Гемофагоцитарный лимфогистиоцитоз
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Исследование частых мутаций в гене UNC13D.
Тип наследования.
Аутосомно - рецессивный.
Гены, ответственные за развитие заболевания.
Ген UNC13D (UNC13, C. ELEGANS, HOMOLOG OF, D) расположен на хромосоме 17 в регионе 17q25.1. Содержит 32 экзона.
К развитию данного заболевания могут привести также мутации в генах PRF1, STX11, STXBP2.
Определение заболевания.
Это редкое и тяжелое гематологическое заболевание. Оно характеризуется аномальным размножением и активацией особых клеток организма – гистиоцитов (тканевых макрофагов). В развитии болезни, кроме гистиоцитов, участвуют также и неправильно функционирующие лимфоциты. При этой болезни макрофаги поглощают клетки крови (эритроциты, лейкоциты, тромбоциты) и их клетки-предшественники в различных органах и тканях. В результате аномальной активности лимфоцитов и макрофагов возникают тяжелые поражения органов, включая печень, селезенку, лимфатические узлы, кожу, легкие, головной мозг и т.п. Одновременно гемофагоцитоз приводит к цитопении – состоянию дефицита клеток крови.
Патогенез и клиническая картина.
Первичные лимфогистиоцитозы (ГЛГ) чаще всего наблюдаются у детей в возрасте до двух лет. Основными клиническими симптомами являются лихорадка, рефрактерная к антибактериальной терапии, прогрессирующая гепатоспленомегалия, поражение ЦНС, лабораторные проявления – би- или панцитопения, гипофибриногенемия, гипертриглицеридемия. Международным обществом по изучению гистиоцитарных заболеваний выдвинуты клинические критерии гемофагоцитарного лимфогистиоцитоза. Клинически диагноз считается подтвержденным при наличии пяти и более из 8 диагностических критериев ГЛГ.
У 56-80% детей заболевание развивается на первом году жизни, а у отдельных из них диагностируется при рождении, примерно у 20% детей первые клинические признаки заболевания появляются после 3-х лет жизни. Имеются данные о дебюте заболевания в более позднем возрасте: 6, 8, 12, 25 лет. Важно отметить, что возраст заболевших сибсов очень часто совпадает. Примерно в половине случаев имеется позитивный семейный анамнез - больные сибсы или близкородственный брак.
Клиническое течение заболевания очень тяжелое и без специального лечения быстро приводит к летальному исходу. Единственным эффективным способом лечения первичных ГЛГ является трансплантация костного мозга (ТКМ).
При данном типе заболевания мутация происходит в гене, который кодирует белок, участвующий в процессах экзоцитоза цитотоксических гранул NK-клеток и цитотоксических Т-лимфоцитов.
Частота встречаемости: суммарно 1:50000 новорожденных.
Показания к исследованию
Клинические критерии:
лихорадка свыше 7 дней более > 38,5°С;
спленомегалия ≥ 3 см из-под края реберной дуги.
Лабораторные критерии:
цитопения периферической крови с поражением минимум двух ростков при отсутствии гипоклеточности или миелодиспластических изменений в костном мозге: (гемоглобин менее 90 г/л, тромбоциты менее 100 х 109/л, нейтрофилы менее 1,0 х 109/л);
гипертриглециридемия и/или гипофибриногенемия (триглециридемия > 2,0 ммоль/л; фибриноген Подписаться
Внимание!
У нас появилась возможность предзаказывать список необходимых анализов для сдачи как дома, так и в клинике!
Для этого необходимо добавить интересующие Вас анализы в корзину и оформить заказ.
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз (ГЛГ) — редкое и тяжелое гематологическое заболевание. Оно характеризуется аномальным размножением и активацией особых клеток организма — гистиоцитов (тканевых макрофагов).
Термин «лимфогистиоцитоз» означает, что в развитии болезни, кроме гистиоцитов, участвуют также и неправильно функционирующие лимфоциты. В свою очередь, термин «гемофагоцитарный» означает, что при этой болезни макрофаги поглощают клетки крови (эритроциты, лейкоциты, тромбоциты) и их клетки-предшественники в различных органах и тканях. Этот процесс и называтся гемофагоцитозом.
В результате аномальной активности лимфоцитов и макрофагов возникают тяжелые поражения органов, включая печень, селезенку, лимфатические узлы, кожу, легкие, головной мозг и т.п. Одновременно гемофагоцитоз приводит к цитопении — состоянию дефицита клеток крови.
ГЛГ не является злокачественным заболеванием; в настоящее время считается, что его причина — нарушение регуляции клеточного иммунитета.
Частота встречаемости, факторы риска
ГЛГ — редкое заболевание. По зарубежным данным, частота его врожденной формы — около 1 случая на 50 000 новорожденных.
Различают первичный (семейный, врожденный) и вторичный (приобретенный) ГЛГ. Нарушения при первичном варианте болезни обусловлены наследственными факторами и чаще всего проявляются в очень раннем возрасте, обычно до 12-18 месяцев. Наследование первичного ГЛГ — аутосомно-рецессивное, то есть больной ребенок может родиться (с вероятностью 25%) только тогда, когда оба родителя являются носителями «дефектных» генов. Поэтому ГЛГ чаще встречается при близкородственных браках. Известно несколько разновидностей болезни, которым соответствуют разные генетические дефекты. Мальчики и девочки болеют с одинаковой частотой, зависимости от этнического происхождения также не выявлено. Семьям, где уже были случаи ГЛГ у детей, рекомендована консультация генетика. Возможна пренатальная (то есть до рождения ребенка) ДНК-диагностика.
Вторичный ГЛГ может возникнуть при аномальной активации клеток в результате некоторых инфекций (например, инфицирования вирусом Эпштейна-Барр), злокачественных опухолей, аутоиммунных заболеваний и иммунодефицитных состояний. Эта форма обычно наблюдается в более позднем возрасте, чем первичный ГЛГ.
Признаки и симптомы
Для ГЛГ характерно упорное повышение температуры, причем лихорадка не поддается лечению антибактериальными препаратами. Наблюдается увеличение селезенки (спленомегалия), обычно также увеличение печени (гепатомегалия). Более чем в половине случаев возникает кожная сыпь, поражающая также и волосистую часть головы. Часто встречается увеличение лимфоузлов — лимфаденопатия. Нередки признаки поражения центральной нервной системы (ЦНС): изменения поведения, нарушения координации движений, судороги. Другие возможные симптомы — желтуха, отеки, признаки поражения желудочно-кишечного тракта (боли в животе, рвота, понос, отказ от пищи, потеря веса).
Кроме того, при ГЛГ возникают клинические проявления цитопении, то есть состояния дефицита клеток крови: недостаток эритроцитов проявляется бледностью и слабостью, недостаток тромбоцитов — синяками и кровотечениями (возможны, например, кровотечения из десен), недостаток лейкоцитов — частыми инфекциями.
Диагностика
При выявлении указанных клинических признаков производятся лабораторные исследования с целью подтвердить или исключить диагноз ГЛГ. В общем анализе крови наблюдается цитопения: анемия, тромбоцитопения и/или нейтропения. Присутствуют также характерные изменения в биохимическом анализе крови. Гистологические исследования указывают на накопление лимфоцитов и макрофагов в различных органах, включая селезенку, лимфоузлы, костный мозг, печень и центральная нервная система. В образцах тканей, взятых посредством биопсии, наблюдается явление гемофагоцитоза (см. выше), но не всегда выявленные изменения бывает легко оценить.
Используются иммунологические тесты, позволяющие обнаружить именно те иммунные нарушения, которые обычно приводят к развитию ГЛГ (снижение активности NK-лимфоцитов).
Диагноз первичного ГЛГ подтверждается молекулярно-генетическим анализом, так как известен целый ряд соответствующих ему генетических дефектов. В то же время вторичный ГЛГ может иметь сходные клинические проявления, но диагностироваться только по совокупности перечисленных и некоторых других клинических и лабораторных признаков.
Лечение
При ГЛГ огромную роль играют своевременная диагностика и начало лечения, так как состояние больного может ухудшаться очень быстро.
В лечении ГЛГ используется химиотерапия. Так, протокол HLH-2004 предполагает использование дексаметазона, этопозида, циклоспорина, а также применение метотрексата в случае поражения ЦНС. Сопроводительная терапия обязательно включает в себя профилактику инфекций. При необходимости производятся переливания компонентов крови.
При врожденном ГЛГ после химиотерапии, позволяющей стабилизировать состояние пациента, необходима аллогенная трансплантация костного мозга. Без трансплантации невозможно достичь продолжительной ремиссии. Трансплантация может также понадобиться в некоторых случаях вторичного (приобретенного) ГЛГ, хотя в международной медицинской практике вопрос о лечении таких больных не решен однозначно.
Прогноз
Первичный ГЛГ без лечения приводит к смертельному исходу. Непосредственной причиной смерти может быть, например, полиорганная недостаточность, сепсис или грибковая инфекция. Даже при условии применения химиотерапии ремиссия обычно непродолжительна и долговременная выживаемость низка. Основную надежду на излечение дает аллогенная трансплантация костного мозга, несмотря на тяжесть этой процедуры и связанные с ней опасные осложнения. Так, в одном из зарубежных исследований пятилетняя выживаемость пациентов после трансплантации достигала 66%, тогда как без трансплантации составляла лишь 10%. В случае успешной трансплантации пациенты могут в дальнейшем иметь нормальные качество и продолжительность жизни.
При вторичном ГЛГ прогноз сильно варьирует в зависимости от конкретного случая заболевания: зафиксированы как случаи с крайне тяжелым течением, плохо поддающиеся терапии, так и случаи самопроизвольного улучшения.
Гемофагоцитарный лимфогистиоцитоз в педиатрической практике
Цель статьи: представить клинический случай успешной диагностики и лечения гемофагоцитарного лимфогистиоцитоза (ГЛГ) у девочки 2,5 года, госпитализированной по поводу тяжелой пневмонии.
Основные положения. ГЛГ представляет собой синдром, клинически проявляющийся системной воспалительной реакцией организма, подобной септическому процессу, в большинстве случаев он с трудом диагностируется врачами-педиатрами. Диагностическую значимость имеют развивающиеся панцитопения и гиперферритинемия. Мы представляем клинический случай девочки в возрасте 2,5 года, госпитализированной по поводу тяжелой пневмонии с развившимся ГЛГ. Поскольку нет единого подхода к лечению вторичного ГЛГ, был проведен консилиум и решено начать лечение со стероидной терапии (дексаметазона) и внутривенного введения иммуноглобулина (Октагама), а при отсутствии эффекта в течение первой недели подключить цитостатики. Лечение начато сразу после обнаружения в мазке костного мозга активных макрофагов. Уже на пятые сутки терапии отмечалась положительная клиническая и параклиническая динамика, в связи с чем принято решение воздержаться от агрессивной иммуносупрессии. Практически через месяц при отсутствии признаков дисфункции органов и нормализации показателей ребенок был выписан из стационара.
Заключение. При ГЛГ имеется высокий риск летального исхода, поэтому очень важны ранняя диагностика и своевременно начатая терапия. Особенность данного случая заключается в том, что нам удалось быстро поставить диагноз, рано начать лечение и получить полную ремиссию без агрессивной иммуносупресии.
Вклад авторов: Багдасарян Н.Г. — план обследования и лечения пациентки, интерпретация лабораторных данных, окончательное оформление статьи, утверждение рукописи в печать; Айвазян Г.С. — интерпретация клинических и лабораторных данных и оформление статьи; Аветисян Л.А. — лечение пациентки, обработка анкетных данных; Мнацаканян Н.Р., Алексанян Л.Т. — обзор публикаций по теме статьи.
Конфликт интересов: авторы заявляют об отсутствии возможных конфликтов интересов.
Гемофагоцитарный лимфогистиоцитоз (ГЛГ) характеризует ряд заболеваний врожденного и приобретенного генеза, в основе которых лежит нарушение регуляции иммунного ответа с последующей выработкой цитотоксических Т-лимфоцитов [1] . Как следствие, развивается системная воспалительная реакция с активацией и пролиферацией Т-лимфоцитов и макрофагов, фагоцитозом эритроцитов, лейкоцитов, тромбоцитов и их клеток-предшественников, с мультиорганной дисфункцией [1, 2] , очень похожей на тяжелый сепсис, что затрудняет его диагностику [3] .
ГЛГ может осложнить течение основного заболевания и в случае отсутствия своевременного лечения привести к смертельному исходу [4, 5] . Различают первичный и вторичный ГЛГ [6, 8] . Первичный ГЛГ — это результат мутации генов, кодирующих белки, играющих важную роль в регулировании цитотоксических лимфоцитов [7] . Способствовать развитию вторичного ГЛГ могут вирусы (герпесвирусы, парвовирус В19, аденовирус, энтеровирусы, вирусы Коксаки, вирус гриппа А Н5N1), ГЛГ может также возникнуть при туберкулезе, брюшном тифе, болезни Лайма, бруцеллезе, пневмонии, риккетсиозе, паразитарных и грибковых инфекциях, злокачественных новообразованиях и аутоиммунных заболеваниях [3, 8, 9] .
Мы представляем клинический случай с целью акцентировать внимание педиатров на нередком, однако часто не диагностируемом синдроме и показать возможность его терапии без использования агрессивной иммуносупрессии.
В педиатрическую клинику Университетского больничного комплекса «Мурацан» г. Еревана в мае 2018 года на 5-й день заболевания поступила девочка в возрасте 2 лет 6 месяцев с жалобами на лихорадку, общую слабость, кашель.
Анамнез жизни ребенка без особенностей: родилась доношенной, показатели роста и развития в пределах нормы, профилактические прививки получила по национальному календарю.
Данная госпитализация — третья. Предыдущие две госпитализации были по поводу пневмонии (диагнозы поставлены по критериям ВОЗ и подтверждены рентгенологически). При этом состояние ребенка оценивали как умеренно тяжелое без выраженных клинико-гематологических особенностей.
После второй госпитализации девочку с улучшением выписали с продолжением курса антибиотикотерапии (аминопенициллина) в домашних условиях, а через 6 дней она снова поступила в больницу (все три госпитализации в течение 5 месяцев).
На момент поступления состояние ребенка тяжелое, температура — 40°С, резкая слабость, признаки дыхательной недостаточности (ДН): одышка (ЧДД — 50 в мин), ЧСС — 140 в мин, сатурация SpO2 — 92%, втяжения нижних отделов грудной клетки; локальная физикальная симптоматика справа. Рентгенологически вновь подтвердилась пневмония — наддиафрагмальный инфильтрат справа. Был поставлен диагноз пневмонии, и началась антибиотикотерапия с резервного препарата моксифлоксацина (с учетом предыдущего анамнеза).
В общем анализе крови в день поступления в клинику уровень гемоглобина — 118 г/л, СРБ — 48 мг/л, эритроциты — 4,7 × 10 12 /л, тромбоциты — 1491 × 10 9 /л, лейкоциты — 6,1 × 10 9 . На третий день госпитализации и антибиотикотерапии при отсутствии положительной динамики (сохранялись температура до 38,3°С, слабость, усугубились признаки ДН (ЧДД — 54 в мин, ЧСС — 164 в мин, SpO2 — 86%), на повторной рентгенограмме — картина двусторонней базальной полисегментарной пневмонии справа с реакцией плевры с обеих сторон) больную перевели в отделение интенсивной терапии. У девочки отмечалась гепатоспленомегалия.
В этот же день в связи с ухудшением состояния больной повторно проведен общий анализ крови, в котором выявилась панцитопения, усугубившаяся в последующие дни.
Наглядно изменения показателей периферической крови пациентки представлены в таблице 1 , динамика биохимических показателей — в таблице 2 .
Динамика показателей общего анализа крови пациентки в отделении интенсивной терапии
Динамика показателей биохимического анализа крови пациентки в отделении интенсивной терапии
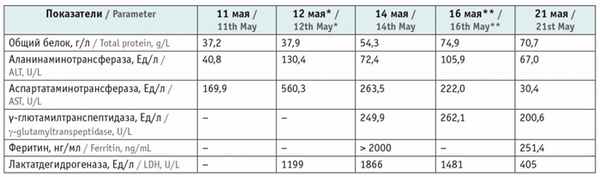
* Свежезамороженная плазма внутривенно.
** Иммуноглобулин (Октагам) 5% внутривенно, дексаметазон внутривенно.
Отмечались повышение уровня СРБ до 96 мг/л, высокие показатели амилазы (547,2 Ед/л) и липазы (733 Ед/л), диастазы в моче (6071,7 Ед/л), триглицеридов (2,1 ммоль/л). По данным коагулограммы, гипофибриногенемия не выявлена.
На сонографии органов брюшной полости — гепатомегалия, увеличение головки и хвостового отдела панкреаса, спленомегалия, наличие свободной жидкости в брюшной полости. По данным ЭхоКГ, в полости перикарда — небольшое количество жидкости в передней камере. На КТ легких — картина полисегментарной пневмонии, наличие внутрилегочных лимфопролиферативных утолщений.
С целью выявления всех возможных причин панцитопении были проведены консультации гематолога, инфекциониста, фтизиатра. Исключены лейшманиоз (метод быстрой иммунохроматографии, rК39 тест), ВИЧ (сывороточный тест), вирусные гепатиты В и С. Методом ИФА обнаружены антитела класса IgM к капсидному антигену вируса Эпштейна — Барр (EBV VCA-IgM). В результате исследования найден также парвовирус В19 (B19V).
В связи с отсутствием эффекта моксифлоксацин был заменен на меропенем тригидрат, проводились инфузионная терапия, переливания плазмы и эритроцитарной массы. Так как в условиях угнетения костного мозга высок риск развития пневмоцистоза, в схему лечения добавили ко-тримоксазол.
С учетом наличия у ребенка мультиорганной дисфункции, отсутствия клинического улучшения, усугубляющейся панцитопении и выраженных измененных биохимических показателей крови проведено исследование костного мозга, выявившее гипоклеточный костный мозг с единичными мегакариоцитами, раздражение моноцитарного ростка кроветворения с визуализацией промоноцитов и монобластов, повышенное количество макрофагальных клеток (7%), сужение красного ростка кроветворения.
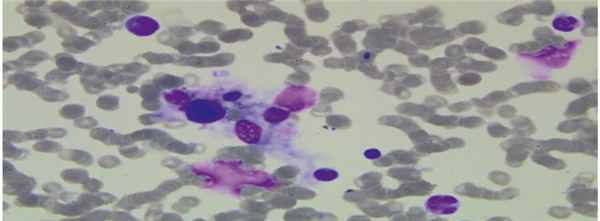
По морфологическим данным больной выставлен диагноз ГЛГ, который подтвержден в клинике педиатрической онкологии, гематологии и клинической иммунологии г. Дюссельдорфа ( рис. ).
Рис. Процесс гемофагоцитоза в мазке костного мозга больной
Согласно Федеральным клиническим рекомендациям [1] , диагностика ГЛГ основывается на наличии 5 из 8 диагностических признаков (лихорадка, спленомегалия, панцитопения, гипертриглицеридемия или гипофибриногенемия, гемофагоцитоз в мазке костного мозга, лимфатических узлах или ликворе, низкая или отсутствующая активность естественных киллерных клеток, гиперферритинемия (> 500 мкг/л), увеличение уровня растворимого CD25 в сыворотке > 2500 Ед/л). Для диагностики первичного ГЛГ в дополнение к определению диагностических критериев проводится молекулярно-генетическое исследование.
В случае нашей больной диагноз ГЛГ был поставлен на основании наличия у нее лихорадки, спленомегалии, гиперферритинемии (уровень ферритина > 2000 нг/мл), панцитопении и гемофагоцитоза в мазке костного мозга. По балльной шкале точность диагностики гемофагоцитарного синдрома в случае нашей больной составила 98% [10] .
В описываемом случае диагноз ГЛГ рассматривался нами как вторичный по отношению к тяжелой пневмонии с мультиорганной дисфункцией.
Наши данные совпадают с данными литературы [3, 4, 7, 8] , где указывается, что наиболее часто ГЛГ ассоциируется с вирусными инфекциями. При этом более 50% составляет герпесвирусная инфекция: как первичная, так и реактивация вирусом Эпштейна — Барр (у больной выявлены вирус Эпштейна — Барр и парвовирус В19).
Согласно Федеральным клиническим рекомендациям и HLH-2004 протоколу, лечение ГЛГ основано на назначении кортикостероидной терапии в сочетании с этопозидом и циклоспорином А (при ее эффективности длительность назначения составляет 8 недель) [1, 11] , при отсутствия эффекта показано проведение химиотерапии
или трансплантации костного мозга [9] . В случае вторичного ГЛГ, ассоциированного с вирусной инфекцией или с онкологией, терапия синдрома может быть начата с лечения причины, приведшей к активации иммунной системы, и лишь при необходимости подключается хемоиммунотерапия [1, 11] .
Поскольку нет единого подхода к лечению вторичного ГЛГ, был проведен консилиум и решено начать лечение со стероидной терапии (дексаметазона) и внутривенного введения иммуноглобулина, а при отсутствии эффекта в течение первой недели подключить цитостатики. Лечение начато сразу после обнаружения в мазке костного мозга активных макрофагов.
Уже на 5-е сутки терапии отмечалась положительная клиническая и параклиническая динамика, в связи с чем принято решение воздержаться от агрессивной иммуносупрессии.
Практически через месяц при отсутствии признаков дисфункции органов и нормализации показателей ребенок был выписан из стационара. В катамнезе (10 месяцев) у больной не отмечались гематологические отклонения.
В Армении молекулярно-генетическое исследование не проводится, но родителям даны рекомендации по проведению данного исследования за рубежом.
При гемофагоцитарном лимфогистиоцитозе имеется высокий риск летального исхода, поэтому очень важны ранняя диагностика и своевременно начатая терапия. Особенность данного случая заключается в том, что нам удалось быстро поставить диагноз, рано начать лечение и получить полную ремиссию без агрессивной иммуносупресии.
Гемофагоцитарный лимфогистиоцитоз
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы;
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы;
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова», Минздрава России
Клинико-морфологическая диагностика вторичного гемофагоцитарного лимфогистиоцитоза
Журнал: Архив патологии. 2020;82(6): 44‑49
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы
Вторичный гемофагоцитарный лимфогистиоцитоз (гемофагоцитарный синдром, синдром массивной активации макрофагов) является редким, агрессивным, тяжело протекающим синдромом с частым смертельным исходом. Патогенез синдрома, роль вирусной инфекции, развитие чрезмерной, аберрантной, воспалительной реакции, приводящей к полиорганной недостаточности и сепсису, остаются до конца не выясненными. В статье приведены данные литературы и описано собственное наблюдение вторичного гемофагоцитарного лимфогистиоцитоза у девочки 14 лет, перенесшей вирусную инфекцию Эпштейна—Барр и умершей вследствие тяжелой полиорганной недостаточности. Прижизненная диагностика была основана на типичной клинической картине заболевания, выявлении вируса методом полимеразной цепной реакции, определении уровня ферритина в крови. Показаны критерии современной морфологической диагностики (иммуногистохимические реакции с моноклональными антителами к EBV, CD68; in situ-гибридизация с помощью зонда EBER) и проведены клинико- и патолого-анатомические сопоставления.
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы;
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы;
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России
ГБУЗ «Морозовская детская городская клиническая больница» Департамента здравоохранения Москвы
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова», Минздрава России
Дата принятия в печать:
Первые сведения о гемофагоцитарном лимфогистиоцитозе (ГФЛГ) относятся к 1939 г., когда в журнале «The Lancet» R. Scott и A. Robb-Smith описали пролиферацию гистиоцитов с гемофагоцитозом в четырех секционных наблюдениях заболеваний неясного генеза, поскольку не исключалась гематологическая природа болезней, а в 1938 г. A. Robb-Smith предложил классификацию ретикулезов и ретикулосарком, авторы сочли возможным обозначить обнаруженную ими гистиоцитарную пролиферацию с гемофагоцитозом как «гистиоцитарный медуллярный ретикулез» [1].
Согласно современной классификации, выделяют первичный (семейный) лимфогистиоцитоз и вторичный гемофагоцитарный синдром (вторичный гемофагоцитарный лимфогистиоцитоз). Синонимы ГФЛГ: гемофагоцитарный синдром, синдром массивной активации макрофагов, гипервоспалительный синдром, гиперферритинемический синдром [2].
Первичный ГФЛГ — редкое аутосомно-рецессивное гематологическое заболевание (1 случай на 50 000 новорожденных в год) с установленными генетическими локусами, связанными с дефектами механизмов клеточной цитотоксичности. Заболевание характерно для раннего детского возраста; его дебют, как правило, совпадает с банальной инфекцией [3]. Вторичный ГФЛГ развивается в более позднем детском возрасте, он также является относительно редким, агрессивным, тяжело протекающим синдромом с частым смертельным исходом. ГФЛГ осложняет течение инфекционных, системных аутоиммунных и целого ряда онкологических заболеваний, а также первичных и приобретенных иммунодефицитов. Установлено, что ГФЛГ может быть ассоциирован с некоторыми первичными иммунодефицитами: Х-сцепленным лимфопролиферативным синдромом (XLP 1, 2, 3 типов), синдромами Chédiak—Higashi, Griscelli, Hermansky—Pudlak [4]. Из приведенных первичных иммунодефицитов Х-сцепленные лимфопролиферативные синдромы первого и второго типов могут проявляться как атипичная реакция на инфекцию вирусом Эпштейна—Барр (ЭБВ) с развитием фульминантного мононуклеоза и ГФЛГ; заболевание встречается только у мальчиков с частотой 1—3 случая на 1 000 000 новорожденных [4].
Кроме того, при COVID-19 во время пандемии 2020 г. у взрослых было установлено развитие клинических проявлений, дисбаланса врожденных и приобретенных иммунных реакций с аберрантной активацией макрофагов и со снижением иммунитета, а также и морфологических изменений, похожих на соответствующие признаки ГФЛГ в педиатрической практике [7—9]. При этом C. Bryce и соавт. [8] приходят к заключению о кардинальном значении развития изменений, подобных ГФЛГ у детей, в патогенезе тяжелых форм COVID-19.
Патогенез ГФЛГ, роль вирусной инфекции, развитие чрезмерной, аберрантной, воспалительной реакции, приводящей к полиорганной недостаточности и сепсису, остаются до конца не выясненными. Было установлено, что во время первичного инфицирования ЭБВ поражает В-клетки и реплицируется в них. ЭБВ-специфические цитотоксические Т-клетки необходимы для элиминации инфицированных В-клеток и продукции клеток памяти. Атипическая воспалительная реакция, согласно данным литературы, наиболее часто объясняется нарушениями функции и/или дефицитом активации NK-клеток и цитотоксических T-клеток, что в свою очередь приводит к активации и пролиферации лимфоцитов и макрофагов (гистиоцитов) и соответственно к неконтролируемому гемофагоцитозу и избыточной продукции цитокинов («цитокиновому шторму») и маркеров воспаления [5]. Обнаружение макрофагов (гистиоцитов) с признаками гемофагоцитоза в лимфатических узлах, селезенке, костном мозге и возможных других органах-мишенях в условиях чрезмерной системной воспалительной реакции, ассоциированной с вирусной инфекцией Эпштейна—Барр, является доказательным признаком вторичного ГФЛГ.
Приводим собственное клиническое наблюдение.
У девочки 14 лет через 2 нед после перенесенного инфекционного мононуклеоза появились упорная лихорадка, желтушность кожного покрова и склер, геморрагическая сыпь на туловище и конечностях, увеличение размеров печени, селезенки и лимфатических узлов, гемолитическая анемия. В общем анализе крови: снижение уровня гемоглобина, повышение содержания тромбоцитов, лейкопения. Биохимический анализ крови выявил повышение общего билирубина, трансаминаз, щелочной фосфатазы, мочевины, креатинина, С-реактивного белка, ферритина. При определении антител к капсидному антигену вируса Эпштейна—Барр VCA (IgM) получен положительный результат — 17,6 Ед/мл. При прогрессировании заболевания у ребенка развились холестатический гепатит, острый холецистит, панкреатит, интерстициальный нефрит, двусторонний гидроторакс, асцит, анасарка, образовалась язва луковицы двенадцатиперстной кишки. В дальнейшем заболевание осложнилось двусторонней бронхопневмонией и гастроэнтероколитом. Смерть наступила через 1,5 мес от начала заболевания вследствие полиорганной недостаточности.
Посмертный клинический диагноз
Основное заболевание: D76.2. Гемофагоцитарный синдром, связанный с инфекцией (ЭБВ-инфекция).
Сопутствующие заболевания: язвенная болезнь луковицы двенадцатиперстной кишки; желудочное кровотечение.
Осложнения основного заболевания: двусторонняя полисегментарная пневмония, двусторонний гидроторакс, дыхательная недостаточность III степени. Сердечно-сосудистая недостаточность, гидроперикард. Холестатический гепатит умеренной степени активности смешанного генеза (токсическое поражение печени, острая ЭБВ-инфекция). Острая печеночно-клеточная недостаточность. Острая почечная недостаточность, анурия. Асцит. ДВС-синдром.
При аутопсийном исследовании кожный покров ярко-желтого цвета с распространенными кровоизлияниями в местах инъекций, также ярко-желтого цвета подкожно-жировая клетчатка, слизистые оболочки, интима крупных сосудов и внутренние органы. Распространенный отек подкожно-жировой клетчатки. В серозных полостях свободная прозрачная светло-желтая жидкость: в брюшной полости 950 мл, в правой плевральной полости 300 мл, в левой плевральной полости 150 мл. Серозные оболочки гладкие, блестящие, синюшно-желтые. Селезенка массой 425 г (норма 93 г), фолликулярный рисунок слабо выражен, соскоб пульпы скудный. Лимфатические узлы не увеличены. Печень массой 2565 г (норма 1125 г), плотноэластической консистенции, неоднородного желтовато-коричневого цвета. Почки массой 380 г (норма 190 г) с желтовато-коричневым корковым и мозговым веществом, фиброзная капсула снимается легко. Поджелудочная железа увеличена, массой 115 г (норма 50 г). Масса правого легкого 490 г (норма 330 г), левого легкого 445 г (норма 264 г), легкие однородного темно-красного цвета с желтоватым прокрашиванием, плотноэластической консистенции, мелкие кусочки из разных отделов легких погружаются в воду. В области луковицы двенадцатиперстной кишки язвенный дефект неправильной формы размером 2,5×1,8 см. Привратник проходим, в просвете двенадцатиперстной, тонкой и толстой кишки жидкая кровь. Слизистая оболочка желудка, тонкой и толстой кишки полнокровная, местами серо-желтого цвета, тусклая, шероховатая.
При морфологическом исследовании аутопсийного материала использовали окраску гистологических препаратов гематоксилином и эозином, ШИК-реакцию; иммуногистохимические реакции с моноклональными антителами к EBV, CD 68, in sit-гибридизацию с помощью зонда EBER. При световой микроскопии выявлены характерные для гемофагоцитарного синдрома, связанного с ЭБВ-инфекцией, изменения: лимфогистиоцитарная инфильтрация с гемофагоцитозом и выраженной макрофагальной реакцией в костном мозге, лимфатических узлах, селезенке, печени, почках, поджелудочной железе с развитием активного интерстициального гепатита с жировой дистрофией печени, интерстициального нефрита, панкреатита (рис. 1, а—е). При иммуногистохимическом исследовании в клеточных инфильтратах положительное иммунное окрашивание с моноклональными антителами к CD68 (рис.1, б); с моноклональными антителами к EBV (рис. 1, в); in situ-гибридизация с помощью зонда EBER выявила клетки, инфицированные ЭБВ (рис. 1, г, е). В вилочковой железе картина выраженной акцидентальной трансформации с исходом в приобретенную атрофию органа. При окраске гематоксилином и эозином и ШИК-реакции обнаружен кандидоз легких и пищеварительного тракта (рис. 2, а—е).
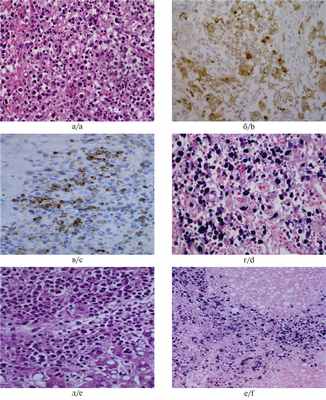
Рис. 1. Лимфогистиоцитарная инфильтрация с выраженной макрофагальной реакцией и гемофагоцитозом в лимфатических узлах (а—г) и печени (д—е).
а, д — окраска гематоксилином и эозином; б — иммунное окрашивание с антителом к CD68; в — иммунное окрашивание с антителом к EBV; г, е — клетки инфицированы вирусом Эпштейна—Барр; in situ-гибридизация с помощью зонда EBER; а—д — ×400; е — ×200.
Fig. 1. Lymphohistiocytic infiltration with a pronounced macrophage response and hemophagocytosis in the lymph nodes (a—d) and liver (e—f).
a, e — H&E; b — immune staining with anti-CD68 antibody; c — immune staining with anti-EBV antibody; d, f — Epstain—Barr encoded RNA (EBER) in situ hybridization; a—e — ×400; f — ×200.
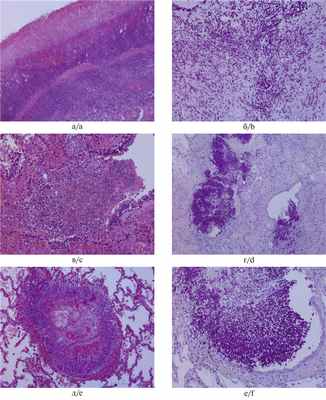
Рис. 2. Кандидозный язвенный гастроэнтероколит (а—б); кандидозная бронхопневмония (в—е).
а, в, д — окраска гематоксилином и эозином; б, г, е — ШИК-реакция; а, в, д, е — ×200; б — ×400; г — ×100.
Fig. 2. Candidal ulcerative gastroenterocolitis (a—b); candidal bronchopneumonia (c—f).
a, c, e — H&E; b, d, f — periodic acid Schiff reaction; a, c, e, f — ×200; b — ×400; d — ×100.
Патолого-анатомический диагноз
Основное заболевание: гемофагоцитарный синдром, связанный с ЭБВ-инфекцией (по данным молекулярно-биологического исследования EBV в крови методом ПЦР: ДНК Epstein—Barr virus — Ig 4,35. 10 5 клеток): макрофагальная реакция и гемофагоцитоз в костном мозге, лимфатических узлах, селезенке; активный интерстициальный гепатит, нефрит, панкреатит; желтуха кожного покрова, слизистых оболочек, интимы крупных сосудов, внутренних органов.
Осложнения основного заболевания: двусторонняя полисегментарная кандидозная бронхопневмония; эрозивно-язвенный кандидозный гастрит, энтерит, колит; язва луковицы двенадцатиперстной кишки; двусторонний гидроторакс, асцит, анасарка; отек мягких мозговых оболочек и вещества головного мозга.
Непосредственная причина смерти: полиорганная недостаточность.
Код по МКБ-Х [D76.2]: гемофагоцитарный синдром, связанный с инфекцией.
Таким образом, клинико-морфологическая диагностика вторичного гемофагоцитарного лимфогистиоцитоза в представленном наблюдении основывается на данных анамнеза — связь дебюта заболевания с острой ЭБВ-инфекцией, наличие типичной клинико-лабораторной картины заболевания, результаты клинико-морфологических сопоставлений. Критериями современной морфологической диагностики представленного заболевания являются использование иммуногистохимических реакций с моноклональными антителами к EBV, CD68; in situ-гибридизация с помощью зонда EBER. Возникшая необратимая тяжелая полиорганная недостаточность обусловлена также развитием висцерального кандидоза, характерного осложнения заболеваний с приобретенным дефектом клеточного звена иммунитета.
Гемофагоцитарный лимфогистиоцитоз
Гемофагоцитарный лимфогистиоцитоз - это заболевания наследственного или приобретённого характера, которые развиваются на фоне нарушения регуляции иммунного ответа с последующей выработкой гистиоцитов и Т—лимфоцитов, обладающих цитотоксическим действием.
Причины заболевания
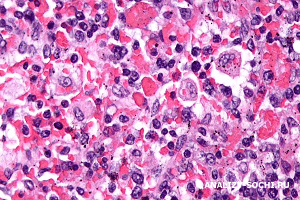
Для первичного патологического процесса характерно нарушение регулярной роли иммунных клеток. Постепенно происходит увеличение объема провоспалительных цитокинов. Лимфоциты накапливаются в различных тканях.
Вторичный процесс развивается на фоне инфекции, роста опухолевых клеток и аутоиммунных патологий. Реже данное заболевание появляется после трансплантации органов.
Симптомы и первые признаки
Заболевание характеризуется многообразием клинических проявлений. Пациенты предъявляют жалобы на ухудшение самочувствия с развитием слабости, недомогания, а также температуры, которая не устраняется после применения лекарственных средств.
Прогрессирует увеличение печени и селезенки. На кожных покровах появляются высыпания полиморфного характера. Нарушение функционирования нервной системы выражается повышенной возбудимостью, раздражительностью или развитием судорожного синдрома.
Постепенно происходит увеличение лимфатических узлов, из-за анемии кожа становится бледной с возможной желтушностью.
Диагностика

Для постановки диагноза важно оценить жалобы, условия их появления, сопутствующие патологии, данные анамнеза, а также данные проведённых исследований.
- Повышенную температуру тела, сохраняющуюся более 7 дней.
- Гепатоспленомегалию.
- Снижение уровня гемоглобина, тромбоцитов, а также нейтрофилов.
- Увеличение ферритина.
- Возрастание количества sCD25.
- Недостаточную активность NK—клеток или их полное отсутствие.
Помимо анализов крови пациентам назначают ультразвуковое сканирование внутренних органов, компьютерную томографию и эндоскопические исследования. В данном случае удается выявить структурные изменения и оценить степень тяжести патологического процесса.
Лечение и профилактика
Лечение заключается в проведении химиотерапии, которая включает применение иммуносупрессивных средств и трансплантацию родственного материала. Для заболеваний, причиной которым послужила инфекция, назначают антибиотики, инфузионные средства, введение иммуноглобулина и иммуносупрессивных препаратов.
Своевременно назначенная терапия позволяет на длительное время улучшить самочувствие и остановить прогрессирование заболевания.
Россия, Краснодарский край,
г. Сочи, ул. Старонасыпная, 22.
Размещенная на сайте информация и прейскурант не являются публичной офертой.
Читайте также: