Генетика фенилкетонурии. Наследование
Добавил пользователь Алексей Ф. Обновлено: 27.01.2026
Фенилкетонурия это нарушение метаболизма аминокислот Обзор нарушений метаболизма аминокислот и органических кислот (Overview of Amino Acid and Organic Acid Metabolism Disorders) Почки активно реабсорбируют значительные количества аминокислот. Дефекты переноса аминокислот в почечной канальце включают цистинурию и болезнь Хартнупа, которые обсуждаются в другом месте. Прочитайте дополнительные сведения , приводящее к возникновению клинического синдрома умственной отсталости с когнитивными и поведенческими расстройствами, вызванными повышенным уровнем фенилаланина. Основной причиной является недостаточность активности фенилаланин гидроксилазы. Диагноз ставят на основании выявления высокого уровня фенилаланина при нормальном или низком содержании тирозина. Лечение – пожизненная диета без фенилаланина. Прогноз при лечении отличный.
Для получения информации о других нарушениях аминокислот, см. таблицу нарушения обмена фенилаланина и тирозина Нарушения метаболизма фенилаланина и тирозина . См. также Подходы к лечению пациентов с подозрением на наследственное нарушение обмена веществ (Approach to the Patient With a Suspected Inherited Disorder of Metabolism) Подходы к лечению пациентов с подозрением на наличие наследственного заболевания обмена веществ Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения .
Патофизиология фенилкетонурии
Избыточно потребляемый фенилаланин (т.е. не использующийся для синтеза белка), как правило, преобразуется в тирозин фенилаланингидроксилазу; тетрагидробиоптерин (BH4) является необходимым кофактором в данной реакции. Когда одна из нескольких мутаций гена приводит к недостаточности или отсутствию фенилаланингидроксилазы, происходит накопление потребляемого фенилаланина; мозг является основным повреждаемым органом, возможно, из-за нарушения миелинизации.
Некоторый избыток фенилаланина метаболизируется в фенилкетоны, которые выводятся из организма с мочой, что приводит к развитию фенилкетонурии. Степень дефицита фермента и, следовательно, тяжести гиперфенилаланинемии варьируется среди пациентов в зависимости от конкретной мутации.
Варианты заболевания
Хотя почти все случаи (98–99%) ФКУ развиваются в результате дефицита фенилаланингидроксилазы, фенилаланин также может накапливаться, если BH4 не синтезируется из-за недостаточности дигидроптерин синтазы или не регенерируется из-за недостаточности дигидропиридин редуктазы. Кроме того, поскольку BH4 также является кофактором тирозингидроксилазы, участвующей в синтезе дофамина и серотонина, дефицит BH4 изменяет синтез медиаторов, вызывающих неврологические симптомы независимо от накопления фенилаланина.
Симптомы и признаки фенилкетонурии
Большинство детей с фенилкетонурией при рождении нормальные, симптомы и признаки появляются постепенно, в течение нескольких месяцев, по мере накопления фенилаланина. Отличительная черта нелеченой ФКУ – тяжелая умственная отсталость. У детей также возникают крайняя гиперактивность, нарушение походки, психозы, часто возникает неприятный «мышиный» запах тела, обусловленный фенилуксусной кислотой (продукт распада фенилаланина) в моче и поте. Кроме того, дети, как правило, имеют более светлую кожу, волосы и цвет глаза, чем непоражённые члены семьи, и у некоторых из них болезнь может проявляться сыпью, похожей на младенческую экзему.
Диагностика ФКУ
Рутинный неонатальный скрининг
(См. также the American College of Medical Genetics and Genomics Therapeutic Committee's 2013 diagnosis and management guidelines for phenylalanine hydroxylase deficiency.)
В США и многих развитых странах, всем новорожденным проводят скрининг на фенилкетонурию Скрининговые тесты для новорождённых Рекомендации по скринингу новорожденных зависят от клинических условий и государственных стандартов. Определение группы крови показано, если мать имеет кровь группы 0 или резус-отрицательную. Прочитайте дополнительные сведения в течение 24-48 часов после рождения, проводя один из нескольких анализов крови Начальное тестирование Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения ; отклоняющиеся от нормы результаты подтверждают непосредственным измерением уровня фенилаланина. В классической ФКУ уровни фенилаланина у новорожденных часто составляют > 20 мг/дл (1,2 ммоль/л). Те, у кого есть частичный недостаток, как правило, имеют уровни 8–10 мг/дл, в то время как на обычной диете уровни > 6 мг/дл уже требуют лечения; для отличия от классической ФКУ требуется анализ мутаций, идентифицирующий незначительные мутации гена или, реже, измерение активности фенилаланин-гидроксилазы печени, которое показывает уровень активности в 5–15% от нормального.
Дефицит BH4 отличается от других форм ФКУ повышенными концентрациями биоптерина или неоптерина в моче, крови, спинномозговой жидкости или во всех 3 жидкостях; также можно использовать генетическое тестирование. Распознавание важно, биоптериновый профиль мочи при первичной диагностике нужно определять регулярно, поскольку стандартное лечение ФКУ не предотвращает неврологические повреждения.
Диагностику у детей в семьях с положительным семейным анамнезом можно проводить пренатально с помощью прямого исследования мутации после биопсии хориона или амниоцентеза.
Прогноз при ФКУ
Адекватное лечение, начатое в первые дни жизни, предотвращает возникновение тяжелых проявлений заболевания. Тем не менее умеренный когнитивный дефицит и проблемы с психическим здоровьем все еще могут возникать даже при хорошем диетическом контроле. Лечение, начатое после 2–3 лет, может быть эффективно только в контроле крайней гиперактивности и некупируемых судорог.
Дети, рожденные от матерей с плохо контролируемой ФКУ (т.е. имеющих высокий уровень фенилаланина), во время беременности имеют высокий риск микроцефалии и дефектов развития.
Лечение ФКУ
Ограничение потребления фенилаланина
Лечение фенилкетонурии заключается в соблюдении пожизненной диеты без фенилаланина. Все природные белки содержат около 4% фенилаланина. Поэтому основные продукты питания включают
Натуральные продукты с низким содержанием белка (например, фрукты, овощи, некоторые злаки)
Белковые гидролизаты, из которых удален фенилаланин
Смеси аминокислот без фенилаланина
Примерами коммерчески доступных продуктов без фенилаланина являются продукты PKU Anamix ® (для младенцев), XPhe Maxamaid ® (для детей от 1 до 8 лет), XP Maxamum ® (для детей > 8 лет); Phenex ® -1 и Phenex ® -2; Phenyl-Free ® 1 и Phenyl-Free ® 2; PhenylAde ® (различные); PKU Lophlex ® LQ; и Phlexy-10 ® (различные формы). Некоторое количество фенилаланина необходимо для роста и метаболизма; эта потребность удовлетворяется за счет измеряемых количеств природного белка с молоком или фруктами с низким содержанием белка.
Требуется частый мониторинг уровней фенилаланина в плазме; рекомендуемые значения для всех детей располагаются в пределах 2–6 мг/дл (120–360 мкмоль/л). Планирование диеты и управление ею должно войти в привычку у женщин детородного возраста до беременности – так они помогут своим будущим детям. Тирозин используют чаще, поскольку это незаменимая аминокислота у пациентов с ФКУ. Кроме того, всем пациентам с дефицитом фенилаланин-гидроксилазы необходимо провести пробное назначение сапроптерина, назначение которого может иметь преимущества.
Для лиц с BH4-недостаточностью лечение также включает тетрагидробиоптерин по 1–5 мг/кг перорально 3 раза в день, леводопу, карбидопу и 5-OH триптофан и фолиевую кислоту по 10–20 мг перорально 1 раз в день при дефиците дигидроптерин-редуктазы. Тем не менее цели лечения и подход являются такими же, как при ФКУ.
Ключевые моменты
ФКУ возникает как последствие, когда одна из нескольких мутаций гена приводит к недостаточности или отсутствию фенилаланингидроксилазы, вслседствие чего происходит накопление потребляемого с пищей фенилаланина; мозг является основным повреждаемым органом, возможно, из-за нарушения миелинизации.
Фенилкетонурия (ФКУ) вызывает клинический синдром умственной отсталости с когнитивными и поведенческими нарушениями; при отсутствии лечения развивается тяжелая умственная отсталость.
В США и многих развитых странах, всех новорожденных обследуют на фенилкетонурию в течение 24-48 часов после рождения, проводя один из нескольких анализов крови; отклоняющиеся от нормы результаты подтверждают непосредственным измерением уровня фенилаланина.
Лечение заключается в пожизненном ограничении поступления фенилаланина с пищей; адекватное лечение, начатое в первые дни жизни, предотвращает возникновение многих проявлений этого заболевания.
Несмотря на то, что при наличии лечения прогноз благоприятный, требуется частый мониторинг уровней фенилаланина в плазме; рекомендуемые значения располагаются в пределах 2–6 мг/дл (120–360 мкмоль/л) для всех детей.
Дополнительная информация
Ниже следуют англоязычные ресурсы, которые могут быть информативными. Обратите внимание, что The manual не несет ответственности за содержание этих ресурсов.
American College of Medical Genetics and Genomics Therapeutic Committee: Diagnosis and management guidelines for phenylalanine hydroxylase deficiency (2013)
Online Mendelian Inheritance in Man® (OMIM®) database: полная информация о генах и их молекулярной и хромосомной локализации
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Генетика фенилкетонурии. Наследование

Прочтите и возьмите себе на заметку, особенно если вы молодые люди
В России уже много лет проводится массовое обследование новорожденных для выявления у них нескольких наследственных заболеваний. Такое обследование проводится во многих странах и называется скринингом новорожденных или неонаталъным скринингом.
Целью скрининга новорожденных является, конечно, не само выявление новорожденных с еще не проявившимися наследственными заболеваниями, а их лечение, которое позволяет предотвратить появление клинических симптомов, во многих случаях весьма тяжелых, или даже фатальных. В результате рано начатого и аккуратно проводимого лечения вместо тяжело больных детей, а затем подростков и взрослых, получаются здоровые люди, полноценные члены общества, нередко являющиеся гордостью семьи.
Скрининг новорожденных в России ведется в отношении 5 наследственных и врожденных заболеваний: фенилкетонурии, гипотиреоза, галактоземии, адрено-гениталъного синдрома и муковисцидоза.
ЧТО ТАКОЕ ФЕНИЛКЕТОНУРИЯ?
Фенилкетонурия, которую сокращенно называют ФКУ, является одной из наследственных болезней обмена веществ, обусловленной изменением (мутацией) в определенном гене. ФКУ не очень редкое заболевание. В России оно встречается в среднем с частотой 1 на 7000 новорожденных.
КАК НАСЛЕДУЕТСЯ ФЕНИЛКЕТОНУРИЯ?
Наследуется ФКУ по аутосомно-рецессивному типу, т.е. больные накапливаются в семье в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок, больной ФКУ. На родословной мужчины обозначены квадратиком, а женщины - кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома из 23 пар, имеющихся у человека. Эта хромосома несет нормальный или дефектный (мутантный) ген фенилкетонурии, последний помечен черной точкой.
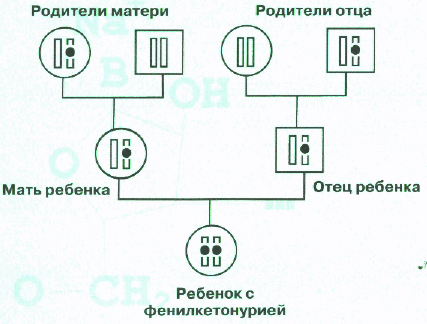
На рисунке для простоты изображена только хромосома, содержащая ген, мутации в котором вызывают ФКУ. У ребенка в обеих хромосомах содержится мутантный ген и поэтому он болен. У каждого из родителей мутантный ген содержится только в одной хромосоме, а вторая хромосома нормальная и поэтому они здоровы. Такие люди, которые имеют один нормальный и один дефектный ген, называются носителями мутантного гена.
У бабки по матери мутантный ген также имеется только в одной хромосоме, как и у деда со стороны отца. Они, как и родители больного ребенка, здоровы, но передали хромосомы, содержащие мутантный ген, своим детям. У вторых деда и бабки обе хромосомы содержат только нормальный ген. Таким образом, при рецессивном наследовании болен только тот член семьи, который получил от своих родителей обе хромосомы, несущие мутантный ген. Все остальные члены семьи здоровы, в том числе и те, кто является носителем мутантного гена. На представленном фрагменте родословной видно, что у родителей больного ребенка могут еще появиться больные дети. Вероятность появления больного ребенка в семьях, в которых родители являются носителями мутантного гена, составляет 1/4 или 25%. Эта вероятность не меняется от числа больных или здоровых детей в семье: для каждого следующего ребенка риск, что он будет болен, составляет 25%. Вероятность рождения здорового ребенка, обе хромосомы которого содержат только нормальный ген, составляет также 25%. Вероятность появления детей, у которых будет один нормальный и один дефектный ген, составляет 50%, они, как и их родители, будут здоровыми носителями мутантного гена. Многие родители больных ФКУ детей и их родственники, первый раз встретившись с врачом-генетиком, настойчиво повторяют, что у их ребенка не наследственное заболевание, так как в их семье ни у кого из родственников никогда не было такого заболевания. В этом случае только доступное объяснение о том, что правила наследования бывают разные, и не редко больной с наследственным заболеванием бывает единственным в семье, позволяют родителям понять, с какой ситуацией они столкнулись.
ПОЧЕМУ РЕБЕНОК МОЖЕТ ЗАБОЛЕТЬ ФЕНИЛКЕТОНУРИЕЙ?
ФКУ обусловлена мутацией в гене, который отвечает за синтез фермента, участвующего в превращении аминокислоты фенилаланина в тирозин. В результате мутации в гене фермент оказывается дефектным, фенилаланин не может превратиться в тирозин и накапливается в крови. Возникает так называемый метаболический блок. Фенилаланин, как и тирозин, являются аминокислотами, а аминокислоты - это те кирпичики, из которых построены все белки. Так как новорожденный постоянно получает с пищей белки (в молоке матери основным белком является казеин), то уровень фенилаланина постоянно растет и, наконец, достигает таких концентраций, при которых он становится токсичным, в первую очередь, для развивающегося мозга младенца.
КАКИЕ НАРУШЕНИЯ В ОРГАНИЗМЕ ВЫЗЫВАЕТ ФЕНИЛКЕТОНУРИЯ?
Без лечения у 95% младенцев с ФКУ развивается тяжелая умственная отсталость и задержка в моторном развитии. Дети не могут сидеть, стоять, ходить, и умственное развитие с возрастом продолжает снижаться. Кроме того, у больного могут появиться судороги, экзема на коже, а в старшем возрасте присоединяются грубые нарушения в поведении. У некоторых больных отмечается маленький размер головы и сердечные пороки.
ЧТО ТАКОЕ СКРИНИНГ НОВОРОЖДЕННЫХ НА ФЕНИЛКЕТОНУРИЮ?
Для того, чтобы избежать развития таких тяжелых клинических проявлений ФКУ нужно, чтобы новорожденный с повышенным содержанием фенилаланина в крови был выявлен в первые дни жизни. Для этого практически во всех странах мира существуют программы скрининга на ФКУ. В России скрининг новорожденных проводится в абсолютном большинстве территорий. Он заключается в том, что у новорожденного на 4 -5 день жизни перед выпиской из родильного дома берут из пятки несколько капель крови, которую наносят на специальную фильтровальную бумагу. Кровь высушивается, и такой бланк, на котором указана фамилия новорожденного и ряд других сведений, необходимых для его идентификации, переправляется в лабораторию региональной медико-генетической консультации. В лаборатории определяют содержание в крови уровня фенилаланина. Если уровень фенилаланина оказывается низким, то это означает, что у ребенка нет ФКУ. Если же уровень фенилаланина в крови высокий, то возникает подозрение нафенилкетонурию. В этом случае лаборатория запрашивает теперь уже в педиатрической службе, так как новорожденный выписан из родильного дома, повторное взятие крови у младенца. В этой связи родители узнают от педиатра, что первый тест на ФКУ у их ребенка оказался ненормальным. У них появляется повод для беспокойства. Повторное тестирование образца крови у ребенка является решающим. В большинстве случаев при повторном исследовании уровень фенилаланина оказывается нормальным. Это означает, что результат первого исследования был неверный (его называют ложноположительным). Причины этого могут быть связаны как с состоянием младенца на момент взятия крови, так и с какой-то ошибкой в анализе. Этот результат, свидетельствующий о том, что у ребенка нет ФКУ, сразу сообщается родителям, и они могут успокоиться.
ЧТО ДЕЛАТЬ, ЕСЛИ ДИАГНОЗ ФЕНИЛКЕТОНУРИИ ПОДТВЕРДИЛСЯ?
Если и при втором тестировании уровень фенилаланина остается высоким, то это означает, что ребенок болен ФКУ, и семья немедленно приглашается в медико-генетическую консультацию. Здесь родителям объясняют, что такое ФКУ, почему она возникла у их ребенка, и назначают лечение. Если лечение начато рано, то клинические симптомы ФКУ у ребенка не проявятся, и он будет расти здоровым, практически не отличаясь от сверстников. Смысл лечения заключается в уменьшении содержания фенилаланина в пище, которую получает ребенок. Обычно это достигается за счет специальных смесей, содержащих в необходимых количествах все незаменимые аминокислоты, за исключением фенилаланина. Успех лечения во многом определяется тем, насколько родители больного ребенка осознали важность диетотерапии, и насколько строго они ее выполняют. Обо всем этом и о многом другом семье расскажет врач-генетик во время первого визита семьи в медико-генетическую консультацию. Затем такие визиты станут регулярными. У ребенка будут постоянно контролировать содержание фенилаланина в крови, и, в зависимости от лабораторных показателей, корректировать состав тех продуктов, которые, с одной стороны, не будут повышать уровень фенилаланина, а с другой, обеспечивать нормальный рост и развитие ребенка. Постоянный контакт семьи с врачом-генетиком является залогом успешного лечения фенилкетонурии. В России диетотерапия для больных ФКУ проводится до возраста ребенка 7 - 8 лет, но многие родители продолжают ее и дольше.
МОЖНО ЛИ ПОМОЧЬ СЕМЬЕ, В КОТОРОЙ ПОЯВИЛСЯ БОЛЬНОЙ ФКУ, ИМЕТЬ ЗДОРОВЫХ ДЕТЕЙ?
Да, и довольно успешно. Для ФКУ возможна дородовая диагностика. Первым шагом в этом направлении является обращение в медико-генетическую консультацию, где врач-генетик определяет показания и возможные методические подходы к дородовой диагностике. В каждом конкретном случае решается вопрос о необходимости молекулярно-генетического обследования больного ребенка или родителей, а затем плода. Сама процедура заключается в том, что во время беременности в сроке 9-11 недель или 16-18 недель врач акушер-гинеколог проводит забор очень небольшого количества клеток плода, находящихся в околоплодной жидкости, плодных оболочках или крови плода, и направляет этот материал в специальную лабораторию пренатальной диагностики. В этой лаборатории врачи лаборанты-генетики проводят молекулярную диагностику, т.е. определяют наличие или отсутствие мутации в гене, отвечающем за фенилкетонурию. В случае положительного результата семья решает вопрос о прерывании беременности больным плодом или настраивается на появление еще одного больного ребенка. Это право выбора остается за семьей.
© 2022 краевое государственное бюджетное учреждение здравоохранения "Красноярский краевой медико-генетический центр" (КГБУЗ "ККМГЦ")
Фенилкетонурия
Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез фенилкетонурии
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко - врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси - Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет - Максамум-ХР и др. Основу диеты составляют низкобелковые продукты - фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям - ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия

Это наследственное заболевание также называют болезнью Феллинга и относят к группе ферментопатий, обусловленных нарушением метаболизма аминокислот, преимущественно фенилаланина. В случае несоблюдения низкобелковой диеты больные фенилкетонурией могут страдать от тяжелых поражений ЦНС вследствие накопления в организме фенилаланина и его токсических метаболитов.
Причины заболевания
Фенилкетонурия — генетическая патология с аутосомно-рецессивным характером наследования. Чаще всего болезнь Феллинга возникает при мутации гена, который кодирует фермент фенилаланин-4-гидроксилазу и расположен на длинном плече хромосомы 12 (локус12q22-q24). Эта мутация является причиной фенилкетонурии I типа, которая встречается в 98 % всех случаев.
Существуют также атипичные формы заболевания. Это фенилкетонурия II и III типов. При схожей симптоматике они не поддаются коррекции посредством диетотерапии.
Патогенез фенилкетонурии
Дефицит фермента фенилалаиин-4-гидроксилазы лежит в основе классической формы заболевания, он приводит к нарушению окисления поступающего с пищей фенилаланина. Его концентрация в крови и спинномозговой жидкости значительно возрастает, а уровень тирозина падает. Это приводит к нарушению миелинизации нервных волокон, снижению образования нейромедиаторов и запускает патогенетические механизмы задержки умственного развития и вызывает прогредиентное слабоумие.
Симптоматика фенилкетонурии
Манифестация болезни Феллинга происходит в возрасте 2–6 мес. Развиваются первые неспецифические симптомы: вялость, беспокойство и гипервозбудимость, мышечная дистония, срыгивание, судорожный синдром. Патогномоничным симптомом фенилкетонурии считается упорная рвота.
После 6 мес. наблюдается отставание ребенка в психомоторном развитии. Это проявляется как снижение активности, безучастность к окружающему, ребенок перестает узнавать родных, не делает попыток сесть или встать на ноги. Могут появляться шелушение кожи, экзема, дерматит, склеродермия.
Дети с фенилкетонурией, не получающие лечения, страдают от микроцефалии, прогнатии, позднего прорезывания зубов, гипоплазии эмали. Для них характерна задержка речевого развития, к трем–четырем годам развивается олигофрения с практически полным отсутствием речи.
Больные имеют диспластическое телосложение, отличаются светлой кожей, волосами и глазами. Верхние и нижние конечности согнуты в суставах (так называемая «поза портного»), походка шаткая, семенящая, может быть тремор рук, гиперкинезы.
Диагностика фенилкетонурии
Исследование на наличие болезни Феллинга входит в программу неонатального скрининга, который обязателен для всех новорожденных. Тест проводят на 3–5 днях жизни ребенка. При обнаружении гиперфенилаланинемии дают направление на генетическое исследование.
Для подтверждения наличия фенилкетонурии проводят:
- определение концентрации фенилаланина и тирозина в крови;
- анализ активности печеночных ферментов;
- биохимическое исследование мочи;
- МРТ головного мозга;
- ЭЭГ;
- обследование у детского невролога.
Генетический дефект может быть обнаружен еще до рождения в рамках неинвазивного пренатального ДНК-теста Panorama или неинвазивного пренатального ДНК-скрининга, которые можно пройти в медико-генетическом центре «Геномед». После рождения ребенка можно провести поиск мутаций в гене РАН (в том числе расширенный), сделать «Скрининг на наследственные заболевания», панель «Наследственные нарушения обмена веществ».
Лечение, прогноз и профилактика фенилкетонурии
Основа терапии — соблюдение диеты с низким содержанием белков. Комплексное лечение может включать в себя прием витаминно-минеральных комплексов, ноотропных средств, антиконвульсантов, а также ЛФК, массаж, иглорефлексотерапию.
При раннем обнаружении фенилкетонурии и назначении элиминационной диеты прогноз благоприятный. Поздно начатая терапия имеет неблагоприятный прогноз по отношению к умственному развитию.
Профилактика фенилкетонурии включает в себя пренатальное генетическое исследование. Женщины с болезнью Феллинга должны тщательно соблюдать диету до начала и во время беременности.
Фенилкетонурия
Фенилкетонурия (ФКУ) – распространенная в Европе и России врожденная ферментопатия, связанная с дефектом обмена фенилаланина (ФА). При дефекте фенилаланилгидроксилазы (ФАГ), расщепляющей ФА, активируются побочные пути его обмена, в результате чего образуются токсичные метаболиты. Их накопление приводит к поражению ЦНС и задержке умственного развития. Фенилкетонурия является одним из немногих наследственных заболеваний, клинические проявления которого можно свести к минимуму модификацией факторов среды. Соблюдение строгой безбелковой диеты с раннего возраста позволяет предотвратить развитие умственной отсталости у детей с диагнозом «ФКУ». Введение неонатального биохимического скрининга позволило выявлять больных детей в роддомах в течение первой недели с момента рождения и перенаправлять их для наблюдения у врачей-генетиков.
Гиперфенилаланинемия (ГФА) - группа нозологий, основным общим симптомом которых является повышение уровня ФА в крови (>2мг/дл) и при отсутствии специфического лечения задержка умственного и психомоторного развития. Основной причиной гиперфенилаланинемий является дефект гена PAH, кодирующего фермент ФАГ. Патология наследуется по аутосомно-рецессивному типу и обеспечивает 98% всех ГФА, ее называют фенилкетонурией или РАН-зависимой ГФА.
Диагностика патогенных вариантов гена РАН является актуальной для пациентов, так как позволяет делать прогнозы относительно клинического течения заболевания, чувствительности к кофакторной терапии, и необходимо для планирования деторождения в отягощенных семьях. В процессе изучения фенилкетонурии были выявлены клинические формы тетрагидробиоптеринзависимой ГФА, которые ранее называли «атипичной ФКУ». Мутации в генах, кодирующих белки синтеза и обмена кофактора ФАГ тетрагидробиоптерина (BH4), приводят к схожей с ФКУ клинике. Такие больные не реагируют на введение диеты, им необходима кофакторная терапия.
Больным с мутациями в генах синтеза и обмена BH4 показана заместительная терапия сапроптерином (фармакологический аналог BH4). В течение последних лет было показано, что такое лечение эффективно и для больных с мутациями в гене PAH, при наличии остаточной активности фермента ФАГ. Показано достоверное снижение уровня фенилаланина в крови больных ФКУ, использующих препараты синтетических аналогов кофактора фенилаланингидроксилазы. Малые дозы препарата позволяют существенно расширить диету и улучшить качество жизни больных ФКУ. Причем, чем больше остаточная активность фермента, тем больше эффект от лечения. С другой стороны, при классической ФКУ у больных с двумя тяжелыми мутациями, например, гомозигот по мутации R408W, лечение не эффективно. При остаточной активности ФАГ близкой к нулю, введение кофактора не дает клинически значимого эффекта. Ввиду высокой аллельной частоты мутации R408W и других тяжелых мутаций в России, а также того факта, что ДНК-диагностика является более дешевым и легко осуществимым мероприятием, чем нагрузочные тесты тетрагидробиоптерином, генотипирование с целью выявления пациентов-неответчиков и потенциальных ответчиков является актуальным.
В таблице представлена классификация частых мутаций гена PAH в зависимости от остаточной активности фермента. Красным выделены тяжелые мутации, зеленым – мягкие. При наличии в генотипе двух тяжелых мутаций в гомозиготном или в компаунд-гетерозиготном состоянии, лечение препаратами BH4 не эффективно. При наличии в генотипе хотя бы одной мягкой мутации, показаны нагрузочные тесты препаратами тетрагидробиоптерина.
Читайте также: