частоты эпилептических приступов при одновременном прогрессировании неврологических расстройств, нарушений высших психических функций и зрения.
В 25% случаев возможно присоединение нейроэндокринных нарушений: ожирение, преждевременное половое развитие [Калинина Л.В. и соавт., 1996].
Атипичные случаи СКР включают ранний (до 1 года) или поздний (после 14 лет) дебют заболевания, начало с прогрессирующего гемипареза с последующим присоединением эпилептических приступов, преобладание симптомов поражения затылочной доли, двухсторонний характер процесса.
При неврологическом осмотре в развернутой стадии заболевания выявляется центральный гемипарез, выпадение чувствительности по проводниковому типу, квадрантная гемианопсия, дизартрия, диспраксия; в терминальной стадии – псевдобульбарные нарушения, расстройство высших психических функций.
ЭЭГ исследование выявляет нарушения в 100% случаев в развернутой стадии заболевания. Наблюдается прогрессирующее замедление основной активности фона в сочетании с появлением продолженного регионального замедления (обычно в лобно – височных отведениях). Также констатируется продолженная пик – волновая активность, возникающая латерализовано от пораженной гемисферы. По мере прогрессирования, эпилептиформная активность захватывает обе гемисферы асинхронно.
Нейровизуализация имеет решающее значение в верификации СКР. Характерно при МРТ исследовании головного мозга в динамике появление прогрессирующей гемиатрофии [Алиханов А.А., Петрухин А.С., 2001]. Атрофия обычно начинается в теменно - височной области в виде локального расширения сильвиевой щели и с течением времени расползается по конвекситальным отделам коры «подобно масляному пятну по листу пергаментной бумаги». В 3 стадии наблюдается распространение атрофии с возможностью захвата «здорового» полушария.
СКР следует дифференцировать, прежде всего, с симптоматическими и предположительно симптоматическими неокортикальными формами эпилепсии; опухолями головного мозга; с другими воспалительными заболеваниями головного мозга и наследственно – дегенеративными заболеваниями с поражением ЦНС. Ранняя диагностика СКР важна для своевременного назначения иммуносупрессивной терапии. Диагноз СКР устанавливается на основании 3 основных критериев: клинического (анамнез, клиника заболевания, течение), нейрофизиологического (ЭЭГ) и анатомического (нейровизуализация, биопсия мозга) (табл. 5.4). При этом, лабораторные тесты (анализы крови, мочи и спинномозговой жидкости) имеют значение лишь для исключения других заболеваний. Специфических изменений лабораторных показателей при СКР нет. У отдельных пациентов описано повышение титра антител к глютаматным GluR3 рецепторам в сыворотке крови; белково – клеточная ассоциация в спинномозговой жидкости [Granata и соавт., 2003], что не имеет существенного диагностического значения. Диагностические критерии СКР представлены Bien и соавт. (2005) в таблице 9. Диагноз может быть установлен только при наличии всех 3 критериев части А (первый этап) или любых 2 критериев части В (второй этап). Второй этап включает проведение церебральной биопсии. При установлении диагноза СКР важно обследование пациентов в динамике: сравнение клинических данных, результатов ЭЭГ и МРТ исследования.
Инвалидизация при СР всегда выражена. Она обусловлена высокой частотой эпилептических приступов, наличием гемипареза и нарушения высших психических функций.
Терапия. Терапевтическая тактика имеет два слагаемых: лечение самого энцефалита и лечение кожевниковской эпилепсии. Кожевниковская эпилепсия, возникающая при ЭКР, относится к резистентным эпилептическим синдромам. Терапия АЭП имеет симптоматический поддерживающий характер.
Стартовая терапия – вальпроаты (конвульсофин, конвулекс, депакин). Назначается конвульсофин в дозе 1000-2500 мг/сут (30-80 мг/кг/сут). Вальпроаты особенно эффективны при вторично – генерализованных судорожных приступах.
Препарат второго выбора – топирамат. Назначается топамакс с постепенной титрацией дозы до 100-400 мг/сут (3-10 мг/кг/сут) в 2 приема. Топамакс особенно эффективен при фокальных моторных и вторично – генерализованных приступах.
Препарат третьего выбора – леветирацетам. Кеппра назначается с постепенной титрацией дозы до 1000-4000 мг/сут (30-60 мг/кг/сут) в 2 приема. Кеппра хорошо переносится и эффективна при фокальных моторных приступах и фокальном эпилептическом миоклонусе.
Препарат четвертого выбора – барбитуровая кислота. Назначается фенобарбитал в дозе 100-300 мг/сут (4-7 мг/кг/сут) в 2 приема. Фенобарбитал эффективен при вторично – генерализованных судорожных приступах.
Ввиду прогрессирования заболевания и резистентности приступов к АЭП, приходится переходить на политерапию. Рекомендуются комбинации: вальпроаты + топирамат; вальпроаты + леветирацетам; топирамат + леветирацетам. Назначение препаратов карбамазепина (финлепсин, тегретол) в монотерапии не рекомендовано ввиду возможной аггравации миоклонических приступов. Однако комбинация вальпроатов с карбамазепином может быть эффективна в урежении частоты фокальных моторных, диалептических и вторично – генерализованных приступов. С подключением бензодиазепинов спешить не следует, оставляя их для помощи пациентам с серийными приступами и статусным течением. Временным хорошим эффектом может обладать комбинация вальпроатов или фенобарбитала с бензодиазепинами (фризиум около 1 мг/кг/сут).
Нами с успехом апробировано лечение эпилептических приступов при СКР высокими дозами ноотропила: в дозе до 1 мг/кг/сут (максимум 40 г) при внутривенном капельном введении (10-20 инфузий на курс) [Мухин К.Ю. и соавт., 2006]. Отмечен хороший временный эффект в отношении широкого спектра приступов, особенно – миоклонических пароксизмов.
Задача медикаментозной терапии самого синдрома Кожевникова – Расмуссена заключается не только в облегчении эпилептических приступов, но и в попытке замедлить прогрессирующий характер атрофического процесса в головном мозге. Терапия включает применение иммуномодуляторов, иммуносупрессоров, стероидных гормонов, противовирусных, антиэпилептических, ноотропных препаратов и др. [Петрухин А.С. и соавт., 2000].
Терапевтическая схема с различными этапами разработана Bien и соавт. (2005). Стартовая терапия при установлении диагноза СКР включает назначение иммуноглобулинов (октагам, человеческий иммуноглобулин – IVIG). IVIG применяется в дозе 400 мг/кг/сут в течение 3-5 дней. Затем рекомендована поддерживающая доза 400 мг/кг однократно каждый месяц. При тяжелом течении заболевания возможно введение иммуноглобулинов каждые 5 дней каждого месяца в суммарной дозе 2000 мг/кг. Необходимо как можно более раннее назначение иммуноглобулинов.
При дальнейшем прогрессировании заболевания (ухудшение клинических, ЭЭГ и МТР данных при динамическом наблюдении) рекомендуется к иммуноглобулинам добавить кортикостероидные гормоны (метипред, преднизолон, дексаметазон). Стероидная терапия начинается с внутривенного введения метилпреднизолона в средней дозе 20 мг/кг/сут в течение 3 дней. Затем переходят на пероральный прием гормонов. Например, преднизолон в дозе 1-2 мг/кг/сут. Продолжительность перорального применения стероидов зависит от «злокачественности» процесса, и главным образом, от частоты эпилептических приступов. Для купирования серийных приступов и при тенденции к статусному течению возможен короткий курс – около 2-х недель с последующей постепенной отменой. Стероидные гормоны особенно эффективны у детей младшего возраста. Ограничивает их применение высокая частота (практически – 100%) побочных эффектов.
В последние годы при неэффективности иммуноглобулинов и стероидов, и при дальнейшем прогрессировании процесса, рекомендуется длительное пероральное назначение препарата такролимус. Данный препарат представляет собой иммуносупрессор, селективно блокирующий Т лимфоциты, непосредственно задействованные в патогенезе СКР. Констатировано урежение эпилептических приступов, уменьшение моторного дефицита и улучшение когнитивных функций у больных СКР при применении такролимуса [Bien и соавт., 2005].
В отдельных исследованиях отмечена эффективность азатиоприна, циклофосфана, плазмофереза (каждые 8 недель), а также иммуноадсорбента белка А IgG [Калинина Л.В. и соавт., 1996; Granata и соавт., 2003; Bien и соавт., 2005]. Эффективность различных противовирусных препаратов (зидовудин, ацикловир, ганцикловир) и препаратов интерферона сомнительна.
Единственным реальным методом помощи больным СКР является нейрохирургическое вмешательство: функциональная гемисферотомия, которая должна быть выполнена на возможно ранних стадиях заболевания. Частота стойкой ремиссии после операции составляет 23-52% [Rasmussen & Andermann, 1991]. В России первая радикальная операция при синдроме Кожевникова – Расмуссена была проведена в марте 2007 в институте нейрохирургии им. Н.Н. Бурденко при нашем предоперационном обследовании. При отсутствии хирургического вмешательства, СКР прогрессирует и заканчивается тяжелейшей инвалидизацией или летальным исходом в течение 2-15 лет (в среднем, - 3 года) с момента дебюта. Описаны случаи спонтанной стабилизации процесса.
Энцефалит Расмуссена как причина эпилепсии
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
Уроки 101 гемисферотомии у детей с полушарной симптоматической эпилепсией. Часть I. Исходы лечения приступов
Журнал: Журнал «Вопросы нейрохирургии» имени Н.Н. Бурденко. 2021;85(5): 15‑21
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ЦЕЛЬ ИССЛЕДОВАНИЯ
Проанализировать влияние некоторых клинических факторов (семиология и частота приступов, возраст дебюта эпилепсии и ее длительность, этиология поражения и наличие аномалии в «здоровом» полушарии по данным магнитно-резонансной томографии) и способа дисконнекции пораженного полушария на исходы после гемисферотомии.
МАТЕРИАЛ И МЕТОДЫ
Оперирован 101 ребенок. Медиана возраста на момент операции 43 мес, длительности эпилепсии 30 мес. У большинства (78) детей имелась задержка в развитии. В 25 наблюдениях на магнитно-резонансных томограммах имелись структурные нарушения и в противоположной, «здоровой», гемисфере. В 55 случаях произведена латеральная периинсулярная деафферентация, в 46 — вертикальная парасагиттальная дисконнекция (в 54 — слева, в 47 — справа). Повторно оперированы 8 пациентов в связи с продолжающимися приступами или их возобновлением (у всех — из-за неполной дисконнекции).
РЕЗУЛЬТАТЫ
ЗАКЛЮЧЕНИЕ
У детей с эпилепсией, обусловленной структурным анатомическим поражением одного из полушарий мозга, гемисферотомия — единственная возможность помощи, если лечение антиэпилептическими препаратами неэффективно.
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
ФГАУ «Национальный медицинский исследовательский центр нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России
Классификация эпилепсии
Эпилепсия — заболевание головного мозга, соответствующее любому из следующих состояний:
- Не менее двух неспровоцированных (или рефлекторных) эпилептических приступов с интервалом > 24 ч.
- Один неспровоцированный (или рефлекторный) эпилептический приступ и вероятность повторных приступов, соответствующая общему риску рецидива (³ 60 %) после двух неспровоцированных эпилептических приступов, в следующие 10 лет.
- Диагноз эпилептического синдрома.
Критерии разрешения эпилепсии включают достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом либо отсутствие эпилептических приступов в течение 10 лет у пациентов, не получавших противосудорожные препараты более 5 лет.
Классификация эпилепсии (Международная противоэпилептическая лига (ILAE) 2017 г. )
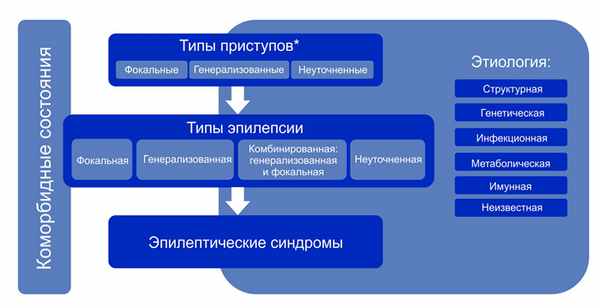
Классификация эпилепсии проводится после определения критериев диагностики эпилепсии (определение выше). Классификация проводится с использованием трехуровнего ранжирования — определение типа приступов, типа эпилепсии и синдрома эпилепсии. Нейроимиджинг, ЭЭГ и другие исследования, если они есть, помогают улучшить классификацию на всех трех уровнях. Где это возможно, следует установить диагноз на всех трех уровнях. Этиологию эпилепсии следует устанавливать с самого начала и на каждом этапе всего диагностического пути. Знание этиологии может способствовать оптимизации классификации и имеет важные лечебные последствия для пациента.
Структура Классификации эпилепсии ILAE 2017 г.
Примечание. * Оценивается по началу приступа.
Авакян Г. Н. Блинов Д. В. Лебедева А. В. Бурд С. Г. Авакян Г. Г. Классификация эпилепсии Международной Противоэпилептической Лиги: пересмотр и обновление 2017 года. Эпилепсия и пароксизмальные состояния. 2017; 9 (1): 6–25. DOI: 10.17749/2077–8333.2017.9. 1.006–025.
Алгоритм классификации эпилепсии:
- На первом этапе (уровне) определяют тип приступа: фокальный, генерализованный или с неизвестным началом.
- На втором этапе (уровне) устанавливают тип эпилепсии: фокальная, генерализованная или сочетанная фокальная и генерализованная, или неизвестная (unknown).
- На третьем этапе (уровне) определяют эпилептический синдром и коморбидность. Эпилептический синдром представляет собой совокупность характеристик, включая тип приступа, данные ЭЭГ и нейровизуализации, часто имеет возрастзависимый характер, провоцирующие факторы, хронозависимость и в ряде случаев определенный прогноз.
- На четвертом этапе (уровне) устанавливают этиологию эпилепсии: структурная, генетическая, инфекционная, метаболическая, иммунная или с неизвестной этиологией.
Генерализованная эпилепсия
Пациенты с генерализованной эпилепсией имеют генерализованные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ паттерны, которые сопровождают генерализованные типы приступов (например, генерализованный спайк-волна). Сопутствуют семейная история генерализованных типов приступов или генерализованная эпилепсия.
Генетическая / идиопатическая генерализованная эпилепсия представляет собой эпилепсию с генерализованными приступами, ассоциированнами с генерализованными эпилептиформными паттернами ЭЭГ, такими как генерализованная спайк-волновая активность, которая, считается, имеет генетическую этиологию. Однако это не всегда означает, что эти эпилепсии унаследованы или могут передаваться потомству, поскольку генетическая этиология может быть спонтанной новой мутацией или наследование может быть сложным. Таким образом, термин «идиопатическая генерализованная эпилепсия» используется как синоним генетической генерализованной эпилепсии, и клиницист может выбрать, какой термин использовать, в зависимости от важности акцента на генетическом наследовании для конкретного пациента. Генетические / идиопатические генерализованные эпилепсии включают детскую абсансную эпилепсию, юношескую абсансную эпилепсию, юношескую миоклоническую эпилепсию, и эпилепсию с генерализованными тонико-клоническими приступами.
Фокальная эпилепсия
Пациенты с фокальной эпилепсией имеют фокальные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ-паттерны, которые сопровождают фокальные типы приступов (такие как фокальные острые волны или фокальное интериктальное замедление). Нейроимиджинг демонстрирует фокальную структурную аномалию мозга и способствует установке диагноза, хотя пациенты с генетической этиологией и нормальной визуализацией могут также иметь фокальную эпилепсию. Фокальные эпилепсии могут быть унифокальными, мультифокальными или полушарными.
Сочетанная генерализованная и фокальная эпилепсия
Пациенты могут иметь как генерализованные, так и фокальные типы приступов, с интериктальными и / или иктальными ЭЭГ-паттернами, которые сопровождают оба типа приступов. Пациенты с синдромом Драве и синдромом Леннокс-Гасто могут иметь генерализованную и фокальную эпилепсию.
Неизвестная эпилепсия
Термин «неизвестный» используется для обозначения в случае, когда понимается, что у пациента есть эпилепсия, но невозможно определить, является ли она фокальной, генерализованной или комбинированной: фокальной и генерализованной. Это бывает при недостаточной информации для классификации эпилепсии, например, если ЭЭГ является нормальной / неинформативной.
Синдромы эпилепсии
В то время как концептуализация эпилепсий по их этиологии очень важна, эпилепсии также могут быть организованы (в соответствии с установленными достоверными общепринятыми клиническими и ЭЭГ — характеристиками) в эпилептические синдромы. Такие синдромы имеют типичный возраст начала приступа, специфические типы приступов и характеристики ЭЭГ и часто другие признаки, которые вместе взятые позволяют диагностировать конкретный эпилептический синдром. Идентификация синдрома эпилепсии полезна, так как она предоставляет информацию о том, какие основные этиологии следует учитывать и какие антиконвульсанты могут быть наиболее полезными. Некоторые эпилептические синдромы демонстрируют аггравацию приступов при определенных антиконвульсантах, чего можно избежать при ранней диагностике синдрома эпилепсии.
Эпилептические синдромы
Неонатальный и младенческий период:
- самокупирующиеся неонатальные приступы и самокупирующаяся семейная неонатальная эпилепсия;
- самокупирующаяся семейная и несемейная младенческая эпилепсия;
- ранняя миоклоническая энцефалопатия;
- синдром Отахара;
- синдром Веста;
- синдром Драве;
- миоклоническая эпилепсия младенчества, младенческая эпилепсия с мигрирующими фокальными приступами;
- миоклоническая энцефалопатия при непрогрессирующих заболеваниях;
- фебрильные приступы, фебрильные приступы плюс, генетическая эпилепсия с фебрильными приступами плюс.
Детский возраст:
- эпилепсия с миоклонически-атоническими (ранее – астатическими) приступами;
- синдром Леннокса–Гасто;
- фебрильные приступы, фебрильные приступы плюс;
- абсансная эпилепсия;
- эпилепсия с миоклоническими абсансами;
- детская затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса);
- детская затылочная эпилепсия с поздним дебютом (синдром Гасто);
- фотосенситивная затылочная эпилепсия;
- самокупирующаяся эпилепсия с центрально-темпоральными спайками;
- атипичная эпилепсия с центрально-темпоральными спайками;
- эпилептическая энцефалопатия с продолженной пик-волновой активностью во время сна;
- синдром Ландау–Клеффнера;
- аутосомно-доминантная ночная лобная эпилепсия.
Подростковый и взрослый возраст:
- юношеская абсансная эпилепсия;
- юношеская миоклоническая эпилепсия;
- эпилепсия с изолированными генерализованными тонико-клоническими приступами;
- аутосомно-доминантная эпилепсия со слуховыми проявлениями;
- другие семейные височные эпилепсии.
Любой возраст:
- семейная фокальная эпилепсия с вариабельным фокусом;
- рефлекторные эпилепсии;
- прогрессирующие миоклонические эпилепсии.
Этиология эпилепсии
В последние годы значительно расширилось наше понимание основополагающих причин эпилепсии, подкрепленное достижениями в области современной нейровизуализации и генетического тестирования. Такая терминология, как «идиопатическая», «криптогенная» и «симптоматическая», больше не используется. Эпилепсии теперь описываются более точно конкретными основополагающими причинами.
Генетическая этиология
Понятие генетической эпилепсии заключается в том, что эпилепсия, насколько мы понимаем, является прямым результатом известного или предполагаемого генетического дефекта (ов), в котором судороги являются основным симптомом расстройства. Генетический дефект может возникать на хромосомном или молекулярном уровне. Важно подчеркнуть, что «генетический» не означает то же, что «унаследовано», поскольку мутации de novo не являются редкостью. Наличие генетической этиологии не исключает экзогенного влияния на возникновение эпилепсии.
Наиболее важные генетические причины эпилепсии, которые могут быть идентифицированы при клиническом тестировании:
Существует много способов, которыми генетические факторы могут способствовать развитию эпилепсии. Определенные генетические факторы, возможно, не были унаследованы и не могут быть переданы потомству. Вот некоторые важные генетические концепции, используемые на этом веб-сайте, и их определения:
- унаследованные аномалии генов, аутосомно-доминантное, аутосомно-рецессивное и менделевское наследование;
- приобретенные аномалии генов — de novo, спорадические, мозаицистические, зародышевые и соматические;
- полигенная / комплексная генетическая этиология.
Структурная этиология
Структурные эпилепсии определяются как имеющие выраженную структурную аномалию мозга, которая связана с существенно повышенным риском эпилепсии. Структурная аномалия мозга может быть приобретена (например, вследствие инсульта, травмы или инфекции) или может быть генетического происхождения; однако, как мы это понимаем в настоящее время, структурная аномалия мозга представляет собой отдельное нарушение, расположенное между приобретенным или генетическим дефектом и эпилепсией.
Нейроимиджинг
Магнитно-резонансная томография (МРТ) с использованием 1.5 Тесла аппарата является минимальным стандартным исследованием для исключения структурной аномалии. При этом исследовании важно выполнять протоколы, специфические для эпилепсии, которые позволяют тщательно изучать специфические приобретенные аномалии (например, склероз гиппокампа ) и тонкие пороки развития коры головного мозга, такие как фокальная дисплазия коры. Изображение с использованием 3 Тесла и использование передовых методов анализа программного обеспечения может помочь в выявлении структурных нарушений, не очевидных при обычной МРТ. Интериктальная и иктальная ЭЭГ вместе с дополнительными функциональными исследованиями нейровизуализации, такими как ПЭТ, ОФЭКТ и МЭГ, помогают внимательно изучить конкретную область мозга и идентифицировать тонкую аномалию. У детей младшего возраста в возрасте до 2 лет тонкие аномалии не могут быть выявлены из-за незаконченной миелинизации, и повторное исследование требуется в динамике.
Общие структурные аномалии мозга, связанные с эпилепсией:
- пороки развития коры головного мозга;
- сосудистые пороки развития;
- гиппокампальный склероз;
- гипоксически-ишемические структурные аномалии;
- травматическая повреждение мозга;
- опухоли;
- порэнцефалическая киста.
Метаболическая этиология
Метаболические эпилепсии определяются как имеющие определенное метаболическое нарушение, связанное с выраженным риском развития эпилепсии. Метаболические расстройства имеют генетическое происхождение; однако, как мы это понимаем в настоящее время, метаболические аномалии представляют собой отдельное нарушение, стоящее между генетическим дефектом и эпилепсией.
Важные метаболические эпилепсии:
- дефицит биотинидазы и голокарбоксилазы-синтазы;
- дефицит церебрального фолата;
- нарушения креатина;
- приступы при нарушении фолатного цикла;
- недостаточность транспортера глюкозы 1 (GLUT1);
- митохондриальные расстройства;
- пероксисомальные расстройства;
- пиридоксинзависимая эпилепсия.
Иммунная этиология
Иммунные эпилепсии определены как имеющие выраженную иммунную опосредованную этиологию с подтверждением воспаления центральной нервной системы, что, как было показано, связано с существенно повышенным риском развития эпилепсии.
Важные иммуноопосредованные эпилепсии:
- Синдром Расмуссена;
- Антителоопосредованная эпилепсия.
Инфекционная этиология
Наиболее распространенная этиология эпилепсии во всем мире является инфекционной, особенно в развивающихся странах. Инфекции в центральной нервной системе могут вызывать как острые симптоматические припадки (которые тесно связаны со сроками первичной инфекции), так и эпилепсией. Инфекционная этиология включает туберкулез, ВИЧ, церебральную малярию, нейроцистицеркоз, подострый склерозирующий панэнцефалит, церебральный токсоплазмоз. Эти инфекции иногда имеют структурный коррелят, однако основная причина эпилепсии определяется как инфекционный процесс. Инфекционная этиология может иметь специфические последствия лечения. Существуют также последствия для общественного здравоохранения, поскольку профилактика таких инфекций может снизить нагрузку на эпилепсию, особенно в развивающихся странах. Наиболее распространенные из таких инфекций следующие:
- бактериальный менингит или менингоэнцефалит;
- церебральная малярия;
- церебральный токсоплазмоз;
- цитомегаловирусная инфекция;
- ВИЧ;
- нейроцистицеркоз;
- туберкулез;
- вирусный энцефалит;
- подострый склерозирующий панэнцефалит;
- прочие инфекции (токсокариоз, шистосомоз, болезнь Лайма (нейроборрелиоз).
Неизвестная этиология
«Неизвестная» этиология должна рассматриваться нейтрально и обозначать, что природа основной причины возникновения эпилепсии пока неизвестна; это может быть фундаментальный генетический дефект или отдельное, пока еще установленное, нарушение.
Имитаторы эпилепсии
Существует ряд состояний, связанных с рецидивирующими пароксизмальными событиями, которые могут имитировать симптомы, и ошибочно диагностироваться как эпилепсия. Важно, чтобы эти расстройства учитывались при оценке пароксизмальных событий, так как частота ошибочных диагнозов при эпилепсии высока во всем мире. История заболевания и видеозапись приступов помогают установить правильный диагноз. Существуют некоторые состояния, при которых могут сосуществовать эпилептические и неэпилептические события.
Читайте также: